The Microbial Production of Polyhydroxyalkanoates from Waste Polystyrene Fragments Attained Using Oxidative Degradation


 ,
, 


 ,
,  and
and
Abstract
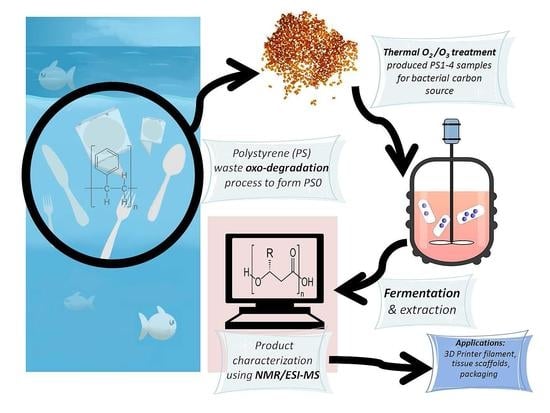
:
1. Introduction
2. Materials and Methods
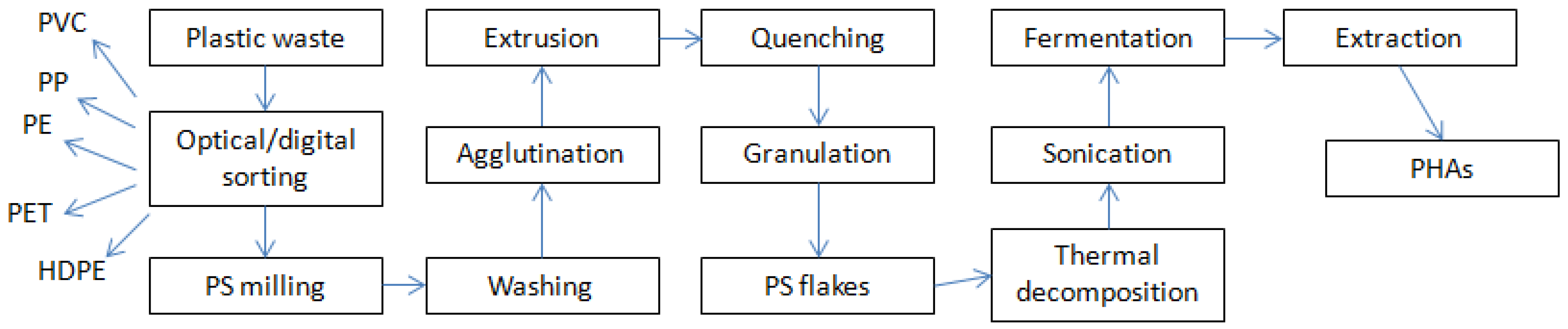
2.1. Carbon Source
2.2. Microorganism
2.3. Growth Media and Chemicals
2.4. Fermentation Procedure
2.5. PHA Extraction Procedure
2.6. Characterization Techniques
2.6.1. GPC
2.6.2. FTIR
2.6.3. NMR
2.6.4. TGA
2.6.5. ESI-MS/MS
2.6.6. PHA Molecular Analysis Procedure
3. Results
3.1. Acid Number of Carbon Sources (PS0–4)
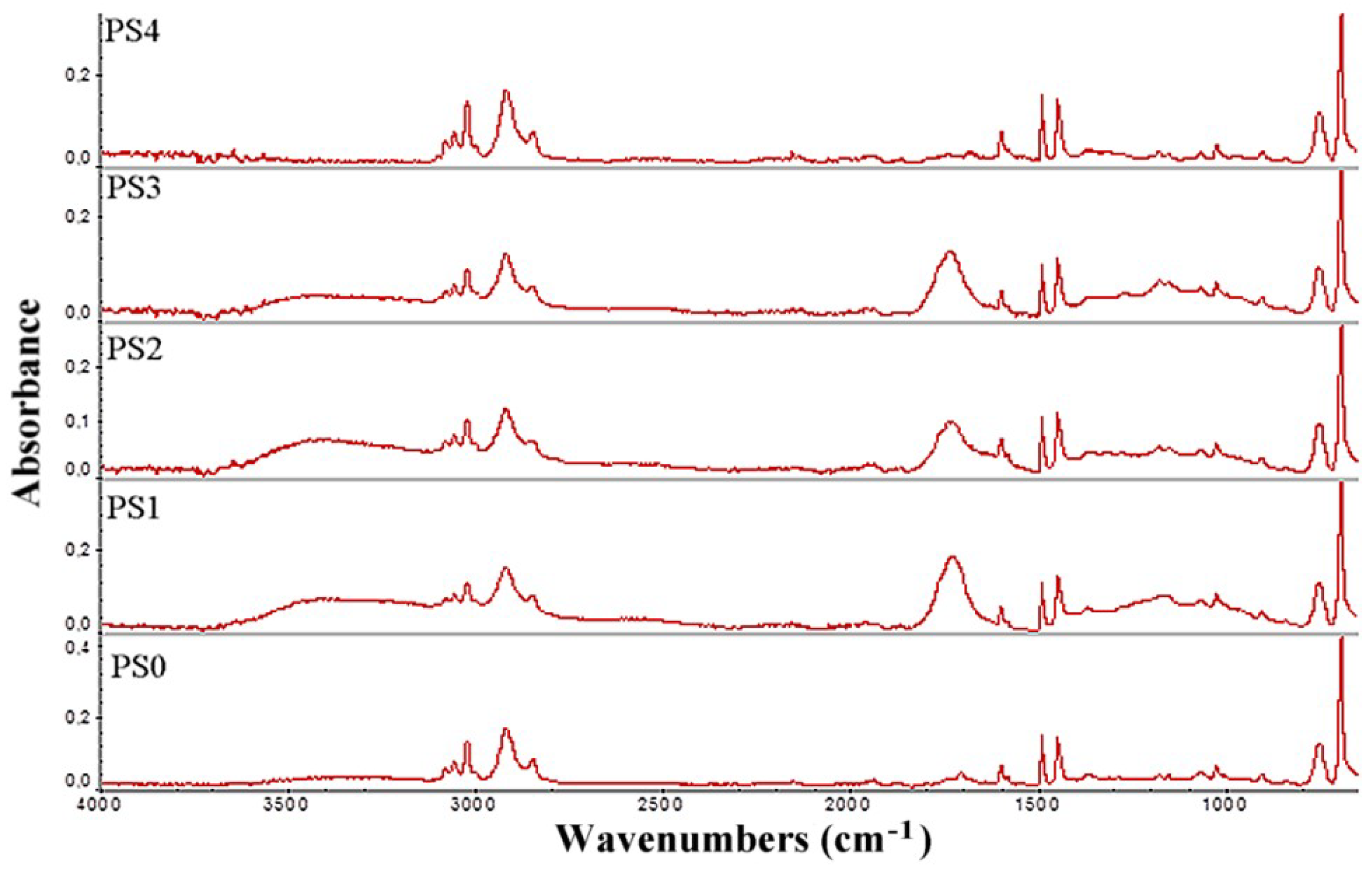
3.2. FTIR Analysis of Carbon Sources
3.3. GPC Analysis of Carbon Sources
3.4. Thermal Analysis of Carbon Sources
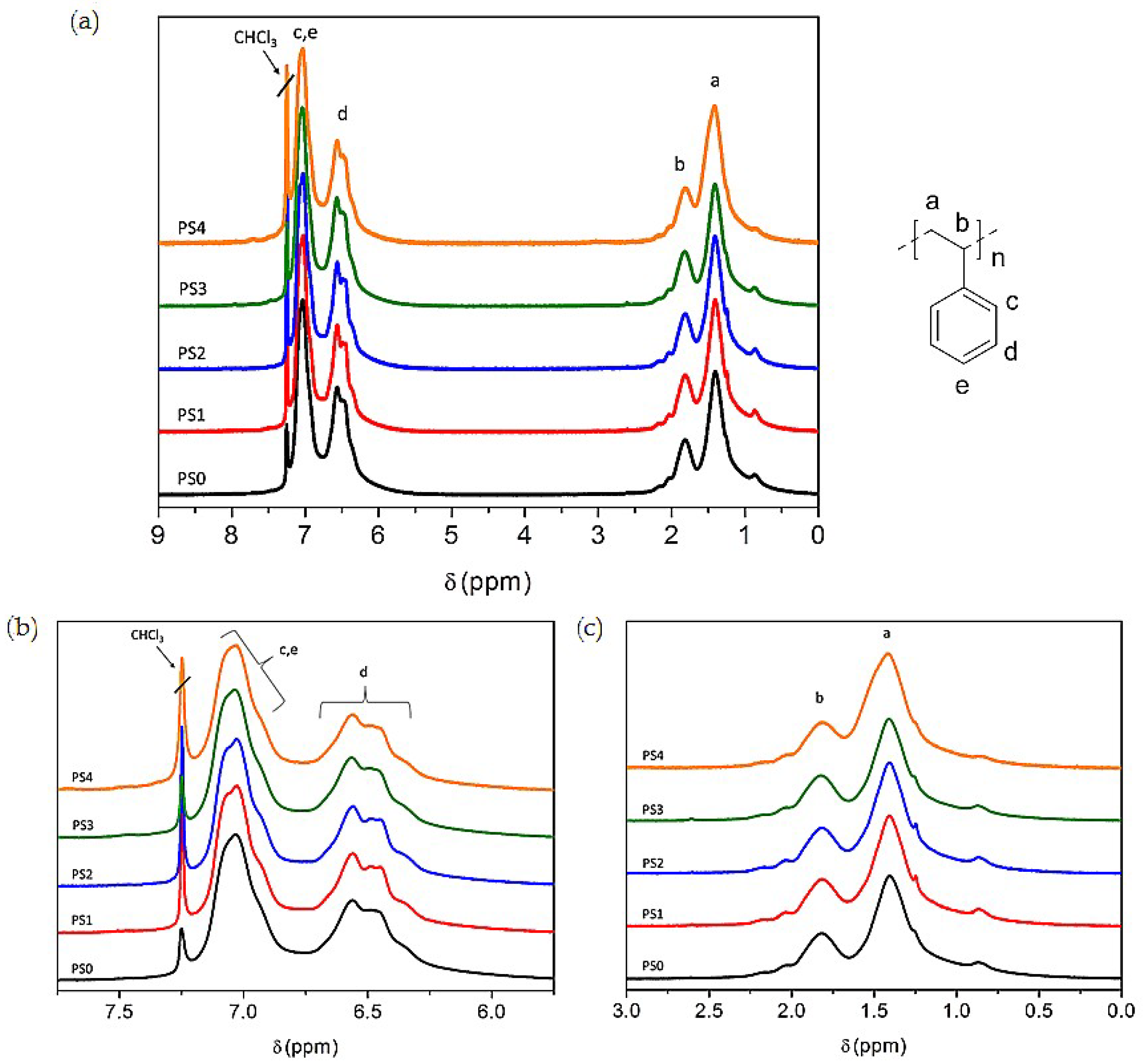
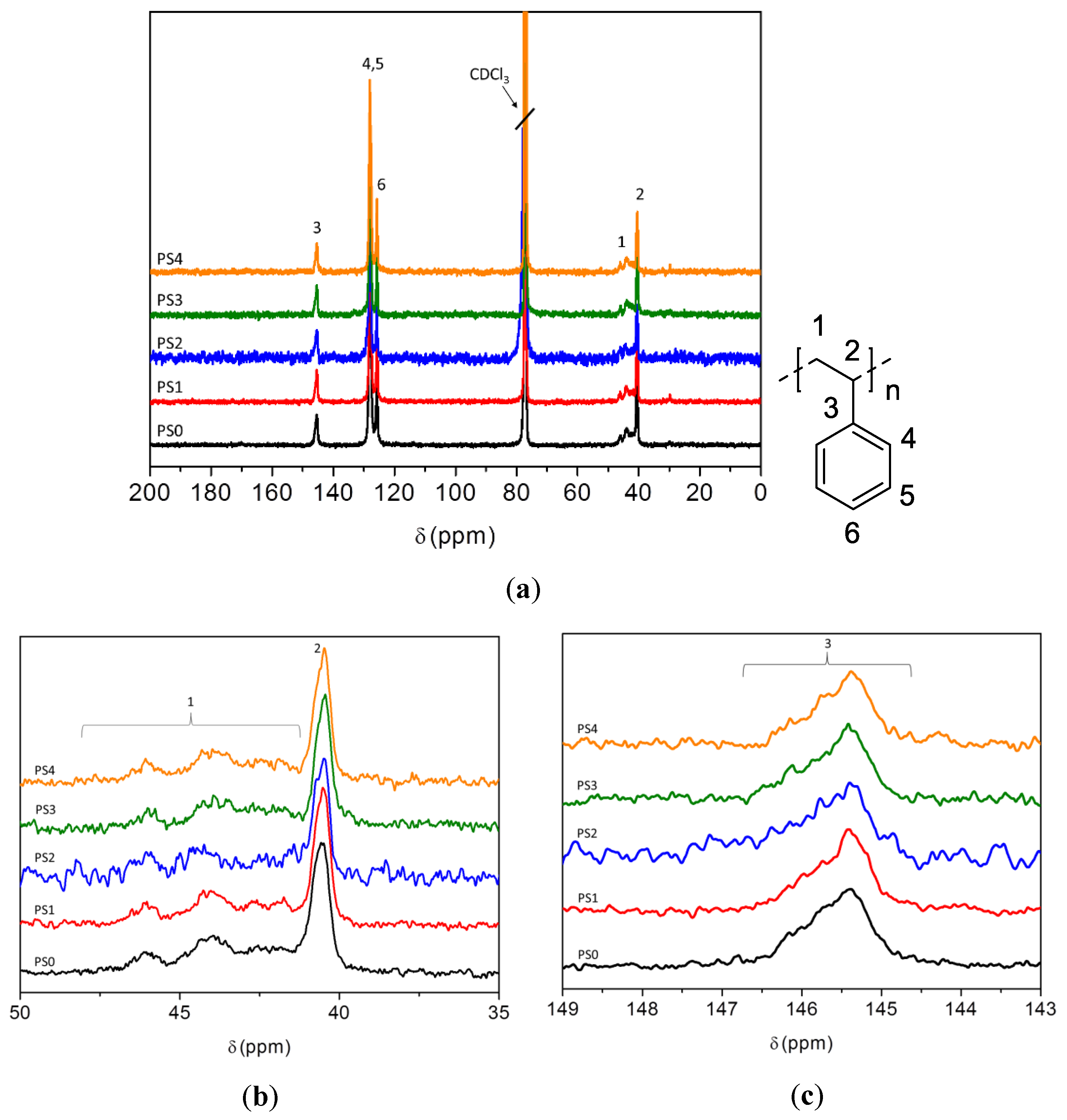
3.5. NMR Analysis of Carbon Sources
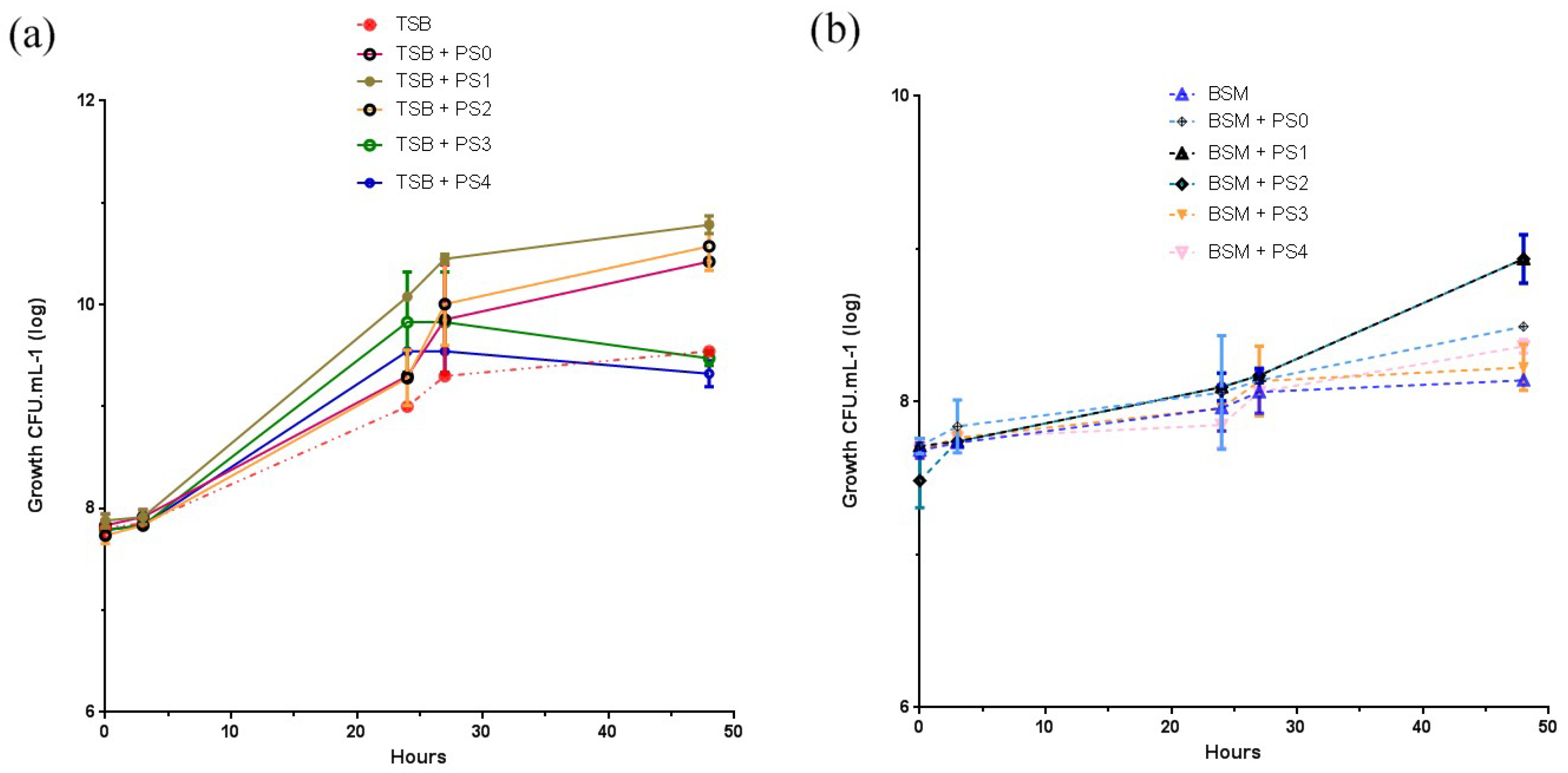
3.6. Bacteria Growth
3.7. PHA Identification and Characterization
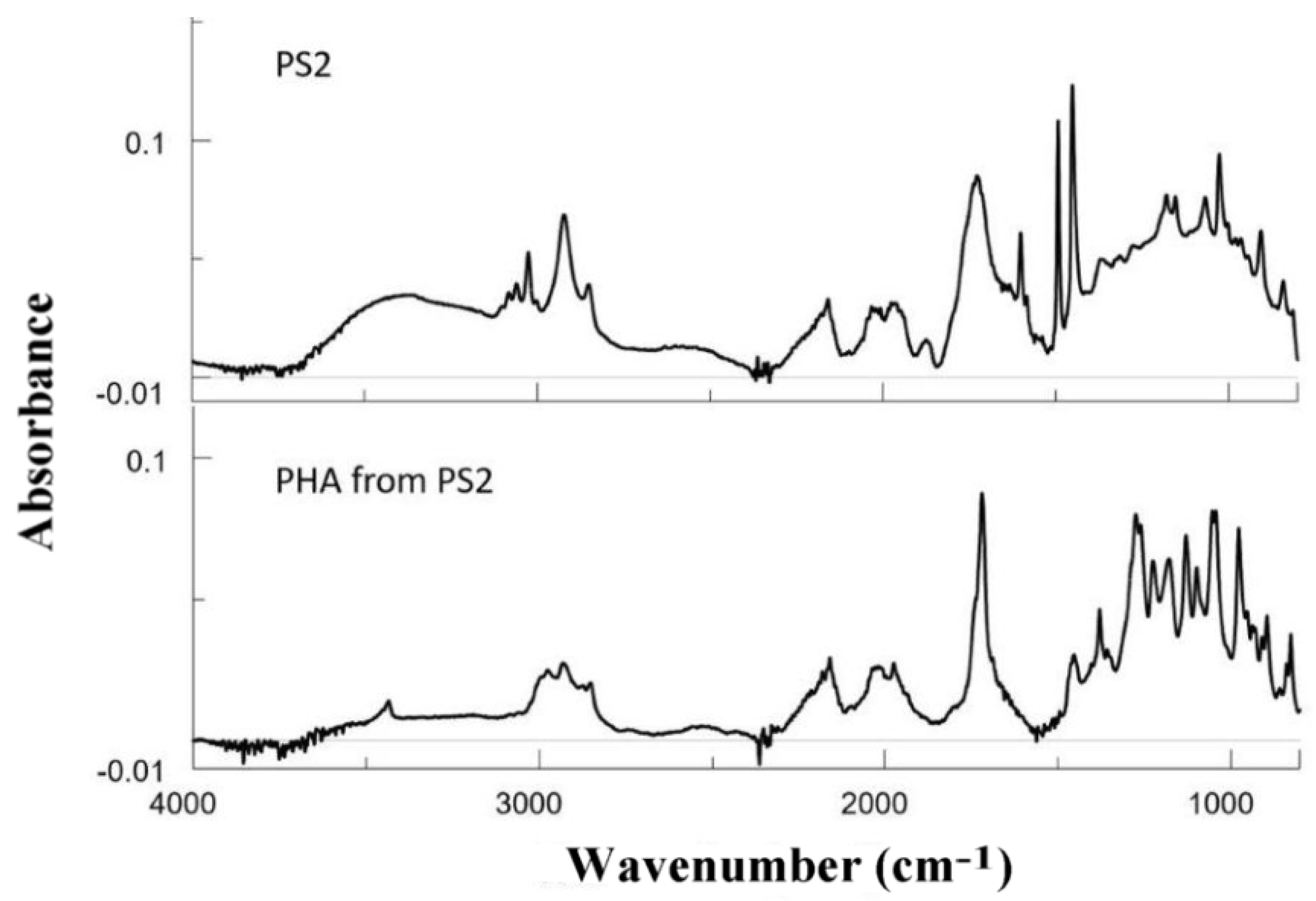
3.7.1. FTIR Results
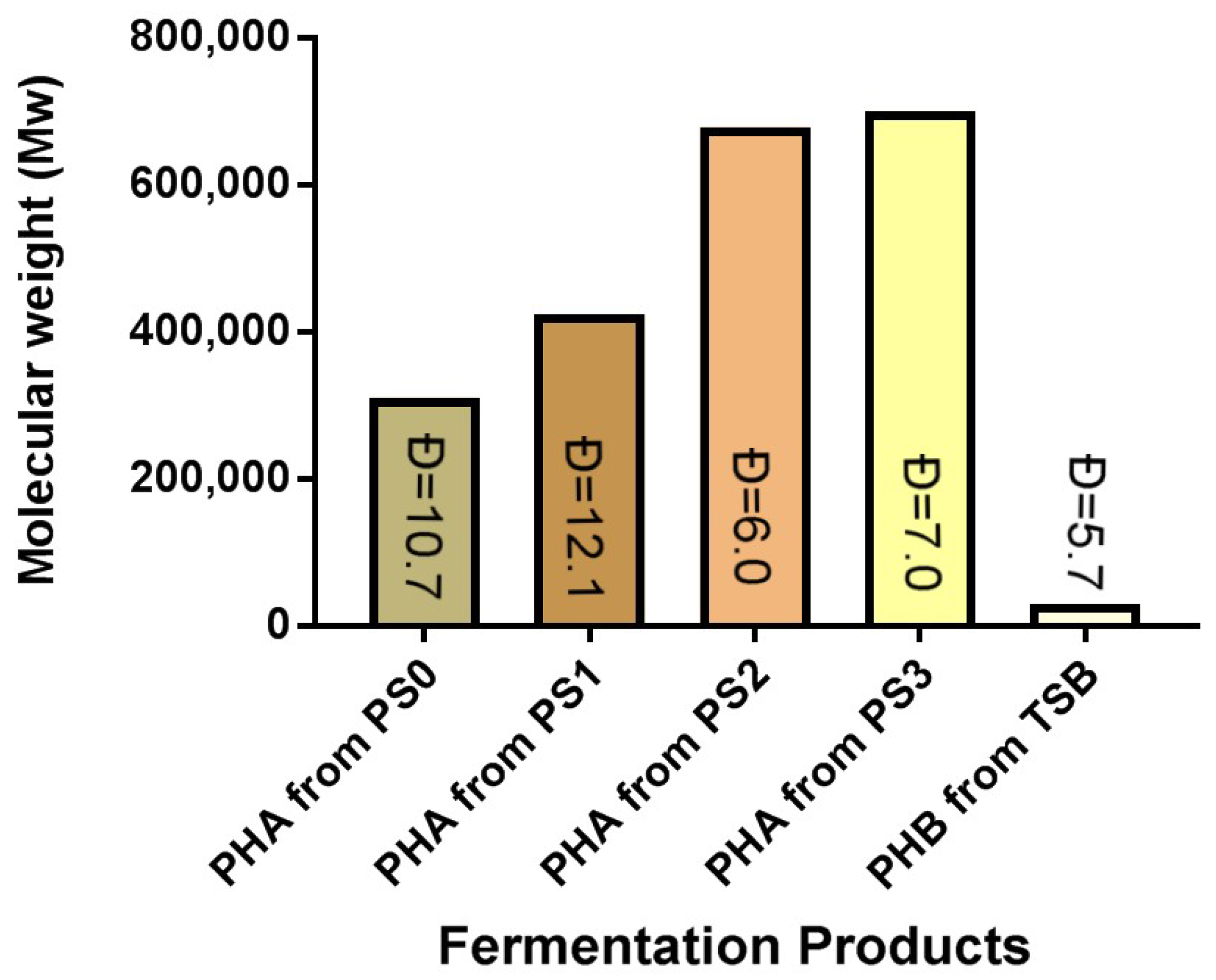
3.7.2. PHA GPC Results
3.7.3. Thermal Analysis of PHA
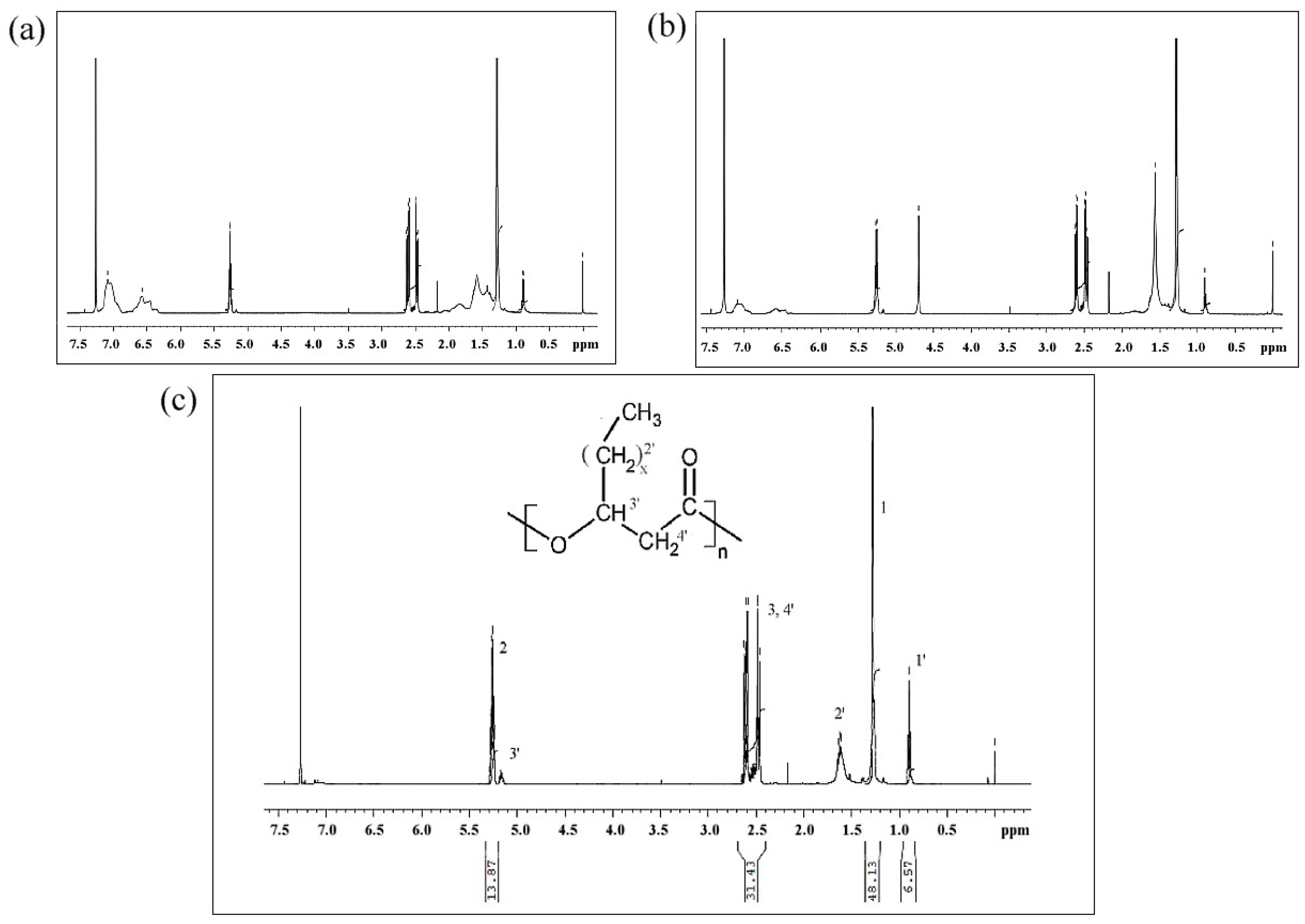
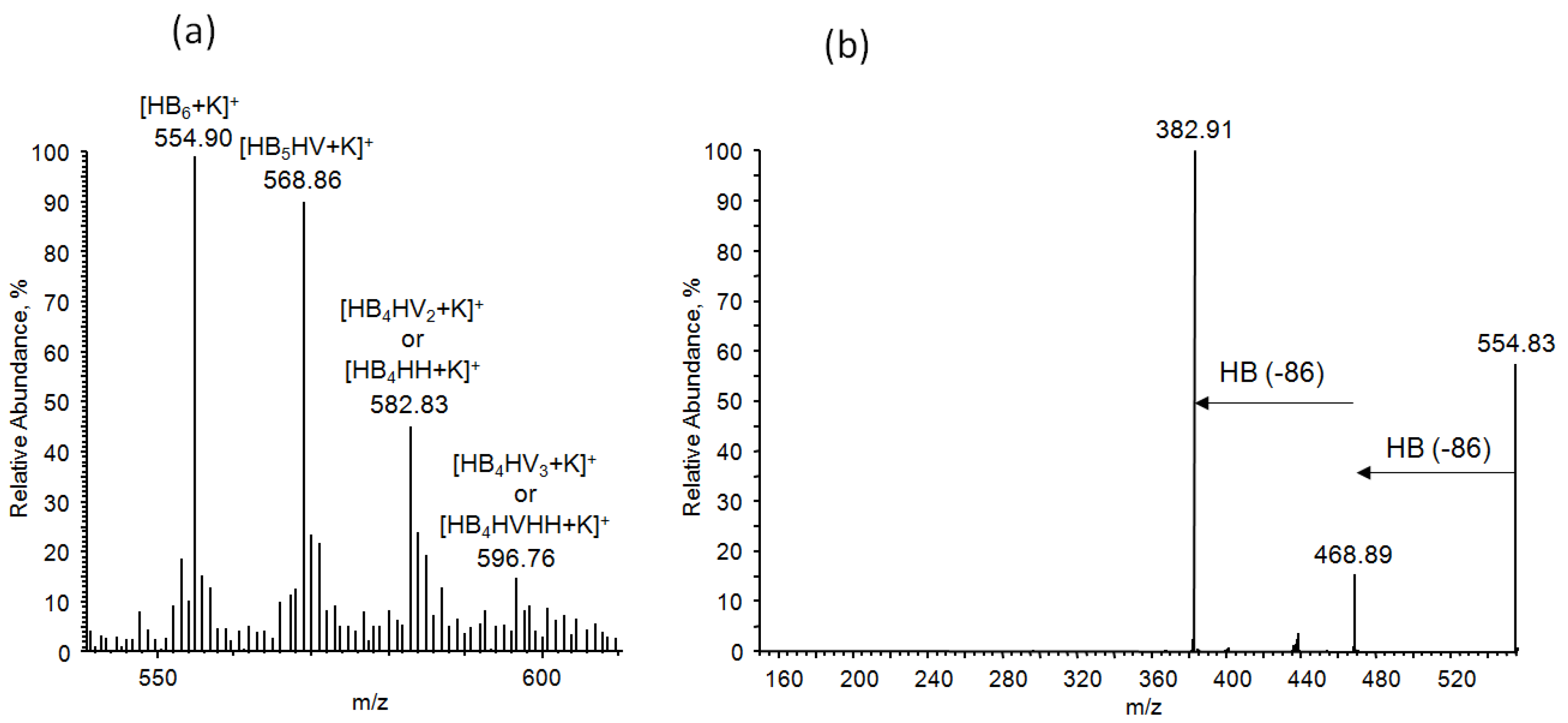
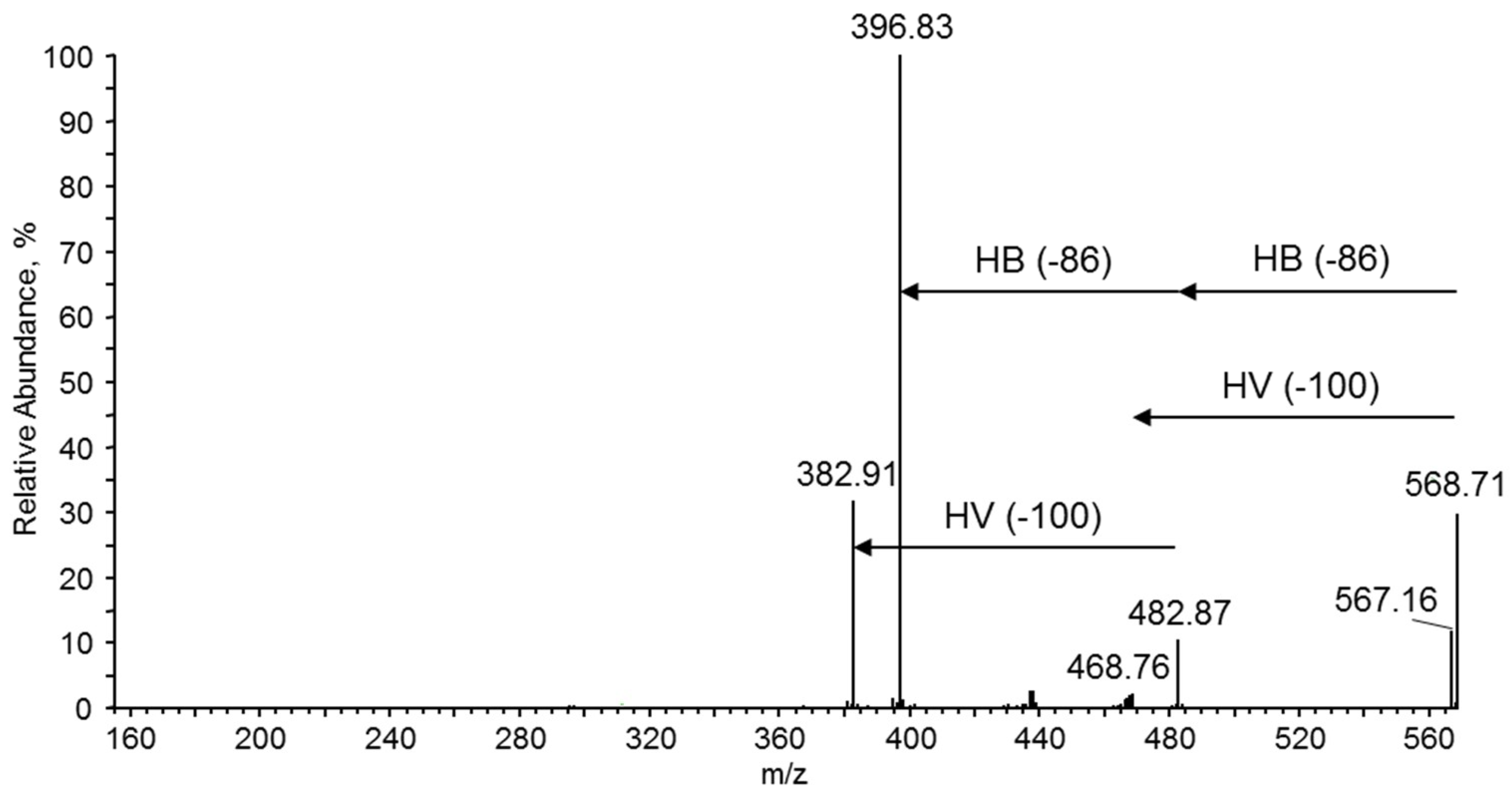
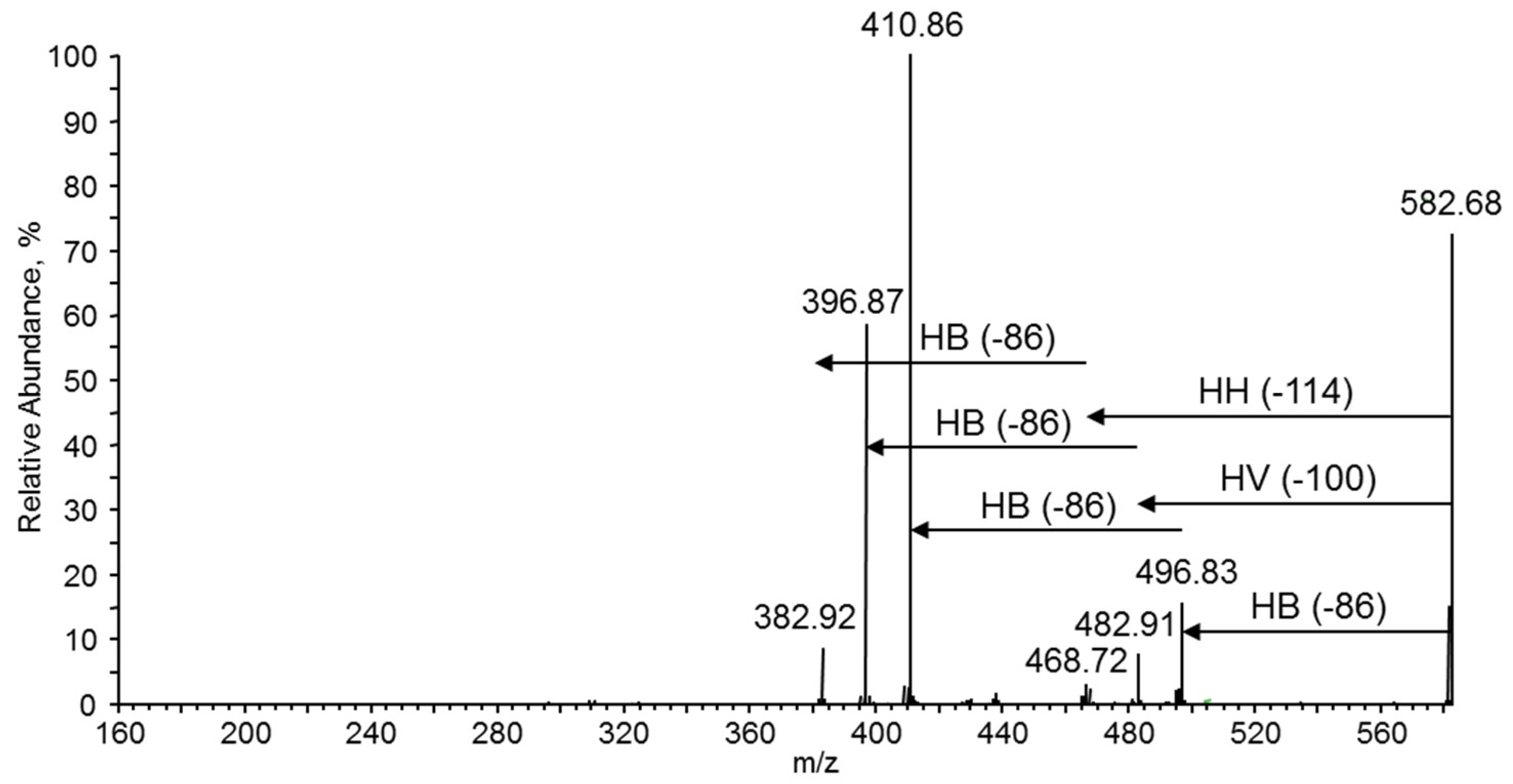
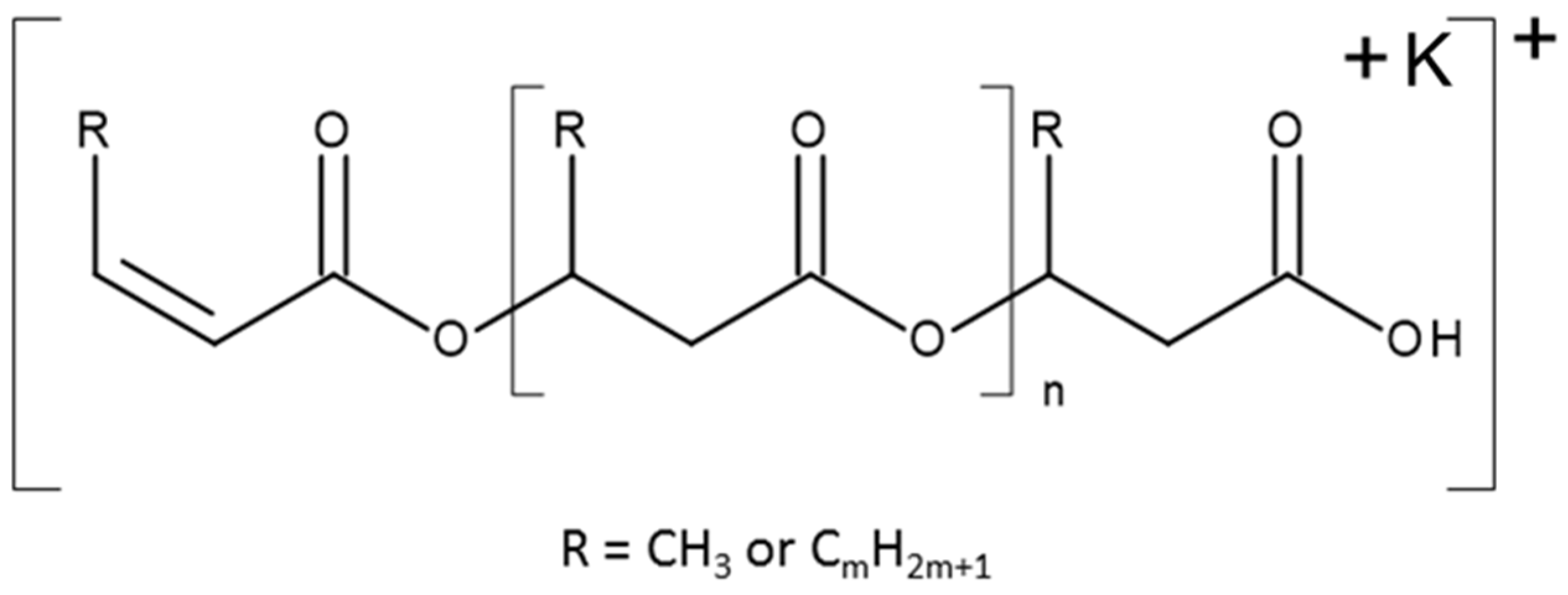
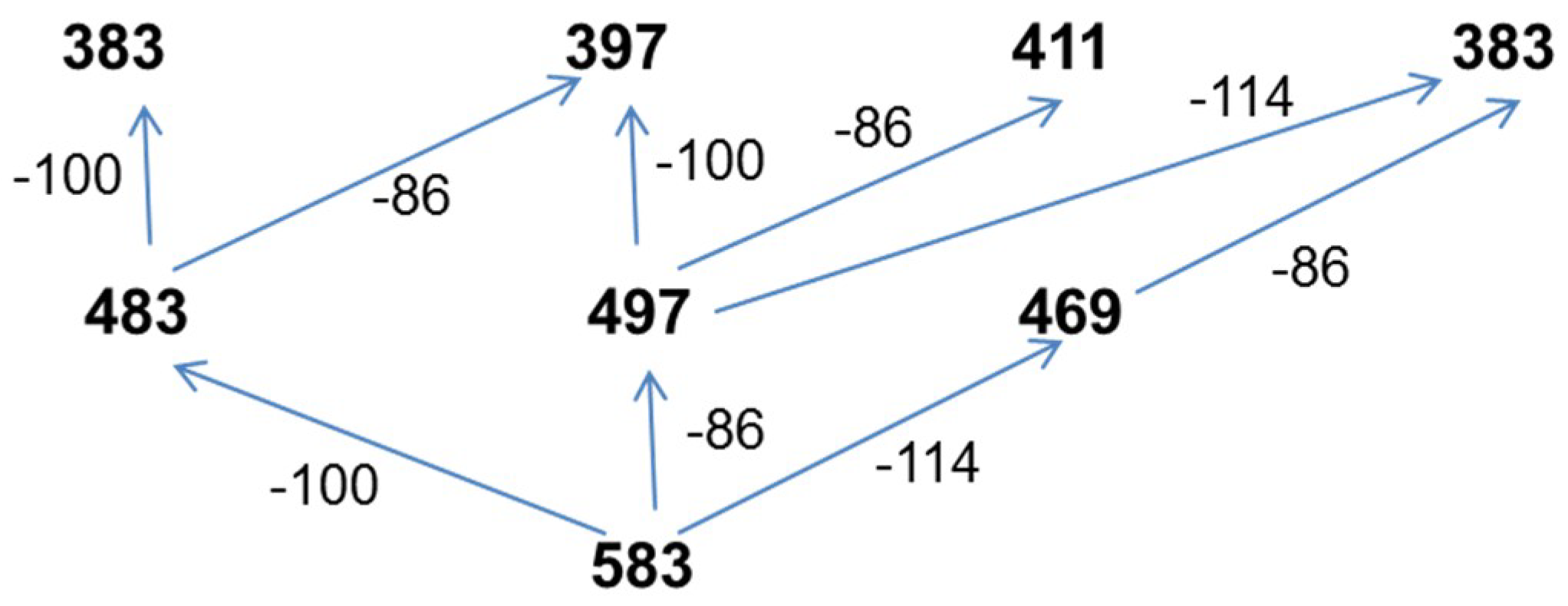
3.7.4. NMR and ESI-MS Results
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verma, R.; Vinoda, K.S.; Papireddy, M.; Gowda, A.N.S. Toxic pollutants from plastic waste: A review. Procedia Environ. Sci. 2016, 35, 701–708. [Google Scholar] [CrossRef]
- North, E.J.; Halden, R.U. Plastics and environmental health: The road ahead. Rev. Environ. Health 2013, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Driedger, A.G.J.; Dürr, H.H.; Mitchell, K.; Cappellen, P.V. Plastic debris in the Laurentian Great Lakes: A review. J. Great Lakes Res. 2015, 41, 9–19. [Google Scholar] [CrossRef]
- Thompson, R.C.; Swan, S.H.; Moore, C.J.; Vom Saal, F.S. Our plastic age. Philos. Trans. R. Soc. B 2009, 364, 1973–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, B.; Jiang, G.; Hill, D.; Adamus, G.; Kwiecien, I.; Zieba, M.; Sikorska, W.; Green, M.; Kowalczuk, M.; Radecka, I. The molecular level characterization of biodegradable polymers originated from polyethylene using non-oxygenated polyethylene wax as a carbon source for polyhydroxyalkanoate production. Bioengineering 2017, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Somleva, M.N.; Peoples, O.P.; Snell, K.D. PHA bioplastics, biochemicals, and energy from crops. Plant Biotechnol. J. 2013, 11, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ausejo, J.; Rydz, J.; Musioł, M.; Sikorska, W.; Sobota, M.; Włodarczyk, J.; Adamus, G.; Janeczek, H.; Kwiecień, I.; Hercog, A.; et al. A comparative study of three-dimensional printing directions: The degradation and toxicological profile of a PLA/PHA blend. Polym. Degrad. Stab. 2018, 152, 191–207. [Google Scholar] [CrossRef]
- Kwiecień, I.; Adamus, G.; Jiang, G.; Radecka, I.; Baldwin, T.C.; Khan, H.R.; Johnston, B.; Pennetta, V.; Hill, D.; Bretz, I.; et al. Biodegradable PBAT/PLA blend with bioactive MCPA-PHBV conjugate suppresses weed growth. Biomacromolecules 2018, 19, 511–520. [Google Scholar] [CrossRef] [PubMed]
- El-Barbary, A.A.; El-Said, K.S.; Betiha, M.; Elkholy, H.M.; Chiellini, E.; El-Magd, M.A. Functionalization of PHB with different thiol compounds inhibits MDM2-p53 interactions in MCF7 cells. J. Appl. Polym. Sci. 2018, 46924. [Google Scholar] [CrossRef]
- Plastics Technology. Available online: http://www.ptonline.com/articles/prices-bottom-out-forpolyolefins-pet-ps-pvc-move-up (accessed on 10 June 2018).
- Eno, R.; Hill, J. Metabolix Bio-Industrial Evolution. In Proceedings of the Jefferies 11th Global Clean Technology Conference, New York, NY, USA, 21 February 2011; pp. 23–24. [Google Scholar]
- Kourmentza, C.; Plácido, J.; Venetsaneas, N.; Burniol-Figols, A.; Varrone, C.; Gavala, H.N.; Reis, M.A. Recent advances and challenges towards sustainable polyhydroxyalkanoate (PHA) production. Bioengineering 2017, 4, 55. [Google Scholar] [CrossRef] [PubMed]
- The Future Potential Economic Impacts of a Bio-Plastics Industry in the UK. Available online: http://bbia.org.uk/wp-content/uploads/2015/11/BBIA-CEBR-Report.compressed.pdf (accessed on 28 May 2018).
- Bio-Based Building Blocks and Polymers. Available online: http://bio-based.eu/media/edd/2017/03/17-02-Bio-based-Building-Blocks-and-Polymers-short-version.pdf (accessed on 13 August 2018).
- American Chemistry Council: Chemical Safety Facts. Polystyrene 2018. Available online: https://www.chemicalsafetyfacts.org/polystyrene-post (accessed on 30 May 2018).
- Kwon, B.G.; Saidoc, K.; Koizumid, K.; Satod, H.; Ogawae, N.; Chung, S.Y.; Kusuif, T.; Koderac, Y.; Kogure, K. Regional distribution of styrene analogues generated from polystyrene degradation along the coastlines of the North-East Pacific Ocean and Hawaii. Environ. Pollut. 2014, 188, 45–49. [Google Scholar] [CrossRef] [PubMed]
- EPA. United States Environmental Protection Agency. Available online: https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data (accessed on 28 April 2018).
- Emadian, S.M.; Onay, T.T.; Demirel, B. Biodegradation of bioplastics in natural environments. Waste Manag. 2017, 59, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Muenmee, S.; Chiemchaisri, W. Enhancement of biodegradation of plastic wastes via methane oxidation in semi-aerobic landfill. Int. Biodeterior. Biodegr. 2016, 113, 244–255. [Google Scholar] [CrossRef]
- 2016 US National Postconsumer Plastic Bottle Recycling Rate Report. Available online: https://plastics.americanchemistry.com/2016-US-National-Postconsumer-Plastic-Bottle-Recycling-Report.pdf (accessed on 28 April 2018).
- Finnveden, G.; Johansson, J.; Lind, P.; Moberg, A. Life cycle assessment of energy from solid waste-Part 1: General methodology and results. J. Clean. Prod. 2005, 13, 213–229. [Google Scholar] [CrossRef]
- Aznar, M.P.; Caballero, M.A.; Sancho, J.A.; Francs, E. Plastic waste elimination by co-gasification with coal and biomass in fluidized bed with air in pilot plant. Fuel Process. Technol. 2006, 87, 409–420. [Google Scholar] [CrossRef]
- Vilaplana, F.; Ribes-Greus, A.; Karlsson, S. Analytical strategies for the quality assessment of recycled high-impact polystyrene: A combination of thermal analysis, vibrational spectroscopy, and chromatography. Anal. Chim. Acta 2007, 604, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Stangenberg, F.; Agren, S.; Karlsson, S. Quality assessments of recycled plastics by spectroscopy and chromatography. Chromatographia 2004, 59, 101–106. [Google Scholar]
- Environment. Available online: http://ec.europa.eu/environment/circular-economy/index_en.htm (accessed on 20 July 2018).
- Aldas, M.; Paladines, A.; Valle, V.; Pazmiño, M.; Quiroz, F. Effect of the prodegradant-additive plastics incorporated on the polyethylene recycling. Int. J. Polym. Sci. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Malykh, N.V.; Petrov, V.M.; Mal’tzev, L.I. Ultrasonic and Hydrodynamic Cavitation and Liquid Hydrocarbon Cracking; XX Session of the Russian Acoustical Society: Moscow, Russia, 2008; p. 345. [Google Scholar]
- Leja, K.; Lewandowicz, G. Polymer biodegradation and biodegradable polymers—A review. Pol. J. Environ. Stud. 2010, 19, 255–266. [Google Scholar]
- Gautam, R.; Bassi, A.S.; Yanful, E.K.; Cullen, E. Biodegradation of automotive waste polyester polyurethane foam using Pseudomonas chlororaphis ATCC55729. Int. Biodeterior. Biodegrad. 2007, 60, 245–249. [Google Scholar] [CrossRef]
- Ward, P.G.; De Roo, G.; O’Connor, K.E. Accumulation of polyhydroxyalkanoate from styrene and phenylacetic acid by pseudomonas putida CA-3. Appl. Environ. Microbiol. 2005, 71, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Hasan, F.; Hameed, A.; Ahme, S. Biological degradation of plastics: A comprehensive review. Biotechnol. Adv. 2008, 26, 246–265. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, R.A.J.; Hill, D.J.; Kenward, M.A.; Williams, C.D.; Radecka, I. Production of polyhydroxyalkanoates from waste frying oil by cupriavidus necator. AMB Express 2011, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, R.A.J.; Hill, D.J.; Kenward, M.A.; Williams, C.D.; Radecka, I. Bacterial synthesis of biodegradable polyhydroxyalkanoates. J. Appl. Microbiol. 2007, 102, 1437–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koller, M.; Braunegg, G. Advanced approaches to produce polyhydroxyalkanoate (PHA) biopolyesters in a sustainable and economic fashion. EuroBiotech J. 2018, 2, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Radecka, I.; Irorere, V.; Jiang, G.; Hill, D.; Williams, C.; Adamus, G.; Kwiecień, M.; Marek, A.A.; Zawadiak, J.; Johnston, B.; et al. Oxidized polyethylene wax as a potential carbon source for PHA production. Materials 2016, 9, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Hill, D.J.; Kowalczuk, M.; Johnston, B.; Adamus, G.; Irorere, V.; Radecka, I. Carbon sources for polyhydroxyalkanoates and an integrated biorefinery. Int. J. Mol. Sci. 2016, 17, 1157. [Google Scholar] [CrossRef] [PubMed]
- Production and Characterization of Polyhydroxybutyrate from Molasses and Corn Steep Liquor Produced by Bacillus Megaterium ATCC 6748. Available online: http://www.cigrjournal.org/index.php/Ejounral/article/view/1216/1074 (accessed on 3 August 2018).
- Koller, M.; Sousa Dias, M.M.; Rodríguez-Contreras, A.; Kunaver, K.; Žagar, E.; Kržan, A.; Braunegg, G. Liquefied wood as inexpensive precursor-feedstock for bio-mediated incorporation of (R)-3-Hydroxyvalerate into polyhydroxyalkanoates. Materials 2015, 8, 6543–6557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, A.A.; Misra, S.S.; Irwin, J.O. The estimation of the bactericidal power of the blood. Epidemiol. Infect. 1938, 38, 732–749. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Kawalec, M.; Sobota, M.; Scandola, M.; Kowalczuk, M.; Kurcok, P. A convenient route to PHB macromonomers via anionically controlled moderate-temperature degradation of PHB. J. Polym. Sci. Polym. Chem. 2010, 48, 5490–5497. [Google Scholar] [CrossRef]
- Yusof, N.A.; Zakaria, N.D.; Maamor, N.A.M.; Abdullah, A.H.; Haron, M.J. Synthesis and characterization of molecularly imprinted polymer membrane for the removal of 2,4-dinitrophenol. Int. J. Mol. Sci. 2013, 14, 3993–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novotnýa, Č.; Malachová, K.; Adamus, G.; Kwiecień, M.; Lotti, N.; Soccio, M.; Verney, V.; Fava, F. Deterioration of irradiation/high-temperature pretreated, linear low-densitypolyethylene (LLDPE) by bacillus amyloliquefaciens. Int. Biodeterior. Biodegrad. 2018, 132, 259–267. [Google Scholar]
- Bevington, J.C.; Huckerby, T.N. Studies of end-groups in polystyrene using 1H NMR. Eur. Polym. J. 2006, 42, 1433–1436. [Google Scholar] [CrossRef]
- London, L.A.; Bolton, L.A.; Samarakoon, D.K.; Sannigrahi, B.S.; Wang, X.Q.; Khan, I.M. Effect of polymer stereoregularity on polystyrene/single-walled carbon nanotube interactions. RSC Adv. 2015, 5, 59186–59193. [Google Scholar] [CrossRef]
- Cheng, H.N.; Lee, G.H. NMR Studies of polystyrene tacticity. Int. J. Polym. Anal. Charact. 1996, 2, 439–455. [Google Scholar] [CrossRef]
- Purohit, V.; Orzel, R.A. Polypropylene: A literature review of the thermal decomposition products and toxicity. J. Am. Coll. Toxicol. 1988, 7, 221–242. [Google Scholar] [CrossRef]
- Seyfriedsberger, G.; Rametsteiner, K.; Kern, W. Polyethylene compounds with antimicrobial surface properties. Eur. Polym. J. 2006, 42, 3383–3389. [Google Scholar] [CrossRef]
- Zhang, W.; Luo, Y.; Wang, H.; Jiang, J.; Pu, S.; Chu, P.K. Ag and Ag/N2 plasma modification of polyethylene for the enhancement of antibacterial properties and cell growth/proliferation. Acta Biomater. 2008, 4, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.R.; Hudson, A.L.; Dunn, S.M.; You, H.; Baker, G.B.; Whittal, R.M.; Martin, J.W.; Jha, A.; Edmondson, D.E.; Holt, A. Bioactive contaminants leach from disposable laboratory plasticware. Science 2008, 322, 917. [Google Scholar] [CrossRef] [PubMed]
- Shamala, T.R.; Divyashree, M.S.; Davis, R.; Latha Kumari, K.S.; Vijayendra, S.V.N.; Raj, B. Production and characterization of bacterial polyhydroxyalkanoate copolymers and evaluation of their blends by fourier transform infrared spectroscopy and scanning electron microscopy. Indian J. Microbiol. 2009, 49, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, M.; Hakkarainen, M.; Albertsson, A. Recycling oxidized model polyethylene powder as a degradation enhancing filler for polyethylene/polycaprolactone blends. ACS Sustain. Chem. Eng. 2016, 4, 129–135. [Google Scholar] [CrossRef]
- Wei, L.; Guho, N.M.; Coats, E.R.; McDonald, A.G. Characterization of poly(3-hydroxybutyrateco-3-hydroxyvalerate) biosynthesized by mixed microbial consortia fed fermented dairy manure. J. Appl. Polym. Sci. 2014, 131, 40333. [Google Scholar] [CrossRef]
- Koller, M. Review: Biodegradable and biocompatible polyhydroxy-alkanoates (PHA): Auspicious microbial macromolecules for pharmaceutical and therapeutic applications. Molecules 2018, 23, 362. [Google Scholar] [CrossRef] [PubMed]
- Adamus, G.; Kurcok, P.; Radecka, I.; Kowalczuk, M. Bioactive oligomers from natural polyhydroxyalkanoates and their synthetic analogues. Polimery 2017, 62, 317–322. [Google Scholar] [CrossRef]
- Vastano, M.; Pellis, A.; Immirzi, B.; Dal Poggetto, G.; Malinconico, M.; Sannia, G.; Guebitz, G.M.; Pezzella, C. Enzymatic production of clickable and PEGylated recombinant polyhydroxyalkanoates. Green Chem. 2017, 19, 5494. [Google Scholar] [CrossRef]
- Winnacker, M.; Rieger, B. Copolymers of polyhydroxyalkanoates and polyethylene glycols: Recent advancements with biological and medical significance, Polym. Int. 2016, 66, 497–503. [Google Scholar] [CrossRef]
- Bonartsev, A.P.; Zharkova, I.I.; Yakovlev, S.G.; Myshkina, V.L.; Makhina, T.K.; Zernov, A.L.; Kudryashova, K.S.; Feofanov, A.V.; Akulina, E.A.; Ivanova, E.V.; et al. 3D-Scaffolds from poly(3-hydroxybutyrate)-poly(ethylene glycol) copolymer for tissue engineering. J. Biomater. Tissue Eng. 2016, 6, 42–52. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Acid Number (mg KOH/g) | Process Conditions |
|---|---|---|
| PS0 | 22.0 | N/A |
| PS1 | 39.0 | 60 °C, O3/O2, 20 h |
| PS2 | 33.7 | 80 °C, O3/O2, 20 h |
| PS3 | 29.5 | 100 °C, O3/O2, 20 h |
| PS4 | 23.6 | 180 °C, O3/O2, 20 h |
| Sample | Mw | Mn | Ð (Mw/Mn) |
|---|---|---|---|
| PS0 | 65,000 | 22,000 | 3.0 |
| PS1 | 57,000 | 19,000 | 3.0 |
| PS2 | 48,000 | 17,000 | 2.8 |
| PS3 | 42,000 | 14,000 | 3.0 |
| PS4 | 30,000 | 8000 | 3.8 |
| Sample Name | Tmax (°C) | Residue (%) |
|---|---|---|
| PS0 | 408/443 | 2.1 |
| PS1 | 411/441 | 3.9 |
| PS2 | 411/441 | 4.2 |
| PS3 | 411/439 | 4.3 |
| PS4 | 411 | 4.6 |
| Media | Average CDW (g/L) | Average PHA (g/L) | PHA (% w/w) |
|---|---|---|---|
| BSM only | 0.08 ± 0.06 | ND | ND |
| TSB only | 1.61 ± 0.60 | 0.38 ± 0.03 | 17% |
| PS0 + BSM | 0.08 ± 0.02 | ND | ND |
| PS0 + TSB | 1.33 ± 0.20 | 0.52 ± 0.05 | 39% |
| PS1 + TSB | 3.56 ± 0.02 | 1.72 ± 0.03 | 48% |
| PS2 + TSB | 3.04 ± 0.03 | 1.28 ± 0.02 | 42% |
| PS3 + TSB | 2.64 ± 0.04 | 0.96 ± 0.03 | 36% |
| PS4 + TSB | 3.96 * ± 0.05 | ND | ND |
| Sample Name | Tmax (°C) | Residue after First Step (%) | Residue after Second Step (%) (Inorganic Part) |
|---|---|---|---|
| PHA from PS0 | 295/435 | 51.8 | 2.3 |
| PHA from PS1 | 294/435 | 27.0 | 1.5 |
| PHA from PS2 | 289/435 | 10.0 | 1.7 |
| Sample Name | Sample Composition |
|---|---|
| Content of Non-HB Units (%) | |
| PS1-PHA0 | 8.2 |
| PS1-PHAD | 8.3 |
| PS2-PHA0 | 12.1 |
| PS2-PHAD | 11.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnston, B.; Radecka, I.; Hill, D.; Chiellini, E.; Ilieva, V.I.; Sikorska, W.; Musioł, M.; Zięba, M.; Marek, A.A.; Keddie, D.; et al. The Microbial Production of Polyhydroxyalkanoates from Waste Polystyrene Fragments Attained Using Oxidative Degradation. Polymers 2018, 10, 957. https://doi.org/10.3390/polym10090957
Johnston B, Radecka I, Hill D, Chiellini E, Ilieva VI, Sikorska W, Musioł M, Zięba M, Marek AA, Keddie D, et al. The Microbial Production of Polyhydroxyalkanoates from Waste Polystyrene Fragments Attained Using Oxidative Degradation. Polymers. 2018; 10(9):957. https://doi.org/10.3390/polym10090957
Chicago/Turabian StyleJohnston, Brian, Iza Radecka, David Hill, Emo Chiellini, Vassilka Ivanova Ilieva, Wanda Sikorska, Marta Musioł, Magdalena Zięba, Adam A. Marek, Daniel Keddie, and et al. 2018. "The Microbial Production of Polyhydroxyalkanoates from Waste Polystyrene Fragments Attained Using Oxidative Degradation" Polymers 10, no. 9: 957. https://doi.org/10.3390/polym10090957
APA StyleJohnston, B., Radecka, I., Hill, D., Chiellini, E., Ilieva, V. I., Sikorska, W., Musioł, M., Zięba, M., Marek, A. A., Keddie, D., Mendrek, B., Darbar, S., Adamus, G., & Kowalczuk, M. (2018). The Microbial Production of Polyhydroxyalkanoates from Waste Polystyrene Fragments Attained Using Oxidative Degradation. Polymers, 10(9), 957. https://doi.org/10.3390/polym10090957