Synthesis of High Molar Mass Poly(phenylene methylene) Catalyzed by Tungsten(II) Compounds

 , and
, and 
Abstract
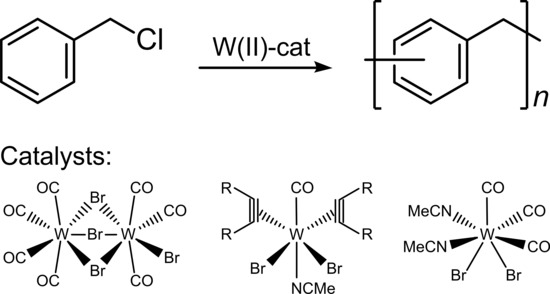
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Tungsten Compounds
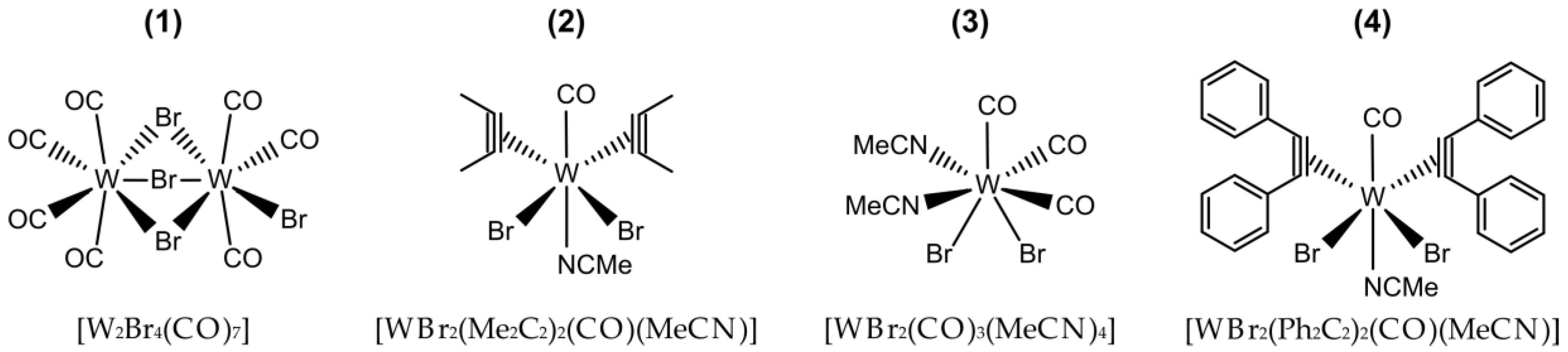
2.2.1. [W2Br4(CO)7]
2.2.2. [WBr2(CO)3(MeCN)2]
2.2.3. [WBr2(Me2C2)2(CO)(MeCN)]
2.2.4. [WBr2(Ph2C2)2(CO)(MeCN)]
2.3. Synthesis of Poly(phenylene methylene) with Tungsten Catalysts
2.4. Synthesis of PPM for Reaction-Time-Dependent Analysis
2.5. Fractionation by Phase Separation
2.6. Characterization
3. Results
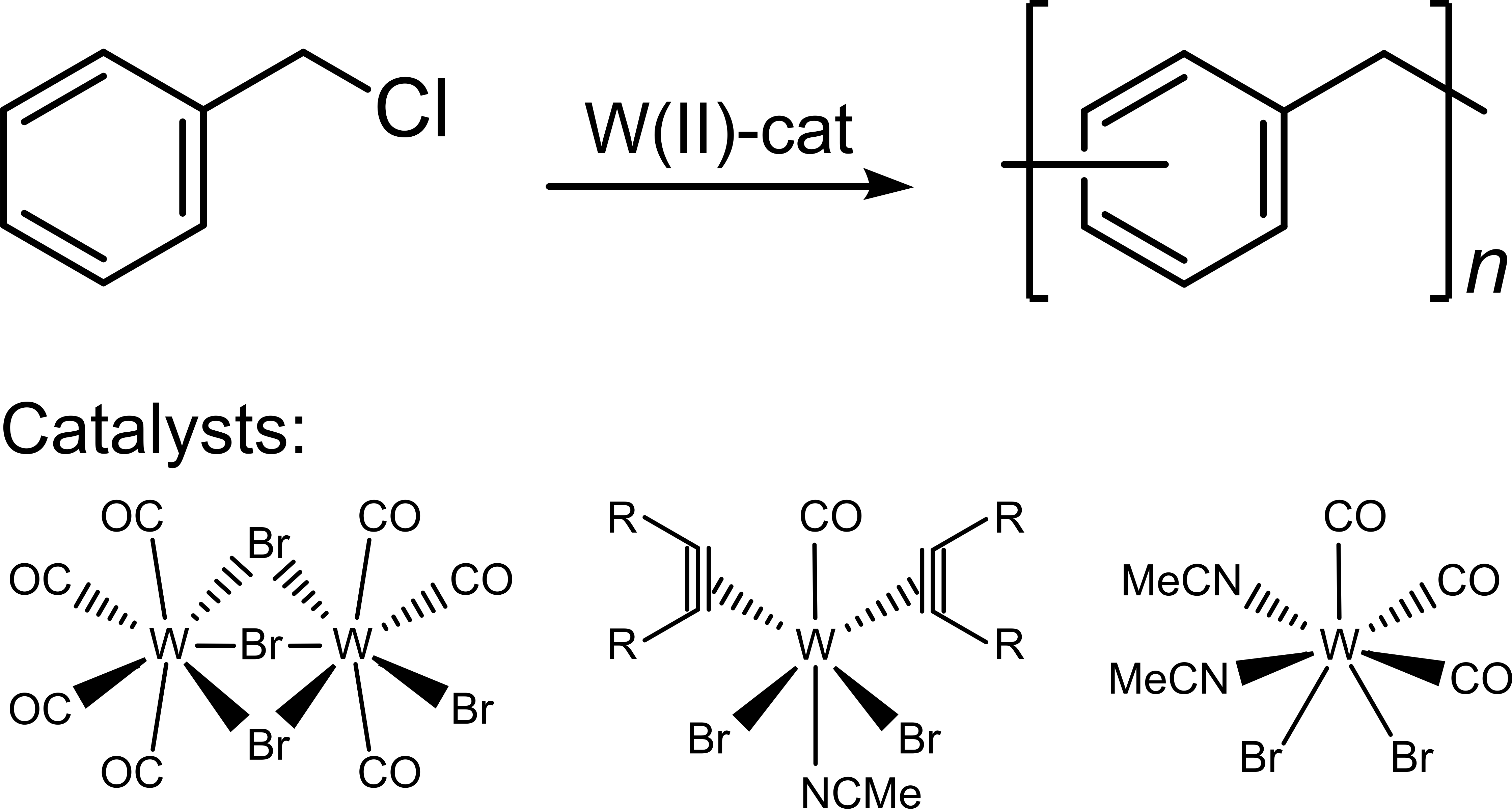
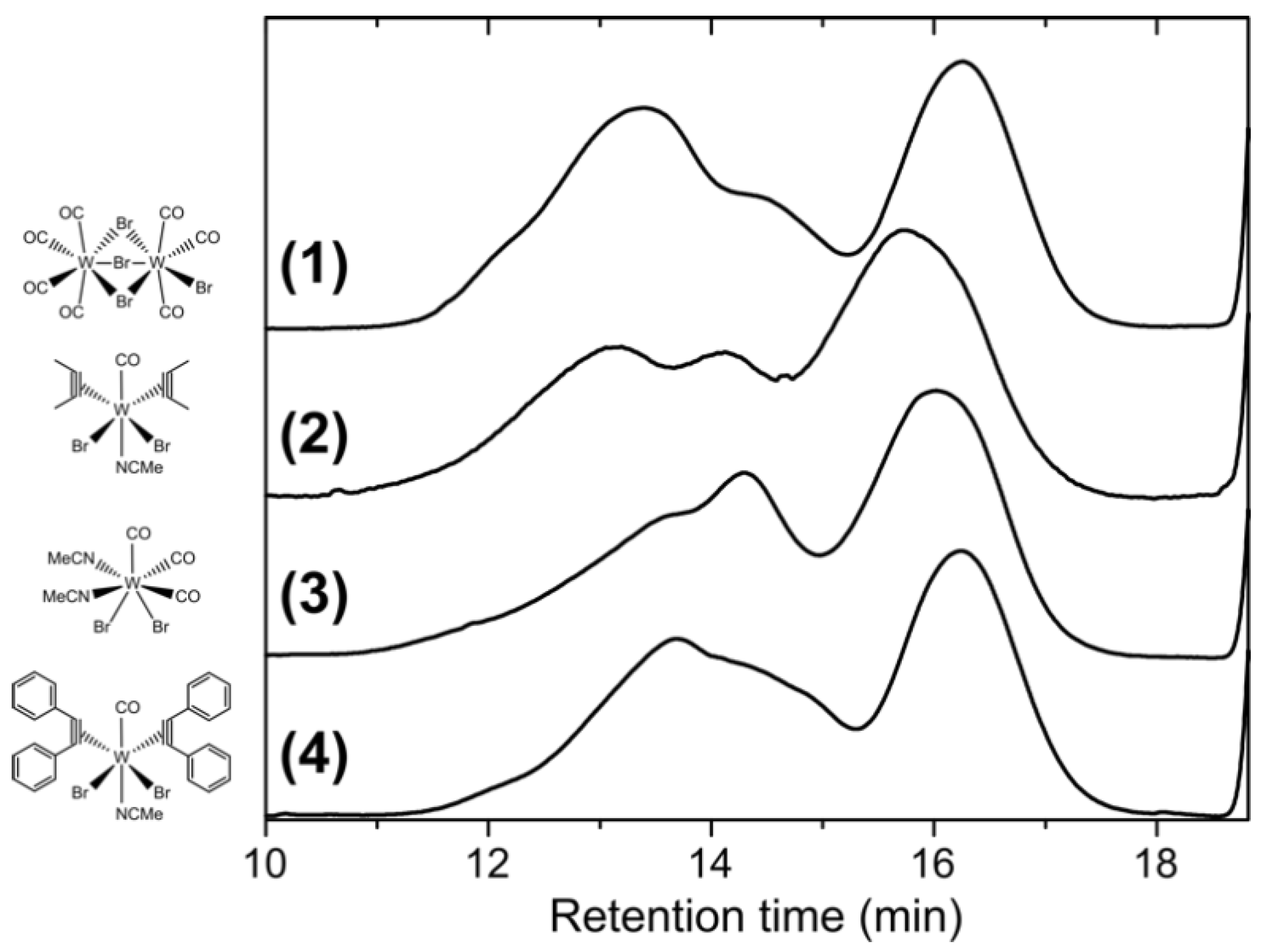
3.1. Polymerization
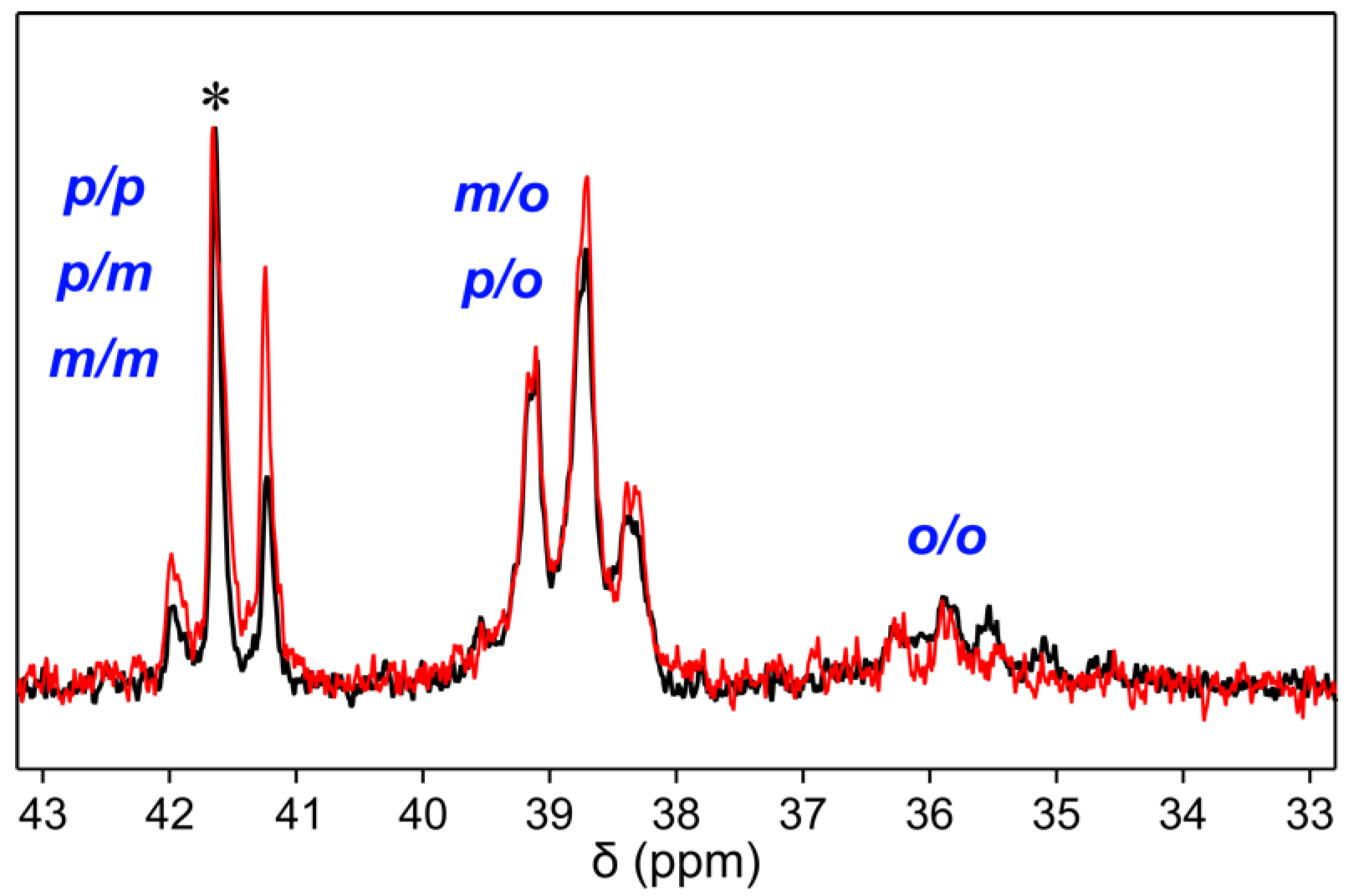
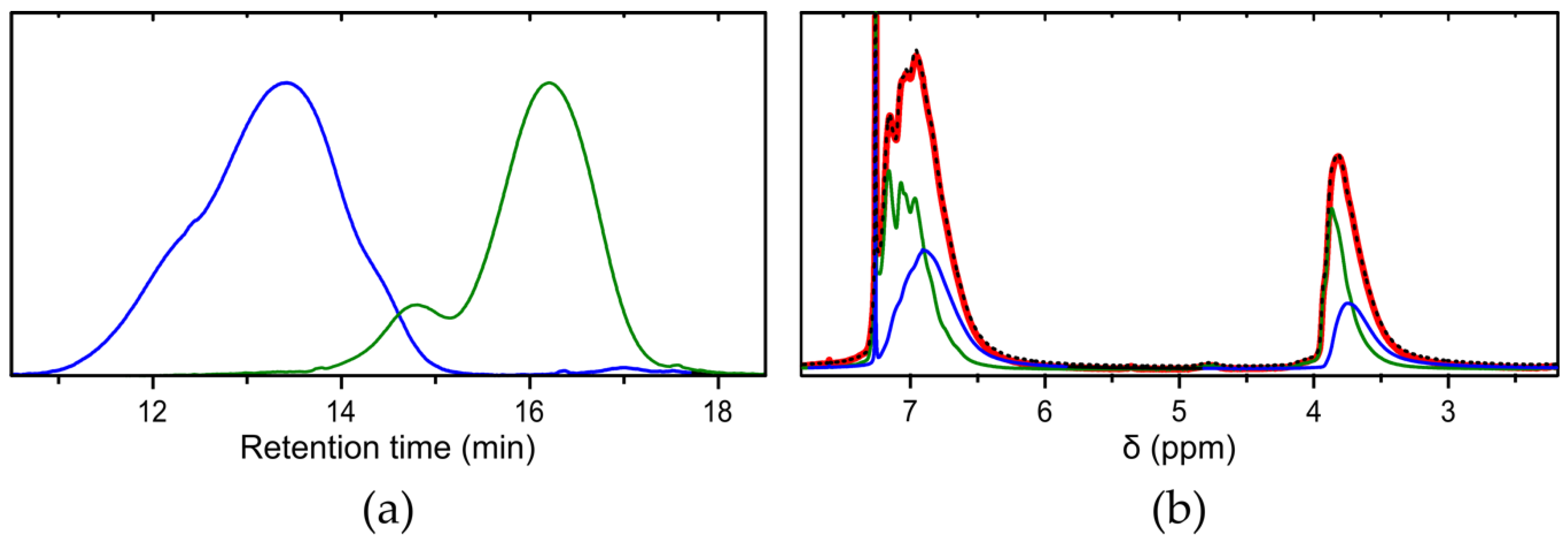
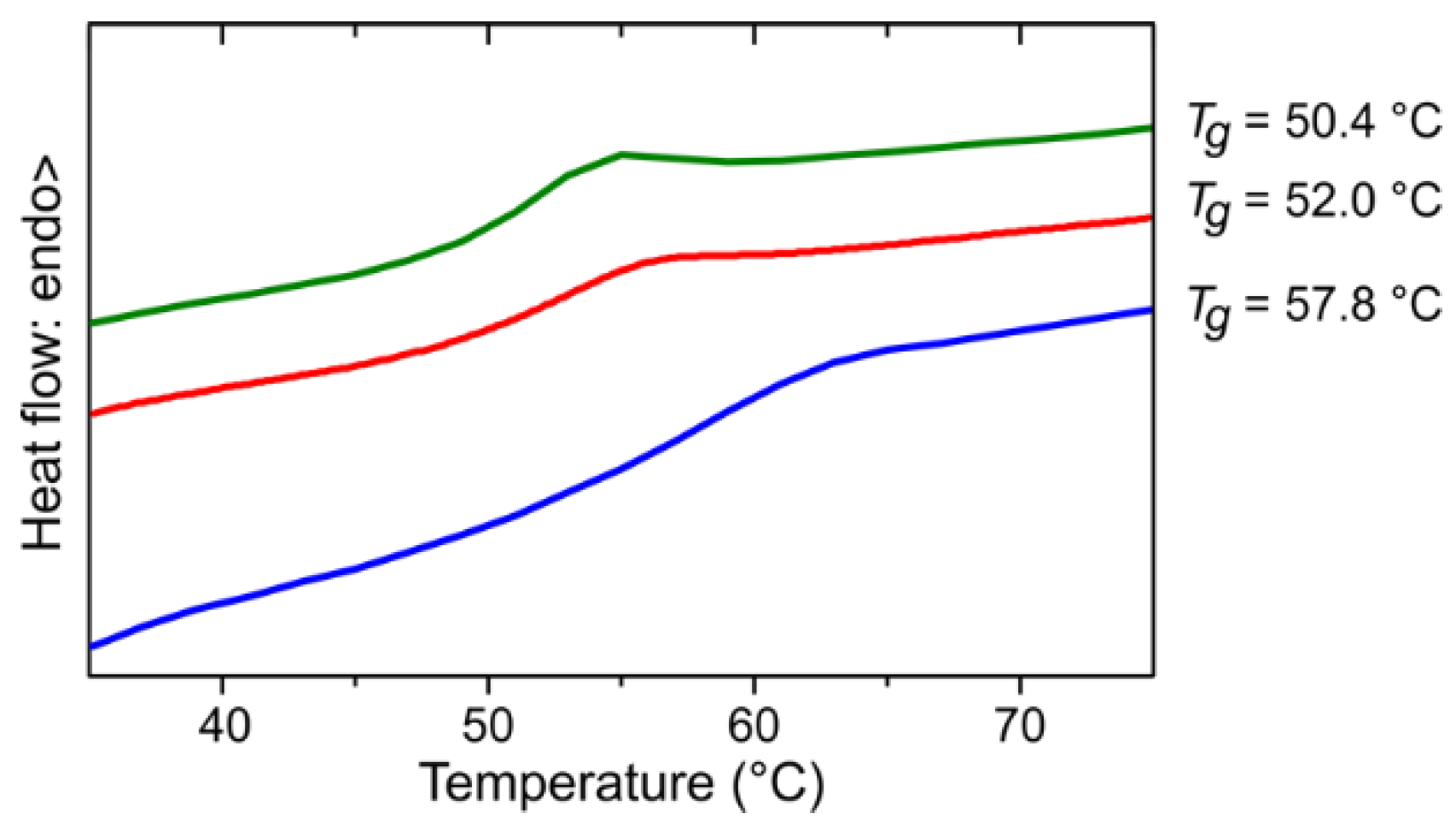
3.2. Fractionation
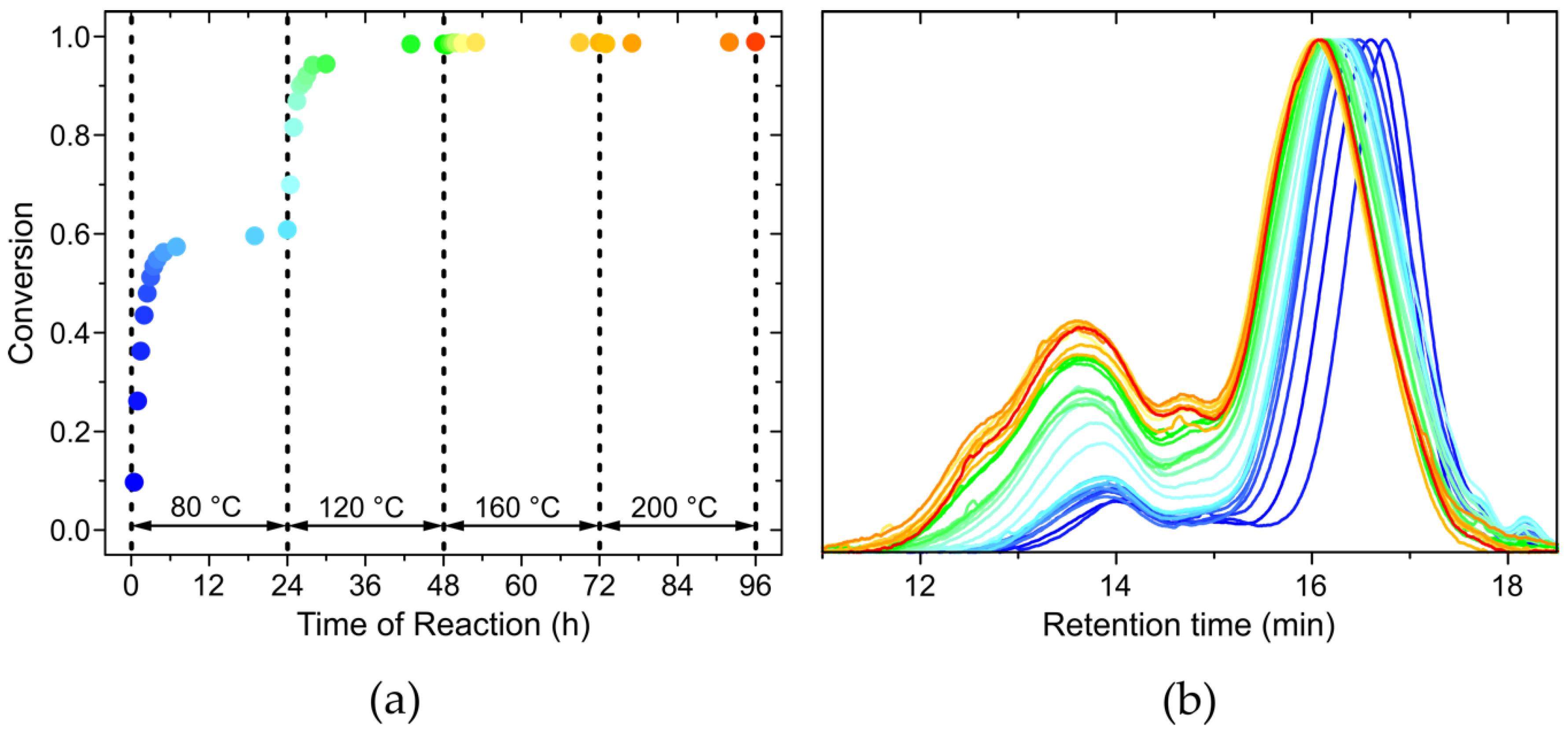
3.3. Evolution of the Course of the Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Braendle, A.; Schwendimann, P.; Niederberger, M.; Caseri, W.R. Synthesis and fractionation of poly(phenylene methylene). J. Polym. Sci. Part A Polym. Chem. 2018, 56, 309–318. [Google Scholar] [CrossRef]
- Ellis, B.; White, P.G. Thermal degradation of polybenzyl. J. Polym. Sci. Polym. Chem. Ed. 1973, 11, 801–821. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Jarvis, K.A.; Ferreira, P.J.; Bielawski, C.W. Graphite Oxide as a Dehydrative Polymerization Catalyst: A One-Step Synthesis of Carbon-Reinforced Poly(phenylene methylene) Composites. Macromolecules 2011, 44, 7659–7667. [Google Scholar] [CrossRef]
- Som, A.; Ramakrishnan, S. Linear soluble polybenzyls. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 2345–2353. [Google Scholar] [CrossRef]
- Valentine, L.; Winter, R.W. 931. The polycondensation of benzyl chloride catalysed by stannic chloride. J. Chem. Soc. 1956, 4768–4779. [Google Scholar] [CrossRef]
- Hino, M.; Arata, K. Iron oxide as an effective catalyst for the polycondensation of benzyl chloride, the formation of para-substituted polybenzyl. Chem. Lett. 1979, 8, 1141–1144. [Google Scholar] [CrossRef]
- Braendle, A.; Perevedentsev, A.; Cheetham, N.J.; Stavrinou, P.N.; Schachner, J.A.; Mösch-Zanetti, N.C.; Niederberger, M.; Caseri, W.R. Homoconjugation in poly(phenylene methylene)s: A case study of non-π-conjugated polymers with unexpected fluorescent properties. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 707–720. [Google Scholar] [CrossRef]
- Braendle, A.; Perevedentsev, A.; Cheetham, N.J.; Stavrinou, P.N.; Schachner, J.A.; Mösch-Zanetti, N.C.; Niederberger, M.; Caseri, W. Polymers with Exceptional Photoluminescence by Homoconjugation. Chimia 2017, 71, 733. [Google Scholar] [CrossRef]
- Martínez, A.G.; Barcina, J.O.; de Fresno Cerezo, A.; Rivas, R.G. Hindered Rotation in Diphenylmethane Derivatives. Electrostatic vs Charge-Transfer and Homoconjugative Aryl−Aryl Interactions. J. Am. Chem. Soc. 1998, 120, 673–679. [Google Scholar] [CrossRef]
- Martínez, A.G.; Barcina, J.O.; Albert, A.; Cano, F.H.; Subramanian, L.R. 7,7-Diphenylnorbornane: The first cofacial diphenylmethane derivative. Tetrahedron Lett. 1993, 34, 6753–6756. [Google Scholar] [CrossRef]
- Martínez, A.G.; Barcina, J.O.; de Fresno Cerezo, A.; Schlüter, A.-D.; Frahn, J. Synthesis of Poly[p-(7-phenylene-7-(2′,5′-dihexyl-4-biphenylene))norbornane]: The First Soluble Polymer with Alternating Conjugation and Homoconjugation. Adv. Mater. 1999, 11, 27–31. [Google Scholar] [CrossRef]
- García Martínez, A.; Osío Barcina, J.; de Fresno Cerezo, Á. Influence of Highly Preorganised 7,7-Diphenylnorbornane in the Free Energy of Edge-to-Face Aromatic Interactions. Chemistry 2001, 7, 1171–1175. [Google Scholar] [CrossRef]
- García Martínez, A.; Osío Barcina, J.; del Rosario Colorado Heras, M.; de Fresno Cerezo, Á.; del Rosario Torres Salvador, M. Complexation Behavior of a Highly Preorganized 7,7-Diphenylnorbornane-Derived Macrocycle: Towards the Design of Molecular Clocks. Chemistry 2003, 9, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Osío Barcina, J.; Colorado Heras, M.D.; Mba, M.; Gómez Aspe, R.; Herrero-García, N. Efficient Electron Delocalization Mediated by Aromatic Homoconjugation in 7,7-Diphenylnorbornane Derivatives. J. Org. Chem. 2009, 74, 7148–7156. [Google Scholar] [CrossRef] [PubMed]
- Osío Barcina, J.; Herrero-García, N.; Cucinotta, F.; De Cola, L.; Contreras-Carballada, P.; Williams, R.M.; Guerrero-Martínez, A. Efficient Photoinduced Energy Transfer Mediated by Aromatic Homoconjugated Bridges. Chemistry 2010, 16, 6033–6040. [Google Scholar] [CrossRef] [PubMed]
- Caraballo-Martínez, N.; del Rosario Colorado Heras, M.; Blázquez, M.M.; Barcina, J.O.; García Martínez, A.; del Rosario Torres Salvador, M. Synthesis of Homoconjugated Oligomers Derived from 7,7-Diphenylnorbornane. Org. Lett. 2007, 9, 2943–2946. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.-D.; Mayer, B. Proximity Effects in Organic Chemistry—The Photoelectron Spectroscopic Investigation of Non-Bonding and Transannular Interactions. Angew. Chem. Int. Ed. Engl. 1983, 22, 283–314. [Google Scholar] [CrossRef]
- Ferguson, L.N.; Nnadi, J.C. Electronic interactions between nonconjugated groups. J. Chem. Educ. 1965, 42, 529–535. [Google Scholar] [CrossRef]
- Simonetta, M.; Winstein, S. Neighboring Carbon and Hydrogen. XVI. 1,3-Interactions and Homoallylic Resonance. J. Am. Chem. Soc. 1954, 76, 18–21. [Google Scholar] [CrossRef]
- Cannizzaro, S. Ueber den der Benzoësäure entsprechenden Alkohol. Justus Liebigs Ann. Chem. 1853, 88, 129–130. [Google Scholar] [CrossRef]
- Parker, D.B.V. The structure of polybenzyls and related polymers. Eur. Polym. J. 1969, 5, 93–104. [Google Scholar] [CrossRef]
- Montaudo, G.; Finocchiaro, P.; Caccamese, S.; Bottino, F. Polycondensation of benzyl chloride and its derivatives: A study of the reaction at different temperatures. J. Polym. Sci. Part A-1 Polym. Chem. 1970, 8, 2475–2490. [Google Scholar] [CrossRef]
- Kuo, J.; Lenz, R.W. Linear polybenzyls. II. Lack of structure control in benzyl chloride polymerization. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 2749–2761. [Google Scholar] [CrossRef]
- Jacobson, R.A. Polymers from benzyl chloride and related compounds. J. Am. Chem. Soc. 1932, 54, 1513–1518. [Google Scholar] [CrossRef]
- Dermer, O.C.; Hooper, E. Catalysts for the Polymerization of Benzyl Chloride. J. Am. Chem. Soc. 1941, 63, 3525–3526. [Google Scholar] [CrossRef]
- Olliges-Stadler, I.; Rossell, M.D.; Niederberger, M. Co-operative Formation of Monolithic Tungsten Oxide-Polybenzylene Hybrids via Polymerization of Benzyl Alcohol and Study of the Catalytic Activity of the Tungsten Oxide Nanoparticles. Small 2010, 6, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Spanier, E.J.; Caropreso, F.E. Condensation of benzyl chloride catalyzed by group II metals. J. Polym. Sci. Part A-1 Polym. Chem. 1969, 7, 2679–2687. [Google Scholar] [CrossRef]
- Ballantine, J.A.; Davies, M.; Patel, I.; Purnell, J.H.; Rayanakorn, M.; Williams, K.J.; Thomas, J.M. Organic reactions catalysed by sheet silicates: Ether formation by the intermolecular dehydration of alcohols and by addition of alcohols to alkenes. J. Mol. Catal. 1984, 26, 37–56. [Google Scholar] [CrossRef]
- Ballantine, J.A.; Davies, M.; Purnell, H.; Rayanakorn, M.; Thomas, J.M.; Williams, K.J. Chemical conversions using sheet silicates: Novel intermolecular dehydrations of alcohols to ethers and polymers. J. Chem. Soc. Chem. Commun. 1981, 427–428. [Google Scholar] [CrossRef]
- Onufrowicz, A. Ueber die Einwirkung von Kupfer auf Benzotrichlorid, Benzal- und Benzylchlorid. Ber. Dtsch. Chem. Ges. 1884, 17, 833–837. [Google Scholar] [CrossRef]
- Tsonis, C.P. Homogeneous catalytic polymerization of benzyl chloride leading to linear high molecular weight polymers: An elusive goal. J. Mol. Catal. 1990, 57, 313–323. [Google Scholar] [CrossRef]
- Tsonis, C. Polymerization of benzyl halides catalyzed by VIB metal carbonyls. Polym. Bull. 1983, 9, 211–216. [Google Scholar] [CrossRef]
- Ul Hasan, M.; Tsonis, C.P. Structural characterization of polybenzyls by high field 13C-NMR spectroscopy. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 1349–1355. [Google Scholar] [CrossRef]
- Ul Hasan, M.; Ali, S.A. Synthesis and high field 13C-NMR spectra of isomeric dibenzylbenzenes. J. Polym. Sci. Polym. Chem. Ed. 1985, 23, 1847–1849. [Google Scholar] [CrossRef] [Green Version]
- Blincow, P.J.; Pritchard, G. Determination of the structure of polyethylarylmethylenes by 13C nuclear magnetic resonance spectroscopy. Polymer 1987, 28, 1824–1828. [Google Scholar] [CrossRef]
- Gunes, D.; Yagci, Y.; Bicak, N. Synthesis of Soluble Poly(p-phenylene methylene) from Tribenzylborate by Acid-Catalyzed Polymerization. Macromolecules 2010, 43, 7993–7997. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Baker, P.K.; Hursthouse, M.B.; Karaulov, A.I.; Lavery, A.J.; Malik, K.M.A.; Muldoon, D.J.; Shawcross, A. Seven-co-ordinate dibromo complexes of molybdenum(II) and tungsten(II) derived from [MBr2(CO)3(NCMe)2]. Crystal structures of the isostructural complexes [WX2(CO)3(NCMe)(SbPh3)] CH2Cl2 (X = Br or I). J. Chem. Soc. Dalton Trans. 1994, 0, 3493–3498. [Google Scholar] [CrossRef]
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.K.; Muldoon, D.J.; Lavery, A.J.; Shawcross, A. Dibromoalkyne complexes of tungsten(II). Polyhedron 1994, 13, 2915–2921. [Google Scholar] [CrossRef]
- Cotton, F.A.; Falvello, L.R.; Meadows, J.H. Structural characterization and infrared studies of tungsten bromo carbonyl compounds. Inorg. Chem. 1985, 24, 514–517. [Google Scholar] [CrossRef]
- Peschel, L.M.; Schachner, J.A.; Sala, C.H.; Belaj, F.; Mösch-Zanetti, N.C. An Update on WII and MoII Carbonyl Precursors and Their Application in the Synthesis of Potentially Bio-Inspired Thiophenolate-Oxazoline Complexes. Z. Anorg. Allg. Chem. 2013, 639, 1559–1567. [Google Scholar] [CrossRef]
- Elias, H.-G. Polykondensationen und Polyadditionen. In Makromoleküle; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2002; pp. 430–434. ISBN 978-3-52-762655-7. [Google Scholar]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation 1. J. Am. Chem. Soc. 1951, 63, 3083–3090. [Google Scholar] [CrossRef]
- Flory, P.J. Kinetics of Polyesterification: A Study of the Effects of Molecular Weight and Viscosity on Reaction Rate. J. Am. Chem. Soc. 1939, 61, 3334–3340. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn [g mol−1] | Mw [g mol−1] | PDI |
|---|---|---|---|
| [W2Br4(CO)7] (P) | 12,600 | 427,300 | 33.9 |
| [WBr2(Me2C2)2(CO)(MeCN)] | 8100 | 135,700 | 16.7 |
| [WBr2(CO)3(MeCN)2] | 11,200 | 355,200 | 31.8 |
| [WBr2(Ph2C2)2(CO)(MeCN)] | 15,000 | 227,900 | 15.1 |
| Fractions of polymer (P) | |||
| F1 | 1600 | 4000 | 2.6 |
| F4 | 167,900 | 1,072,000 | 6.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braendle, A.; Vidovič, C.; Mösch-Zanetti, N.C.; Niederberger, M.; Caseri, W. Synthesis of High Molar Mass Poly(phenylene methylene) Catalyzed by Tungsten(II) Compounds. Polymers 2018, 10, 881. https://doi.org/10.3390/polym10080881
Braendle A, Vidovič C, Mösch-Zanetti NC, Niederberger M, Caseri W. Synthesis of High Molar Mass Poly(phenylene methylene) Catalyzed by Tungsten(II) Compounds. Polymers. 2018; 10(8):881. https://doi.org/10.3390/polym10080881
Chicago/Turabian StyleBraendle, Andreas, Carina Vidovič, Nadia C. Mösch-Zanetti, Markus Niederberger, and Walter Caseri. 2018. "Synthesis of High Molar Mass Poly(phenylene methylene) Catalyzed by Tungsten(II) Compounds" Polymers 10, no. 8: 881. https://doi.org/10.3390/polym10080881
APA StyleBraendle, A., Vidovič, C., Mösch-Zanetti, N. C., Niederberger, M., & Caseri, W. (2018). Synthesis of High Molar Mass Poly(phenylene methylene) Catalyzed by Tungsten(II) Compounds. Polymers, 10(8), 881. https://doi.org/10.3390/polym10080881