Melt-Spun Nanocomposite Fibers Reinforced with Aligned Tunicate Nanocrystals

 and
and
Abstract
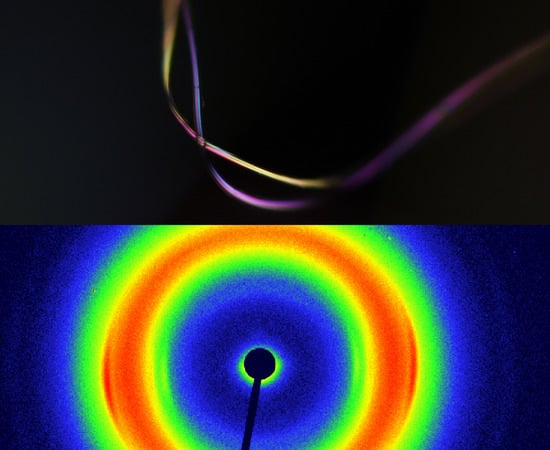
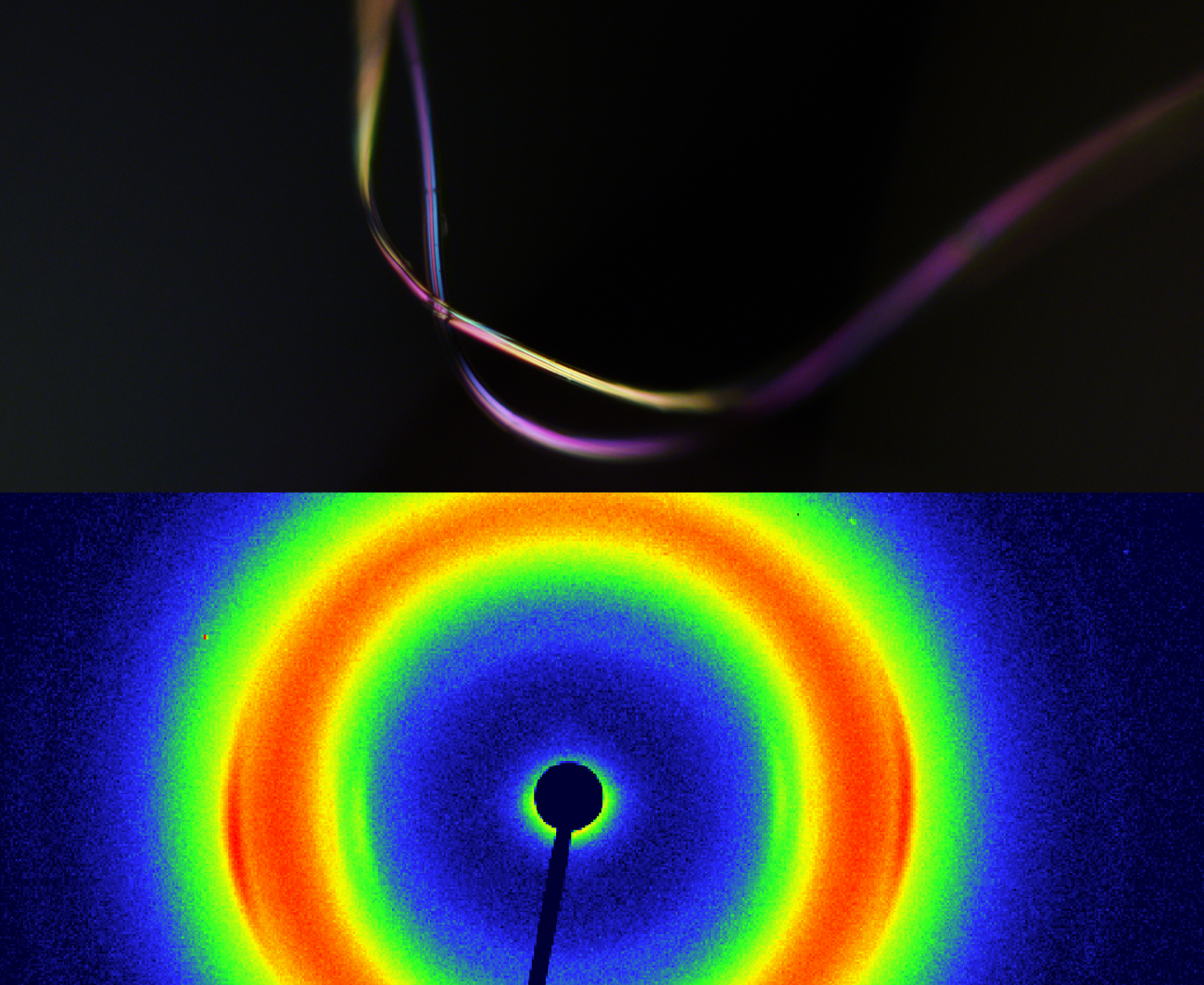
:
1. Introduction
2. Materials and methods
2.1. Materials
2.2. Isolation of Cellulose Nanocrystals (P-tCNCs) Using Phosphoric Acid Hydrolysis
2.3. Isolation of Cellulose Nanocrystals (S-tCNCs) Using Sulfuric Acid Hydrolysis
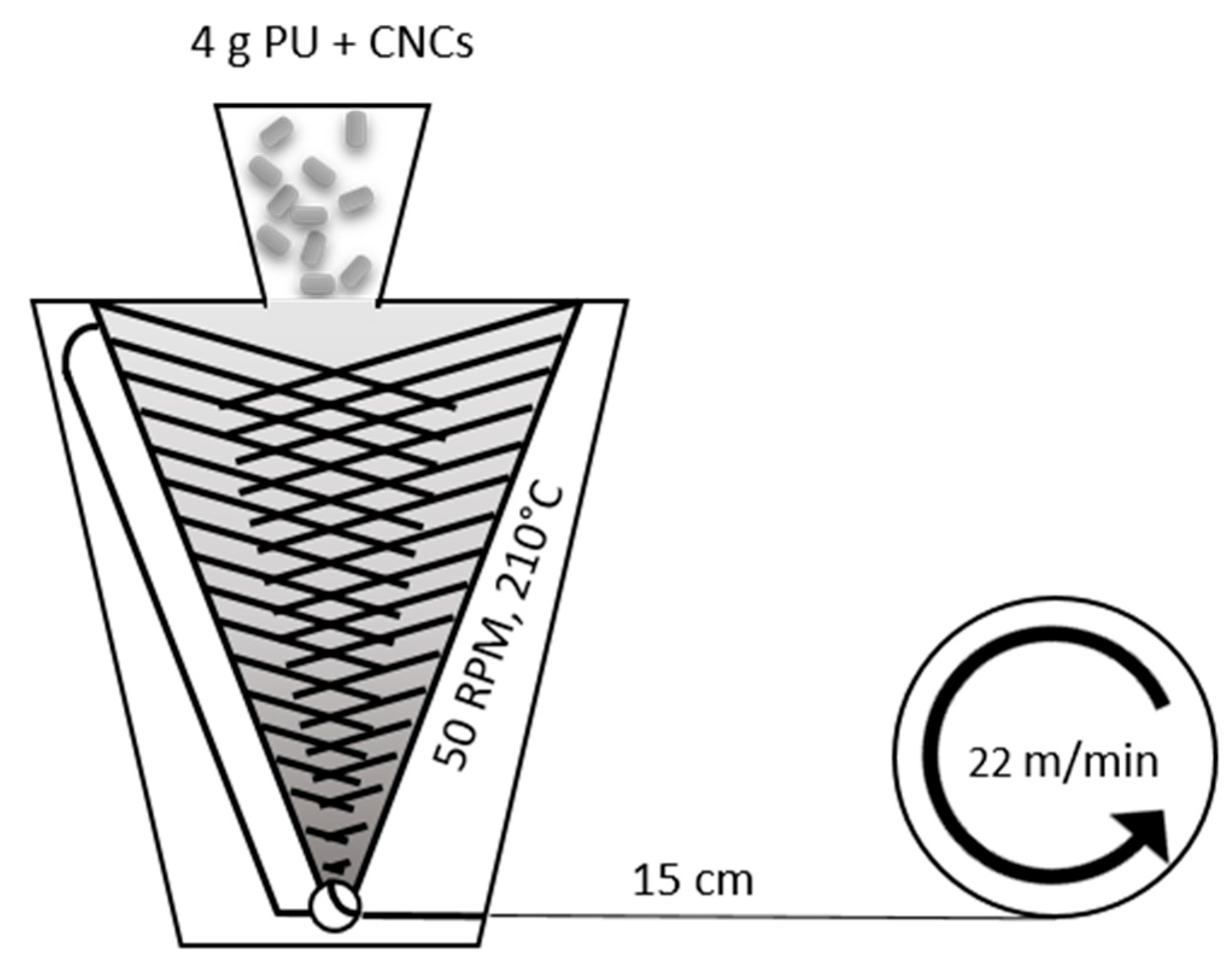
2.4. Nanocomposite Fibers Melt Spinning
2.5. Nanocomposite Film Solvent Casting
2.6. Methods for Characterization
3. Results and Discussion
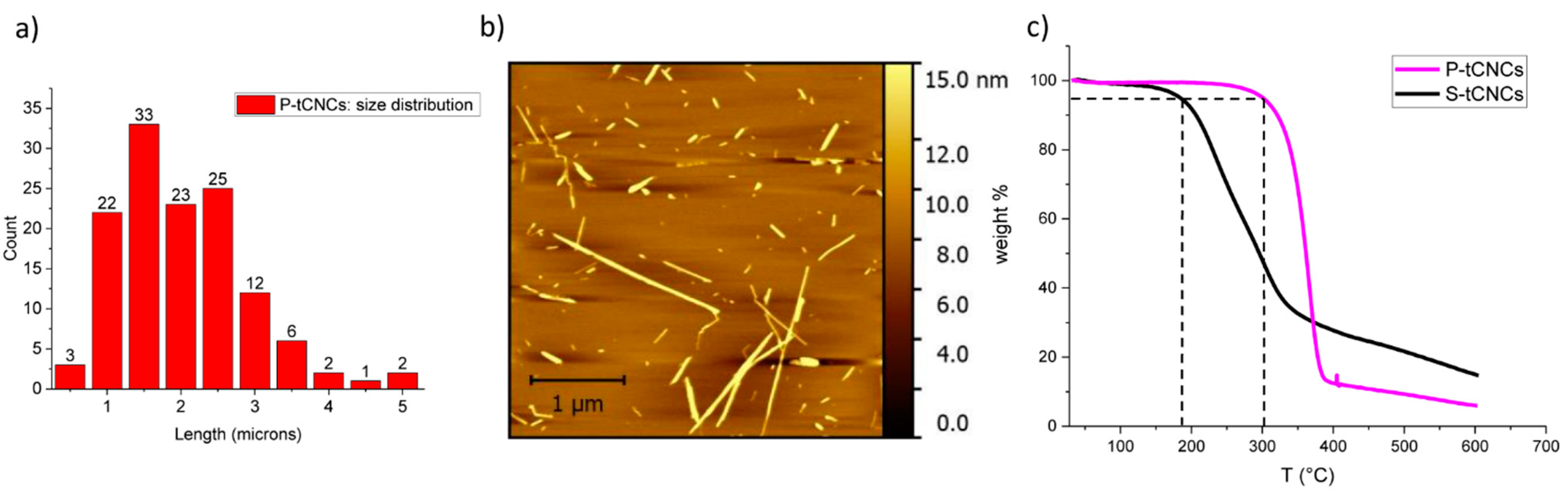
3.1. P-tCNC Extraction and Characterization
3.2. Fiber Processing
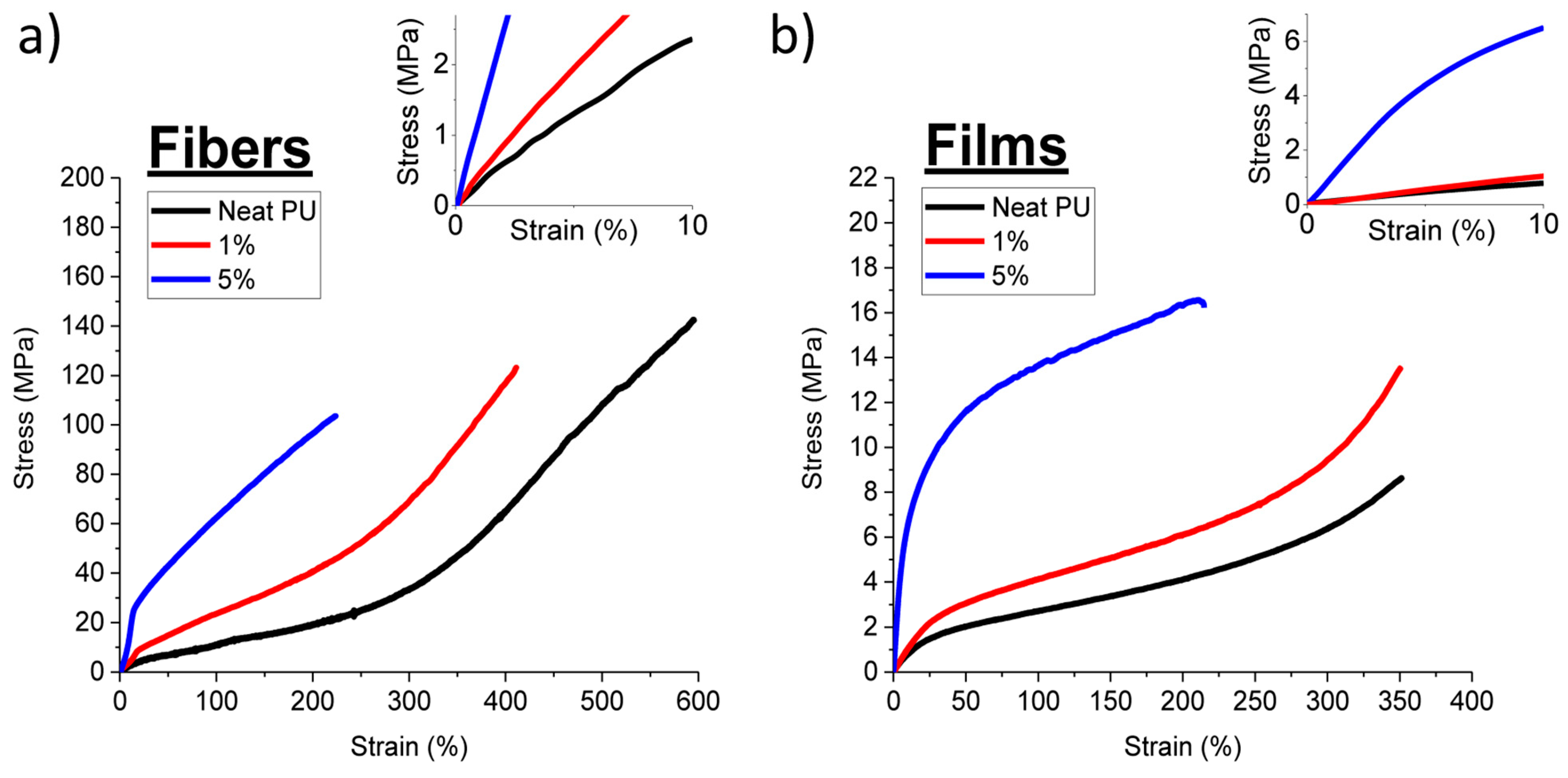
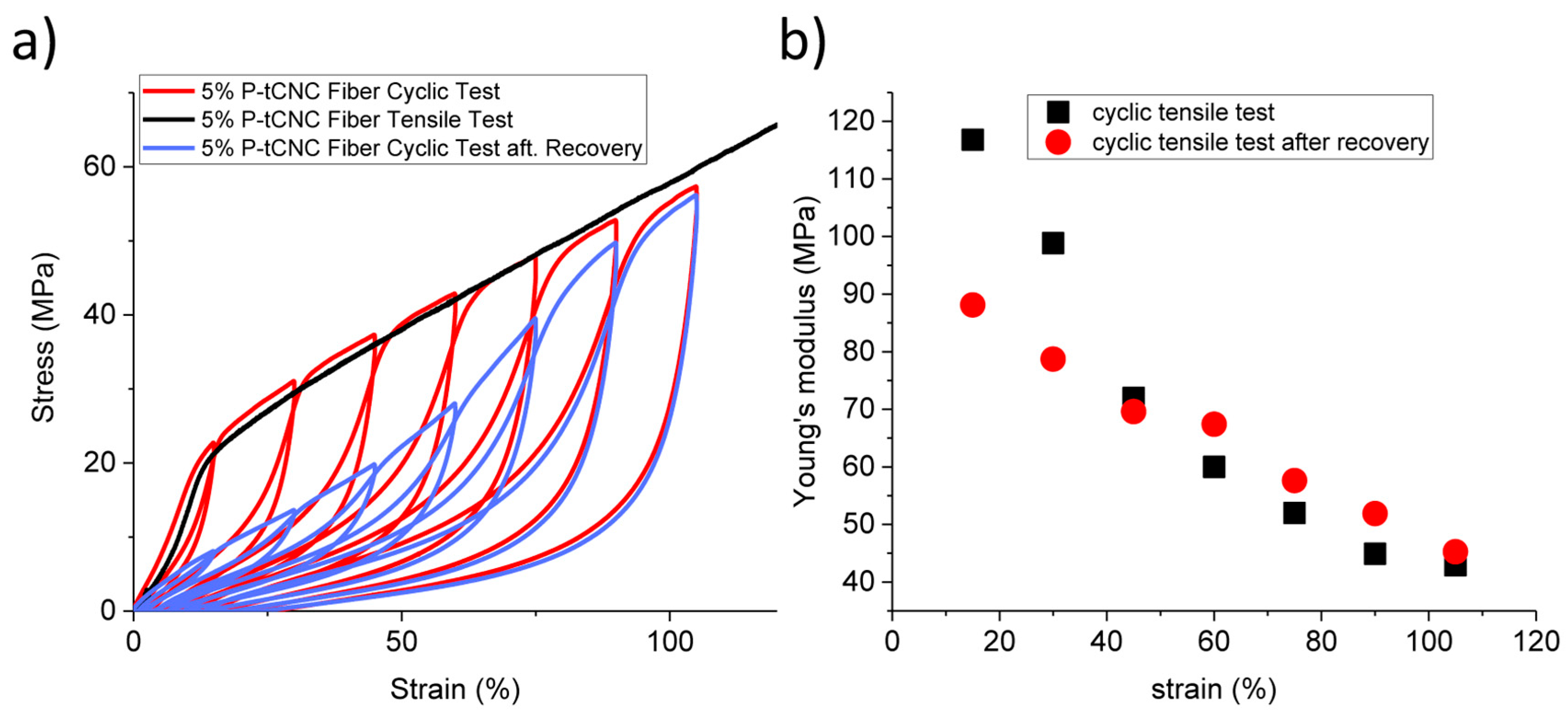
3.3. Mechanical Properties
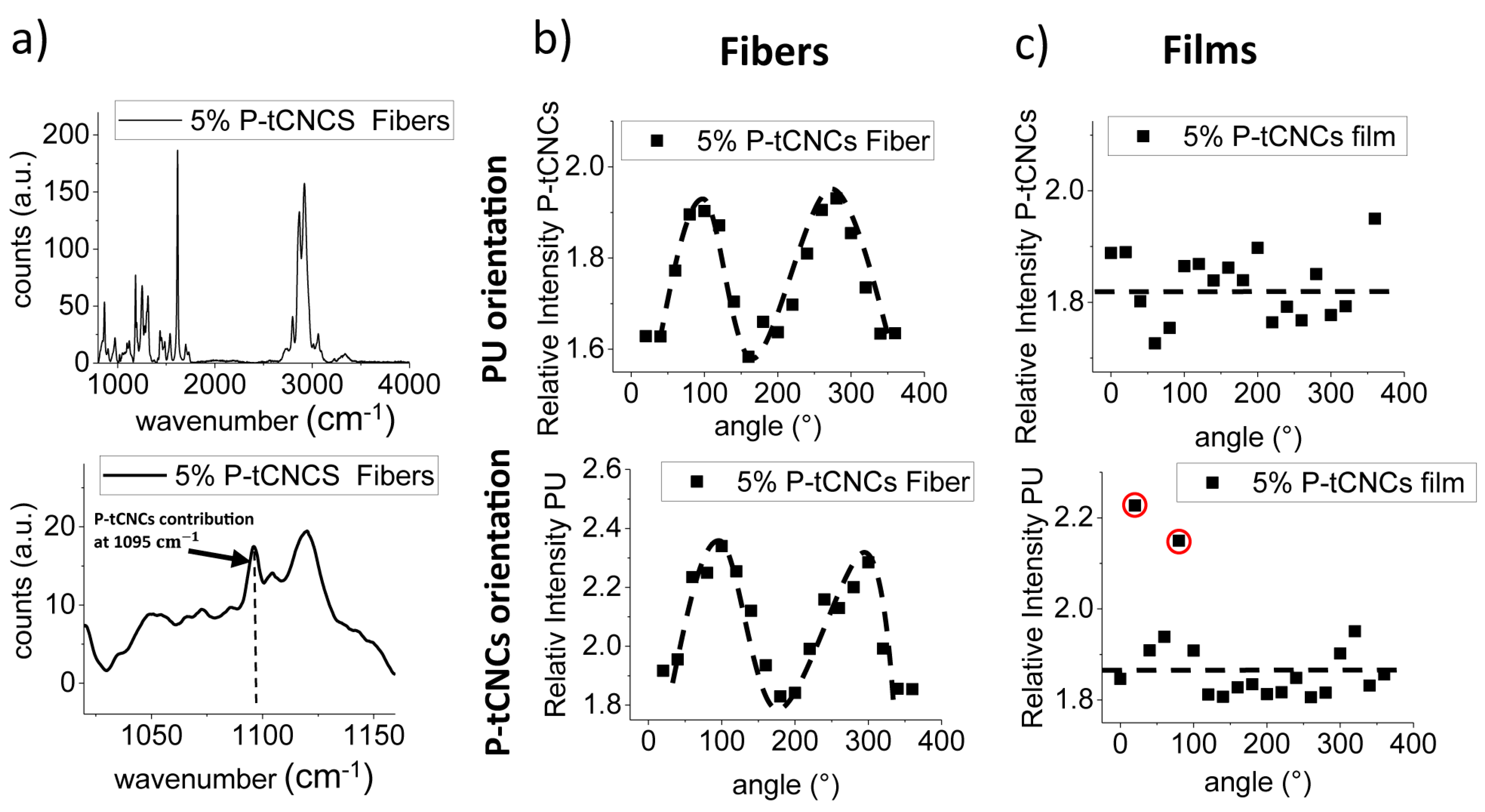
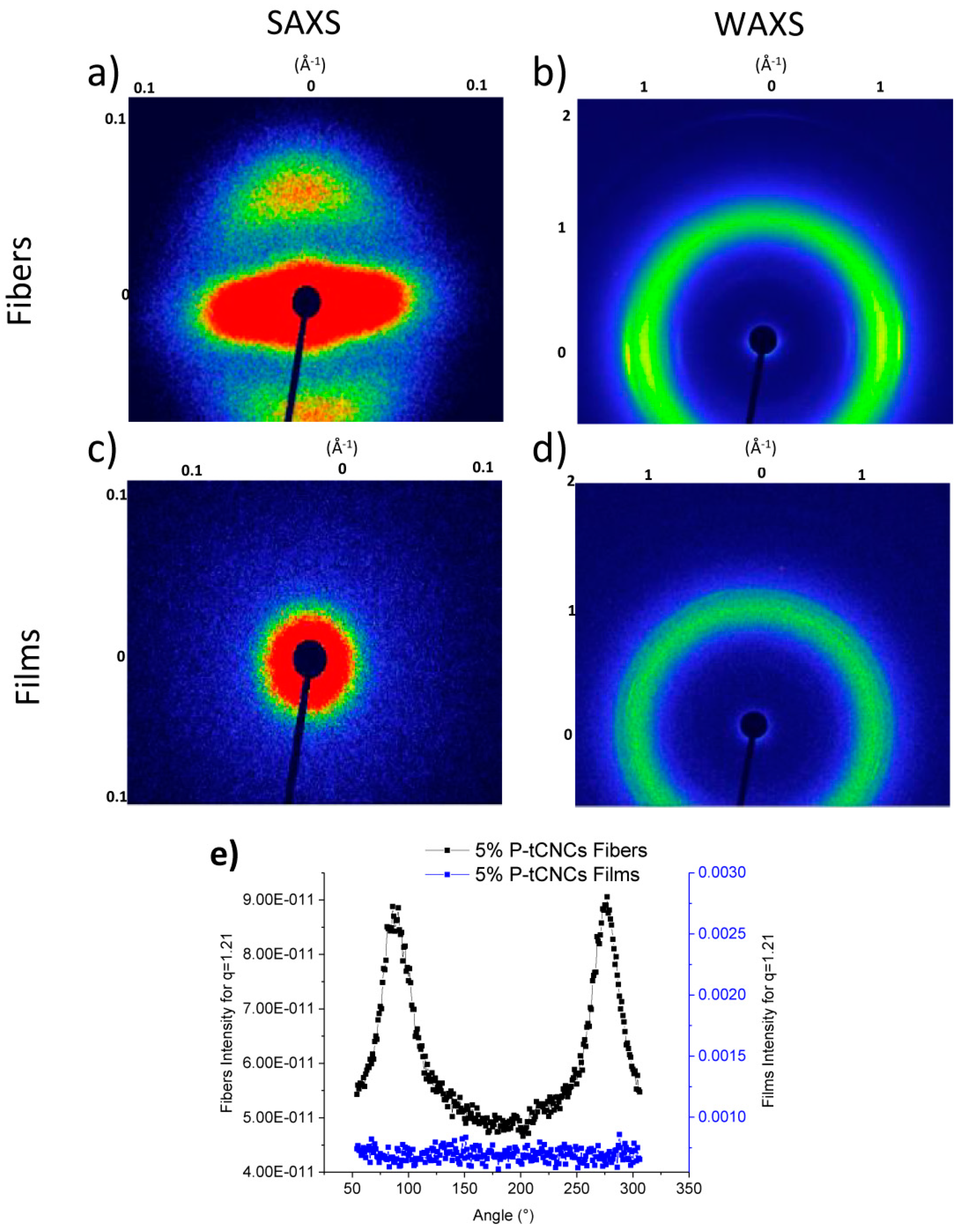
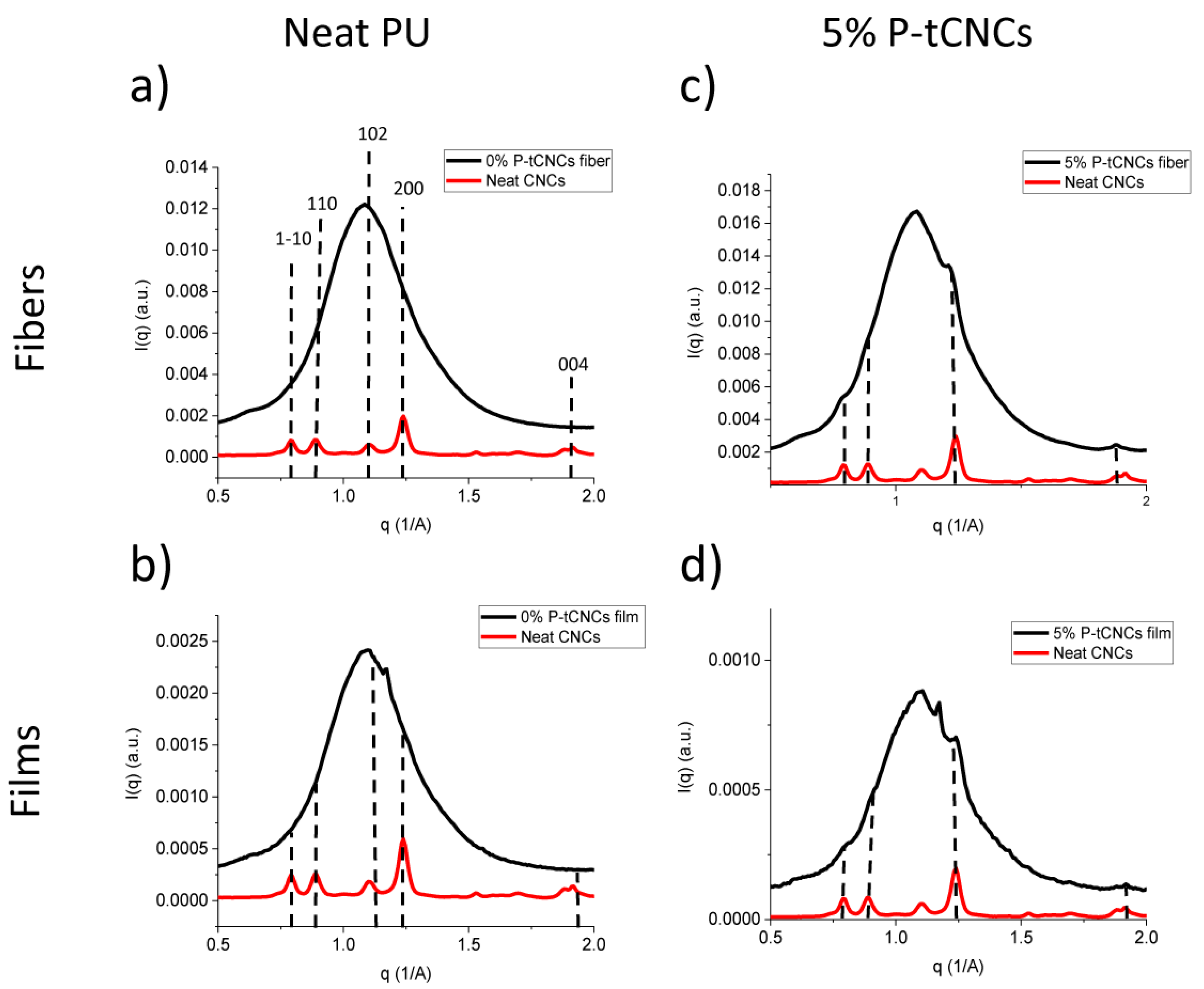
3.4. Orientation of Matrix Polymers and CNCs in Nanocomposite Films and Fibers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mariano, M.; El Kissi, N.; Dufresne, A. Cellulose nanocrystals and related nanocomposites: Review of some properties and challenges. J. Polym. Sci. Part B Polym. Phys. 2014, 52, 791–806. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose Nanocrystals: Chemistry, Self-Assembly, and Applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A. Cellulose nanomaterial reinforced polymer nanocomposites. Curr. Opin. Colloid Interface Sci. 2017, 29, 1–8. [Google Scholar] [CrossRef]
- Dufresne, A. Cellulose nanomaterials as green nanoreinforcements for polymer nanocomposites. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376. [Google Scholar] [CrossRef] [PubMed]
- Endes, C.; Espinosa, S.C.; Mueller, S.; Foster, E.J.; Fink, A.P.; Rutishauser, B.R.; Weder, C. A critical review of the current knowledge regarding the biological impact of nanocellulose. J. Nanobiotechnol. 2016, 14, 78. [Google Scholar] [CrossRef]
- Sapkota, J.; Jorfi, M.; Weder, C.; Foster, E.J. Reinforcing poly(ethylene) with cellulose nanocrystals. Macromol. Rapid Commun. 2014, 35, 1747–1753. [Google Scholar] [CrossRef]
- Sacui, I.A.; Nieuwendaal, R.C.; Burnett, D.J.; Stranick, S.J.; Jorfi, M.; Weder, C.; Foster, E.J.; Olsson, R.T.; Gilman, J.W. Comparison of the properties of cellulose nanocrystals and cellulose nanofibrils isolated from bacteria, tunicate, and wood processed using acid, enzymatic, mechanical, and oxidative methods. ACS Appl. Mater. Interfaces 2014, 6, 6127–6138. [Google Scholar] [CrossRef]
- Oksman, K.; Aitomäki, Y.; Mathew, A.P.; Siqueira, G.; Zhou, Q.; Butylina, S.; Tanpichai, S.; Zhou, X.; Hooshmand, S. Review of the recent developments in cellulose nanocomposite processing. Compos. Part A Appl. Sci. Manuf. 2016, 83, 2–18. [Google Scholar] [CrossRef]
- Nicharat, A.; Shirole, A.; Foster, E.J.; Weder, C. Thermally activated shape memory behavior of melt-mixed polyurethane/cellulose nanocrystal composites. J. Appl. Polym. Sci. 2017, 134, 45033. [Google Scholar] [CrossRef]
- Mendez, J.; Annamalai, P.K.; Eichhorn, S.J.; Rusli, R.; Rowan, S.J.; Foster, E.J.; Weder, C. Bioinspired Mechanically Adaptive Polymer Nanocomposites with Water-Activated Shape-Memory Effect. Macromolecules 2011, 44, 6827–6835. [Google Scholar] [CrossRef]
- Santamaria-Echart, A.; Ugarte, L.; García-Astrain, C.; Arbelaiz, A.; Corcuera, M.A.; Eceiza, A. Cellulose nanocrystals reinforced environmentally-friendly waterborne polyurethane nanocomposites. Carbohydr. Polym. 2016, 151, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Mohd Amin, K.N. Cellulose Nanocrystals Reinforced Thermoplastic Polyurethane Nanocomposites; The University of Queensland: Brisbane, Australia, 2016. [Google Scholar]
- Liu, J.C.; Martin, D.J.; Moon, R.J.; Youngblood, J.P. Enhanced thermal stability of biomedical thermoplastic polyurethane with the addition of cellulose nanocrystals. J. Appl. Polym. Sci. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Wu, Q.; Henriksson, M.; Liu, X.; Berglund, L.A. A high strength nanocomposite based on microcrystalline cellulose and polyurethane. Biomacromolecules 2007, 8, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Dong, H.; Li, C.M. New nanocomposite materials reinforced with flax cellulose nanocrystals in waterborne polyurethane. Biomacromolecules 2007, 8, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Özgür Seydibeyoǧlu, M.; Oksman, K. Novel nanocomposites based on polyurethane and micro fibrillated cellulose. Compos. Sci. Technol. 2008, 68, 908–914. [Google Scholar] [CrossRef]
- Natterodt, J.C.; Shirole, A.; Sapkota, J.; Zoppe, J.O.; Weder, C. Polymer nanocomposites with cellulose nanocrystals made by co-precipitation. J. Appl. Polym. Sci. 2018, 135, 45648. [Google Scholar] [CrossRef]
- Pei, A.; Malho, J.M.; Ruokolainen, J.; Zhou, Q.; Berglund, L.A. Strong nanocomposite reinforcement effects in polyurethane elastomer with low volume fraction of cellulose nanocrystals. Macromolecules 2011, 44, 4422–4427. [Google Scholar] [CrossRef]
- Shirole, A.; Nicharat, A.; Perotto, C.U.; Weder, C. Tailoring the Properties of a Shape-Memory Polyurethane via Nanocomposite Formation and Nucleation. Macromolecules 2018, 51, 1841–1849. [Google Scholar] [CrossRef]
- Nicharat, A.; Sapkota, J.; Weder, C.; Johan Foster, E. Melt processing of polyamide 12 and cellulose nanocrystals nanocomposites. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Marcovich, N.E.; Auad, M.L.; Bellesi, N.E.; Nutt, S.R.; Aranguren, M.I. Cellulose micro/nanocrystals reinforced polyurethane. J. Mater. Res. 2006, 21, 870–881. [Google Scholar] [CrossRef]
- Waletzko, R.S.; Korley, L.T.J.; Pate, B.D.; Thomas, E.L.; Hammond, P.T. Role of Increased Crystallinity in Deformation-Induced Structure of Segmented Thermoplastic Polyurethane Elastomers with PEO and PEO-PPO-PEO Soft Segments and HDI Hard Segments. Macromolecules 2009, 42, 2041–2053. [Google Scholar] [CrossRef]
- James, L.T.; Pate, B.D.; Thomas, E.L.; Hammond, P.T. Effect of the degree of soft and hard segment ordering on the morphology and mechanical behavior of semicrystalline segmented polyurethanes. Polymer 2006, 47, 3073–3082. [Google Scholar]
- Lee, D.; Tsai, H. Properties of Segmented Polyurethanes Derived from Different Diisocyanates. J. Appl. Polym. Sci. 1999, 75, 167–174. [Google Scholar] [CrossRef]
- Xue, B.L.; Wen, J.L.; Zhu, M.Q.; Sun, R.C. Lignin-based polyurethane film reinforced with cellulose nanocrystals. RSC Adv. 2014, 4, 36089–36096. [Google Scholar] [CrossRef]
- Korley, L.T.J.; Liff, S.M.; Kumar, N.; Mckinley, G.H.; Hammond, P.T. Preferential Association of Segment Blocks in Polyurethane Nanocomposites. Macromolecules 2006, 39, 7030–7036. [Google Scholar] [CrossRef]
- Rusli, R.; Shanmuganathan, K.; Rowan, S.J.; Weder, C.; Elchhorn, S.J. Stress-transfer in anisotropic and environmentally adaptive cellulose whisker nanocomposites. Biomacromolecules 2010, 11, 762–768. [Google Scholar] [CrossRef]
- Jalal Uddin, A.; Araki, J.; Gotoh, Y. Extremely oriented tunicin whiskers in poly(vinyl alcohol) nanocomposites. Polym. Int. 2011, 60, 1230–1239. [Google Scholar] [CrossRef]
- Reising, A.B.; Moon, R.J.; Youngblood, J.P. Effect of particle alignment on mechanical properties of neat cellulose nanocrystal films. J. Sci. Technol. For. Prod. Process. 2012, 2, 32–41. [Google Scholar]
- Pullawan, T.; Wilkinson, A.N.; Eichhorn, S.J. Influence of Magnetic Field Alignment of Cellulose Whiskers on the Mechanics of All-Cellulose Nanocomposites. Biomacromolecules 2012, 13, 2528–2536. [Google Scholar] [CrossRef]
- Chen, S.; Schueneman, G.; Pipes, R.B.; Youngblood, J.; Moon, R.J. Effects of crystal orientation on cellulose nanocrystals-cellulose acetate nanocomposite fibers prepared by dry spinning. Biomacromolecules 2014, 15, 3827–3835. [Google Scholar] [CrossRef]
- Yao, J.; Bastiaansen, C.; Peijs, T. High Strength and High Modulus Electrospun Nanofibers. Fibers 2014, 2, 158–186. [Google Scholar] [CrossRef]
- Wanasekara, N.D.; Eichhorn, S.J. Injectable Highly Loaded Cellulose Nanocrystal Fibers and Composites. ACS Macro Lett. 2017, 6, 1066–1070. [Google Scholar] [CrossRef]
- John, M.J.; Anandjiwala, R.; Oksman, K.; Mathew, A.P. Melt-spun polylactic acid fibers: Effect of cellulose nanowhiskers on processing and properties. J. Appl. Polym. Sci. 2013, 127, 274–281. [Google Scholar] [CrossRef]
- Blaker, J.J.; Lee, K.Y.; Walters, M.; Drouet, M.; Bismarck, A. Aligned unidirectional PLA/bacterial cellulose nanocomposite fibre reinforced PDLLA composites. React. Funct. Polym. 2014, 85, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Hooshmand, S.; Aitomäki, Y.; Skrifvars, M.; Mathew, A.P.; Oksman, K. All-cellulose nanocomposite fibers produced by melt spinning cellulose acetate butyrate and cellulose nanocrystals. Cellulose 2014, 21, 2665–2678. [Google Scholar] [CrossRef]
- Clarkson, C.; Youngblood, J. Dry-spinning of cellulose nanocrystal/polylactic acid composite fibers. Green Mater. 2018, 6, 6–14. [Google Scholar] [CrossRef]
- Fallon, J.J.; Kolb, B.Q.; Herwig, C.J.; Foster, E.J.; Bortner, M.J. Mechanically adaptive thermoplastic polyurethane/cellulose nanocrystal composites: Process-driven structure—Property relationships. J. Appl. Polym. Sci. 2019, 136, 46992. [Google Scholar] [CrossRef]
- Brown, P.J.; Kitchens, C.L.; Ure, E.E. Effect of Jet Stretch and Particle Load on Cellulose Nanocrystal-Alginate Nanocomposite Fibers. Langmuir 2010, 26, 14263–14270. [Google Scholar]
- Liu, D.; Li, J.; Sun, F.; Xiao, R.; Song, J. Liquid crystal microphase separation of cellulose nanocrystals in wet-spun PVA composite fibers. RSC Adv. 2014, 4, 30784. [Google Scholar] [CrossRef]
- Wanasekara, N.D.; Santos, R.P.O.; Douch, C.; Frollini, E.; Eichhorn, S.J. Orientation of cellulose nanocrystals in electrospun polymer fibres. J. Mater. Sci. 2016, 51, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Hooshmand, S.; Cho, S.-W.; Skrifvars, M.; Mathew, A.; Oksman, K. Melt spun cellulose nanocomposite fibres: comparison of two dispersion techniques. Plast. Rubber Compos. 2014, 43, 15–24. [Google Scholar] [CrossRef]
- Hooshmand, S.; Skrifvars, M.; Kenny, J.M.; Oksman, K. Poly(lactic acid) melt-spun fibers reinforced with functionalized cellulose nanocrystals. RCS Adv. 2016, 6, 9221–9231. [Google Scholar]
- Camarero Espinosa, S.; Kuhnt, T.; Foster, E.J.; Weder, C. Isolation of thermally stable cellulose nanocrystals by phosphoric acid hydrolysis. Biomacromolecules 2013, 14, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Covestro Texin 985 Data Sheet. Available online: ttps://solutions.covestro.com/en/products/texin/texin-985a_00516217-21873230?SelectedCountry=BM (accessed on 9 October 2019).
- Shanmuganathan, K.; Capadona, J.R.; Rowan, S.J.; Weder, C. Stimuli-Responsive Mechanically Adaptive Polymer Nanocomposites. Appl. Mater. Interfaces 2010, 2, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Weder, C. Cellulose Whisker/Epoxy Resin Nanocomposites. Appl. Mater. Interfaces 2010, 2, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Heux, L.; Chauve, G.; Bonini, C. Nonflocculating and chiral-nematic self-ordering of cellulose microcrystals suspensions in nonpolar solvents. Langmuir 2000, 16, 8210–8212. [Google Scholar] [CrossRef]
- Anglès, M.N.; Dufresne, A. Plasticized Starch/Tunicin Whiskers Nanocomposites. 1. Structural Analysis. Macromolecules 2000, 33, 8344–8353. [Google Scholar] [CrossRef]
- Kaushik, M.; Moores, A. Review: Nanocelluloses as versatile supports for metal nanoparticles and their applications in catalysis. Green Chem. 2016, 18, 622–637. [Google Scholar] [CrossRef] [Green Version]
- Elazzouzi-Hafraoui, S.; Nishiyama, Y.; Putaux, J.; Heux, L.; Dubreuil, F.; Rochas, C. The Shape and Size Distribution of Crystalline Nanoparticles Prepared by Acid Hydrolysis of Native Cellulose. Biomacromolecules 2008, 9, 57–65. [Google Scholar] [CrossRef]
- Capadona, J.R.; Van Den Berg, O.; Capadona, L.A.; Schroeter, M.; Rowan, S.J.; Tyler, D.J.; Weder, C. A versatile approach for the processing of polymer nanocomposites with self-assembled nanofibre templates. Nat. Nanotechnol. 2007, 2, 765–769. [Google Scholar] [CrossRef]
- Annamalai, P.K.; Dagnon, K.L.; Monemian, S.; Foster, E.J.; Rowan, S.J.; Weder, C. Water-Responsive Mechanically Adaptive Nanocomposites Based on Styrene—Butadiene Rubber and Cellulose Nanocrystals—Processing Matters. ACS Appl. Mater. Interfaces 2014, 6, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, J.; Natterodt, J.C.; Shirole, A.; Foster, E.J.; Weder, C. Fabrication and Properties of Polyethylene/Cellulose Nanocrystal Composites. Macromol. Mater. Eng. 2017, 302, 1600300. [Google Scholar] [CrossRef]
- Meesorn, W.; Shirole, A.; Vanhecke, D.; De Espinosa, L.M.; Weder, C. A Simple and Versatile Strategy To Improve the Mechanical Properties of Polymer Nanocomposites with Cellulose Nanocrystals. Macromolecules 2017, 50, 2364–2374. [Google Scholar] [CrossRef]
- Niesten, M.C.E.J.; Krijgsman, J.; Harkema, S.; Gaymans, R.J. Melt Spinnable Spandex Fibers from Segmented Copolyetheresteraramids. J. Appl. Polym. Sci. 2001, 82, 2194–2203. [Google Scholar] [CrossRef]
- Chowdhury, R.A.; Peng, S.X.; Youngblood, J. Improved order parameter (alignment) determination in cellulose nanocrystal (CNC) films by a simple optical birefringence method. Cellulose 2017, 24, 1957–1970. [Google Scholar] [CrossRef]
- Diani, J.; Fayolle, B.; Gilormini, P. A review on the Mullins effect. Eur. Polym. J. 2009, 45, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Mullins, L. Softening Of Rubber Deformation. Rubber Chem. Technol. 1969, 42, 339–362. [Google Scholar] [CrossRef]
- Brandley, E.; Greenhalgh, E.S.; Shaffer, M.S.P.; Li, Q. Mapping carbon nanotube orientation by fast fourier transform of scanning electron micrographs. Carbon 2018, 137, 78–87. [Google Scholar] [CrossRef]
- Wood, J.R.; Zhao, Q.; Wagner, H.D. Orientation of carbon nanotubes in polymers and its detection by Raman spectroscopy. Compos. Part A Appl. Sci. Manuf. 2001, 32, 391–399. [Google Scholar] [CrossRef]
- Pigeon, M.; Prud’homme, R.E.; Pezolet, M. Characterization of Molecular Orientation in Polyethylene by Raman Spectroscopy. Macromolecules 1991, 24, 5687–5694. [Google Scholar] [CrossRef]
- Tanaka, M.; Young, R.J. Review Polarised Raman spectroscopy for the study of molecular orientation distributions in polymers. J. Mater. Sci. 2006, 41, 963–991. [Google Scholar] [CrossRef]
- Adar, F.; Noether, H. Raman microprobe spectra of spin-oriented and drawn filaments of poly(ethylene terephthalate). Polymer 1985, 26, 1935–1943. [Google Scholar] [CrossRef]
- Umesh, P.; Ahmad, I.; Thomas, S.; Dufresne, A. (Eds.) Handbook of Nanocellulose and Cellulose Nanocomposites; Wiley-VCH Verlag GmbH & Company: Weinheim, Germany, 2017. [Google Scholar]
- Chang, H.; Luo, J.; Liu, H.C.; Bakhtiary Davijani, A.A.; Wang, P.H.; Kumar, S. Orientation and interfacial stress transfer of cellulose nanocrystal nanocomposite fibers. Polymer 2017, 110, 228–234. [Google Scholar] [CrossRef]
- Lewandowska, A.E.; Eichhorn, S.J. Raman imaging as a tool for assessing the degree of mixing and the interface between polyethylene and cellulose nanocrystals. IOP Conf. Ser. Mater. Sci. Eng. 2016, 139, 012030. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, U.P.; Sabo, R.; Reiner, R.S.; Clemons, C.M.; Rudie, A.W. Spatially resolved characterization of cellulose nanocrystal—Polypropylene composite by confocal raman microscopy. Appl. Spectrosc. 2012, 66, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Wiley, H.; Atalla, R.H. Band assignments in the Raman spectra of celluloses. Carbohydr. Res. 1987, 160, 113–129. [Google Scholar] [CrossRef]
- Osman, A.F.; Edwards, G.A.; Schiller, T.L.; Andriani, Y.; Jack, K.S.; Morrow, I.C.; Halley, P.J.; Martin, D.J. Structure-property relationships in biomedical thermoplastic polyurethane nanocomposites. Macromolecules 2012, 45, 198–210. [Google Scholar] [CrossRef]
- Desper, C.R.; Schneider, N.S.; Jasinski, J.P.; Lin, J.S. Deformation of Microphase Structures in Segmented Polyurethanes. Macromolecules 1985, 18, 2755–2761. [Google Scholar] [CrossRef]
- Pereira, I.M.; Oréfice, R.L. In situ evaluation of structural changes in poly(ester-urethanes) during shape-memory cycles. Polymer 2010, 51, 1744–1751. [Google Scholar] [CrossRef]
- Koerner, H.; Kelley, J.J.; Vaia, R.A. Transient microstructure of low hard segment thermoplastic polyurethane under uniaxial deformation. Macromolecules 2008, 41, 4709–4716. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tensile Testing | Young’s Modulus (MPa) | Strain-at-Break (%) | Stress-at-Break (MPa) | Toughness (MJ/m3) |
|---|---|---|---|---|
| Neat Fibers | 20 ± 3 | 597 ± 54 | 131 ± 7 | 2.84 ± 0.35 |
| 1 wt% P-tCNC Fibers | 42.5 ± 5.5 | 422 ± 30 | 120 ± 12 | 2 ± 0.2 |
| 5 wt% P-tCNC Fibers | 123.16 ± 10 | 239 ± 22 | 107.5 ± 4 | 1.59 ± 0.16 |
| Neat PU Films | 7.3 ± 0.01 | 361 ± 15 | 9.3 ± 0.4 | 0.14 ± 0.003 |
| 1 wt% P-tCNC Films | 11.8 ± 1 | 340 ± 27 | 12.3 ± 2.5 | 0.19 ± 0.03 |
| 5 wt% P-tCNC Films | 99.5 ± 2 | 224 ± 65 | 17.4 ± 2.5 | 0.3 ± 0.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redondo, A.; Chatterjee, S.; Brodard, P.; Korley, L.T.J.; Weder, C.; Gunkel, I.; Steiner, U. Melt-Spun Nanocomposite Fibers Reinforced with Aligned Tunicate Nanocrystals. Polymers 2019, 11, 1912. https://doi.org/10.3390/polym11121912
Redondo A, Chatterjee S, Brodard P, Korley LTJ, Weder C, Gunkel I, Steiner U. Melt-Spun Nanocomposite Fibers Reinforced with Aligned Tunicate Nanocrystals. Polymers. 2019; 11(12):1912. https://doi.org/10.3390/polym11121912
Chicago/Turabian StyleRedondo, Alexandre, Sourav Chatterjee, Pierre Brodard, LaShanda T. J. Korley, Christoph Weder, Ilja Gunkel, and Ullrich Steiner. 2019. "Melt-Spun Nanocomposite Fibers Reinforced with Aligned Tunicate Nanocrystals" Polymers 11, no. 12: 1912. https://doi.org/10.3390/polym11121912
APA StyleRedondo, A., Chatterjee, S., Brodard, P., Korley, L. T. J., Weder, C., Gunkel, I., & Steiner, U. (2019). Melt-Spun Nanocomposite Fibers Reinforced with Aligned Tunicate Nanocrystals. Polymers, 11(12), 1912. https://doi.org/10.3390/polym11121912