Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin




Abstract
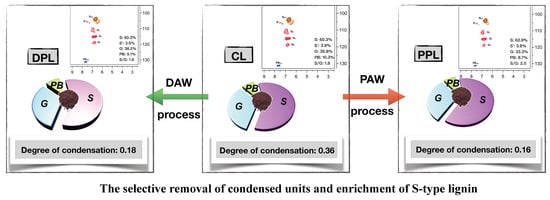
:
1. Introduction
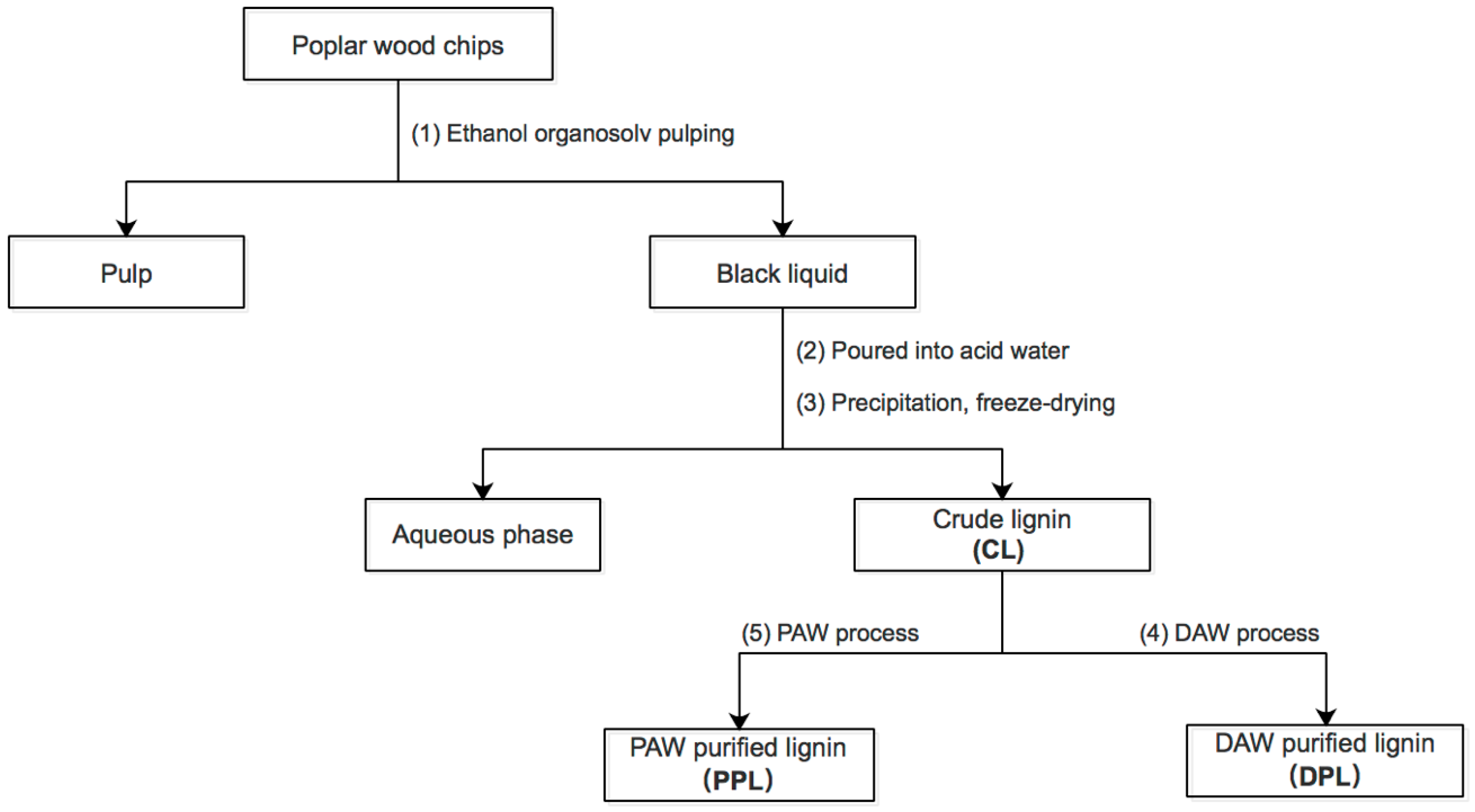
2. Materials and Methods
3. Results and Discussion
3.1. Yield and Composition of Lignin Fractions
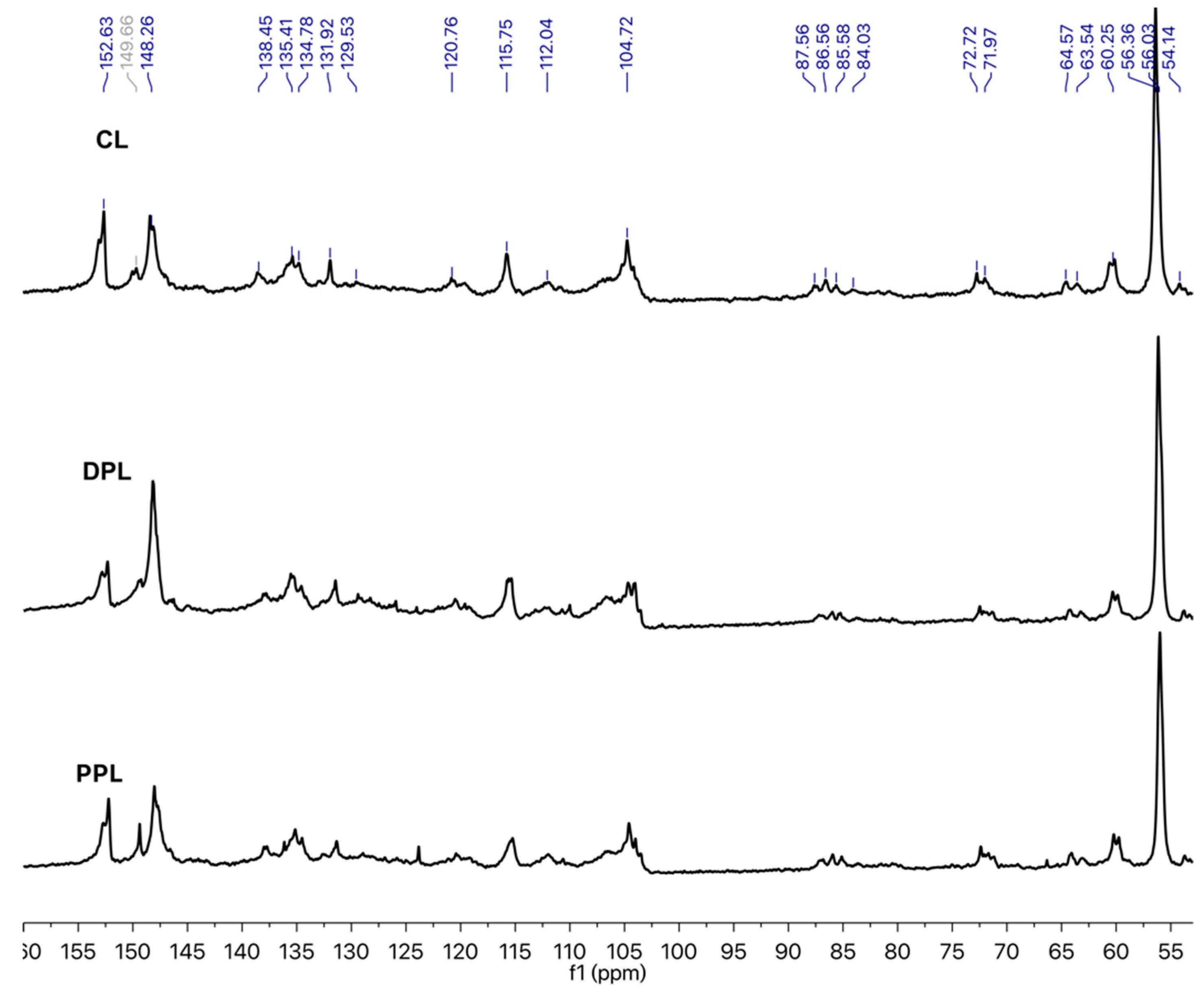
3.2. Internal Linkages and Aromatic Units
3.3. Indentification and Quantification of Functional Groups
3.4. Molecular Weight Distributions
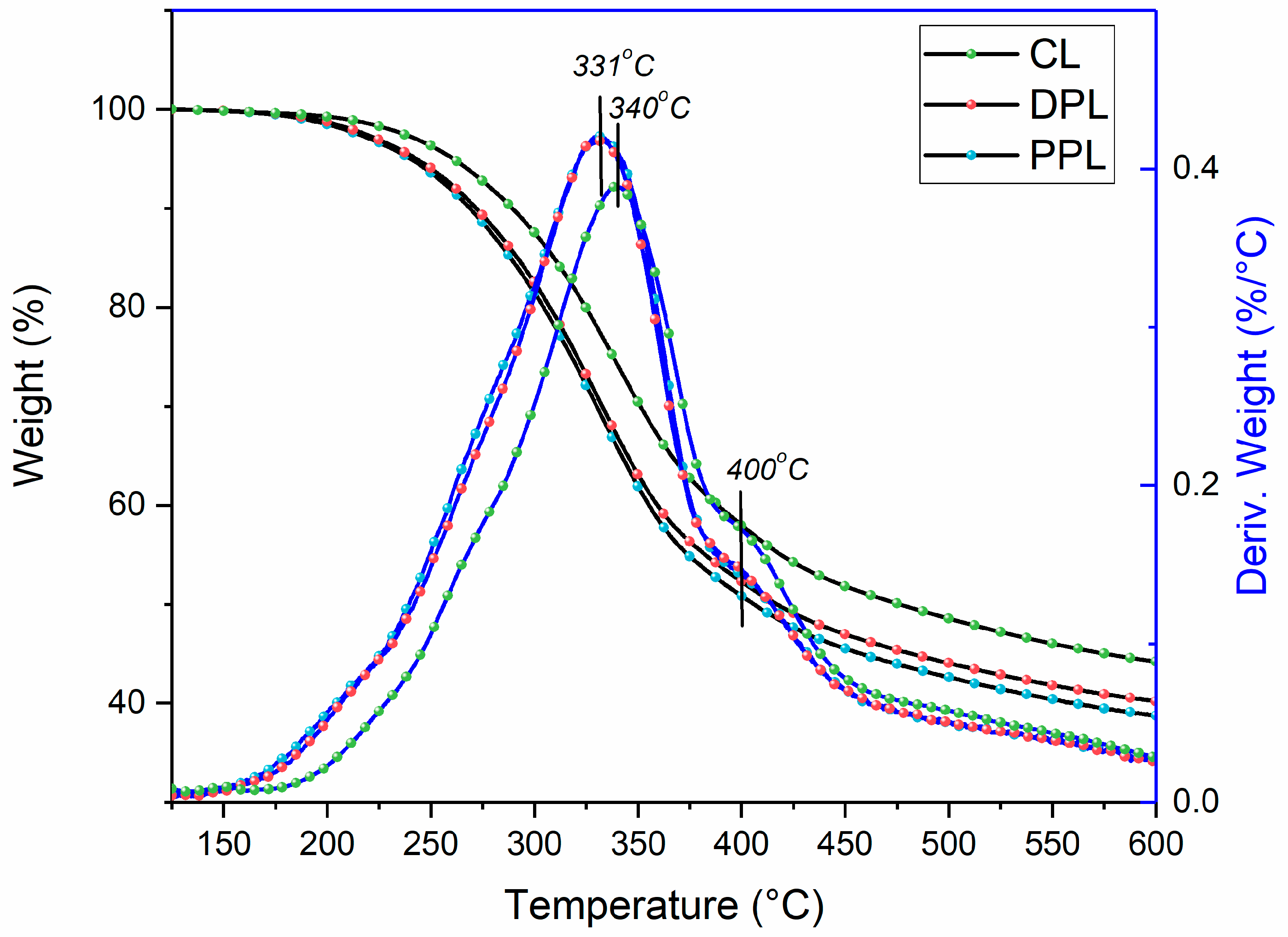
3.5. Thermal Stability
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Faruk, O.; Sain, M.; Farnood, R.; Pan, Y.; Xiao, H. Development of lignin and nanocellulose enhanced bio pu foams for automotive parts. J. Polym. Environ. 2014, 22, 279–288. [Google Scholar] [CrossRef]
- Liu, C.; Xu, J.; Hu, J.; Zhang, H.; Xiao, R. Metal ion-catalyzed hydrothermal liquefaction of calcium lignosulfonate in subcritical water. Chem. Eng. Technol. 2017, 40, 1092–1100. [Google Scholar] [CrossRef]
- Matsushita, Y.; Yasuda, S. Reactivity of a condensed-type lignin model compound in the Mannich reaction and preparation of cationic surfactant from sulfuric acid lignin. J. Wood Sci. 2003, 49, 166–171. [Google Scholar] [CrossRef]
- Lourençon, T.V.; Hansel, F.A.; da Silva, T.A.; Ramos, L.P.; de Muniz, G.I.B.; Magalhães, W.L.E. Hardwood and softwood kraft lignins fractionation by simple sequential acid precipitation. Sep. Purif. Technol. 2015, 154, 82–88. [Google Scholar] [CrossRef]
- Wen, J.-L.; Xue, B.-L.; Sun, S.-L.; Sun, R.-C. Quantitative structural characterization and thermal properties of birch lignins after auto-catalyzed organosolv pretreatment and enzymatic hydrolysis: Quantitative NMR characterization of lignin for biorefinery. J. Chem. Technol. Biotechnol. 2013, 88, 1663–1671. [Google Scholar] [CrossRef]
- Li, H.; McDonald, A.G. Fractionation and characterization of industrial lignins. Ind. Crops Prod. 2014, 62, 67–76. [Google Scholar] [CrossRef]
- Pan, X.; Arato, C.; Gilkes, N.; Gregg, D.; Mabee, W.; Pye, K.; Xiao, Z.; Zhang, X.; Saddler, J. Biorefining of softwoods using ethanol organosolv pulping: Preliminary evaluation of process streams for manufacture of fuel-grade ethanol and co-products. Biotechnol. Bioeng. 2005, 90, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Vvu, S.; Argyropoulos, D. An improved method for lsolating lignin in high yield and purity. J. Pulp. Pap. Sci. 2003, 29, 235–240. [Google Scholar]
- Wang, H.; Chen, W.; Zhang, X.; Wei, Y.; Zhang, A.; Liu, S.; Wang, X.; Liu, C. Structural changes of bagasse dusring the homogeneous esterification with maleic anhydride in ionic liquid 1-allyl-3-methylimidazolium chloride. Polymers 2018, 10, 433. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Garcia, A.; Mondragon, I.; Labidi, J. Comparative study of lignin fractionation by ultrafiltration and selective precipitation. Chem. Eng. J. 2010, 157, 93–99. [Google Scholar] [CrossRef]
- Toledano, A.; García, A.; Mondragon, I.; Labidi, J. Lignin separation and fractionation by ultrafiltration. Sep. Purif. Technol. 2010, 71, 38–43. [Google Scholar] [CrossRef]
- Alekhina, M.; Ershova, O.; Ebert, A.; Heikkinen, S.; Sixta, H. Softwood kraft lignin for value-added applications: Fractionation and structural characterization. Ind. Crops Prod. 2015, 66, 220–228. [Google Scholar] [CrossRef]
- Lundquist, K.; Ohlsson, B.; Simonson, R. Isolation of lignin by means of liquid-liquid extraction. Sven. Papperstid. 1977, 80, 143–144. [Google Scholar]
- Yang, Q.; Wu, S.; Lou, R.; Lv, G. Structural characterization of lignin from wheat straw. Wood Sci. Technol. 2011, 45, 419–431. [Google Scholar] [CrossRef]
- Hu, F.; Jung, S.; Ragauskas, A. Impact of pseudolignin versus dilute acid-pretreated lignin on enzymatic hydrolysis of cellulose. ACS Sustain. Chem. Eng. 2013, 1, 62–65. [Google Scholar] [CrossRef]
- Li, J.-B.; Wu, S.-B.; Li, X.-H. Chemical structure and thermochemical properties of enzymatically acidolyzed lignins from soft and hard wood. BioResources 2013, 8, 5120–5132. [Google Scholar] [CrossRef]
- Lv, G.; Wu, S.; Lou, R.; Yang, Q. Analytical pyrolysis characteristics of enzymatic/mild acidolysis lignin from sugarcane bagasse. Cellul. Chem. Technol. 2010, 44.9, 335. [Google Scholar]
- Xiao, Z.; Li, Y.; Wu, X.; Qi, G.; Li, N.; Zhang, K.; Wang, D.; Sun, X.S. Utilization of sorghum lignin to improve adhesion strength of soy protein adhesives on wood veneer. Ind. Crops Prod. 2013, 50, 501–509. [Google Scholar] [CrossRef]
- Dong, A.; Fan, X.; Wang, Q.; Yu, Y.; Wang, P.; Yuan, J.; Cavaco-Paulo, A. Changes on content, structure and surface distribution of lignin in jute fibers after laccase treatment. J. Nat. Fibers 2018, 15, 384–395. [Google Scholar] [CrossRef]
- Tian, W.; Li, H.; Zhou, J.; Guo, Y. Preparation, characterization and the adsorption characteristics of lignin/silica nanocomposites from cellulosic ethanol residue. RSC Adv. 2017, 7, 41176–41181. [Google Scholar] [CrossRef] [Green Version]
- Sadeghifar, H.; Dickerson, J.P.; Argyropoulos, D.S. Quantitative 31P NMR analysis of solid wood offers an insight into the acetylation of its components. Carbohydr. Polym. 2014, 113, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, J.; Wen, J.; Sun, G.; Sun, Y. Structural transformations of triploid of Populus tomentosa Carr. lignin during auto-catalyzed ethanol organosolv pretreatment. Ind. Crops Prod. 2015, 76, 522–529. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass. Lab. Anal. Proced. 2008, 1617, 1–16. [Google Scholar]
- Santos, J.I.; Martín-Sampedro, R.; Fillat, Ú.; Oliva, J.M.; Negro, M.J.; Ballesteros, M.; Eugenio, M.E.; Ibarra, D. Evaluating lignin-rich residues from biochemical ethanol production of wheat straw and olive tree pruning by FTIR and 2D-NMR. Int. J. Polym. Sci. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Y.; Zhou, J.; Sun, G. Structural changes of poplar wood lignin after supercritical pretreatment using carbon dioxide and ethanol-water as co-solvents. RSC Adv. 2017, 7, 8314–8322. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Lin, F.; Zhang, H.; Xiao, R. Structural elucidation of industrial bioethanol residual lignin from corn stalk: A potential source of vinyl phenolics. Fuel Process. Technol. 2018, 169, 50–57. [Google Scholar] [CrossRef]
- An, Y.-X.; Li, N.; Wu, H.; Lou, W.-Y.; Zong, M.-H. Changes in the structure and the thermal properties of kraft lignin during its dissolution in cholinium ionic liquids. ACS Sustain. Chem. Eng. 2015, 3, 2951–2958. [Google Scholar] [CrossRef]
- Bauer, S.; Sorek, H.; Mitchell, V.D.; Ibáñez, A.B.; Wemmer, D.E. Characterization of Miscanthus giganteus lignin isolated by ethanol organosolv process under reflux condition. J. Agric. Food Chem. 2012, 60, 8203–8212. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.-L.; Sun, S.-L.; Xue, B.-L.; Sun, R.-C. Recent advances in characterization of lignin polymer by solution-state nuclear magnetic resonance (NMR) methodology. Materials 2013, 6, 359–391. [Google Scholar] [CrossRef] [PubMed]
- García, A.; González Alriols, M.; Spigno, G.; Labidi, J. Lignin as natural radical scavenger. Effect of the obtaining and purification processes on the antioxidant behaviour of lignin. Biochem. Eng. J. 2012, 67, 173–185. [Google Scholar] [CrossRef]
- Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 2005, 53, 9639–9649. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, X.; Tang, B.; Dai, Z.; Chen, K.; Zhou, J. Impact of lignin extraction methods on microstructure and mechanical properties of lignin-based carbon fibers. J. Appl. Polym. Sci. 2018, 135, 45580. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Yields (wt %) 1 | Sugar Compositions (wt %) 2 | Ash (wt %) | |||||
|---|---|---|---|---|---|---|---|---|
| Ara | Gal | Glu | Xyl | Man | Total | |||
| CL | - | 0.02 | 0.07 | 0.20 | 0.92 | 0.9 | 2.11 | 1.34 |
| DPL | 77.3 | 0.01 | 0.03 | 0.11 | 0.63 | 0.04 | 0.82 | 0.37 |
| PPL | 43.9 | ND 3 | ND | 0.01 | 0.2 | ND | 0.21 | ND |
| δ (ppm) | Assignments | Quantitative Results (/Ar) | ||
|---|---|---|---|---|
| CL | DPL | PPL | ||
| 140–160 | Aromatic C–O | 2.20 | 2.39 | 2.53 |
| 123–140 | Aromatic C–C | 2.02 | 1.90 | 1.82 |
| 103–123 | Aromatic C–H | 2.03 | 1.69 | 1.75 |
| 58–91 | Alkyl–O | 2.83 | 1.21 | 1.01 |
| 58–61 | β-O-4 | 0.28 | 0.38 | 0.42 |
| 54–58 | OCH3 | 0.73 | 0.90 | 1.55 |
| Degree of condensation | 0.36 | 0.18 | 0.16 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Zhao, J.; Wang, X.; Guo, Y.; Han, Y.; Zhou, J. Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin. Polymers 2018, 10, 967. https://doi.org/10.3390/polym10090967
Gao S, Zhao J, Wang X, Guo Y, Han Y, Zhou J. Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin. Polymers. 2018; 10(9):967. https://doi.org/10.3390/polym10090967
Chicago/Turabian StyleGao, Si, Ji Zhao, Xing Wang, Yanzhu Guo, Ying Han, and Jinghui Zhou. 2018. "Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin" Polymers 10, no. 9: 967. https://doi.org/10.3390/polym10090967
APA StyleGao, S., Zhao, J., Wang, X., Guo, Y., Han, Y., & Zhou, J. (2018). Lignin Structure and Solvent Effects on the Selective Removal of Condensed Units and Enrichment of S-Type Lignin. Polymers, 10(9), 967. https://doi.org/10.3390/polym10090967