Enhanced Foamability with Shrinking Microfibers in Linear Polymer




Abstract
:
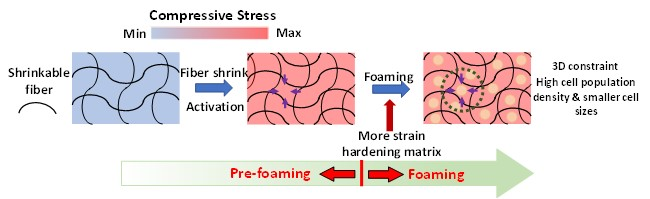
1. Introduction
2. Materials and Methods
2.1. Materials
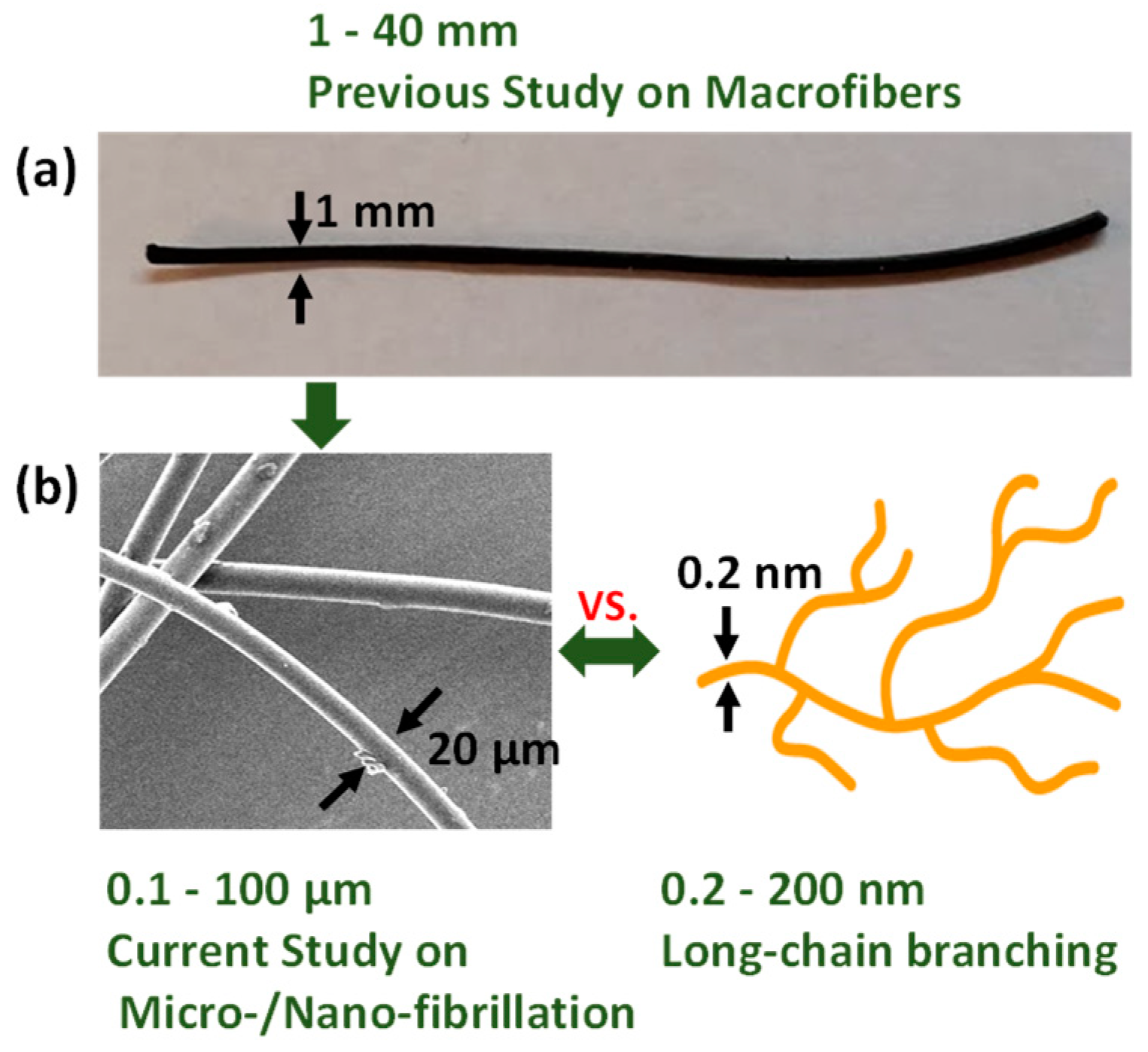
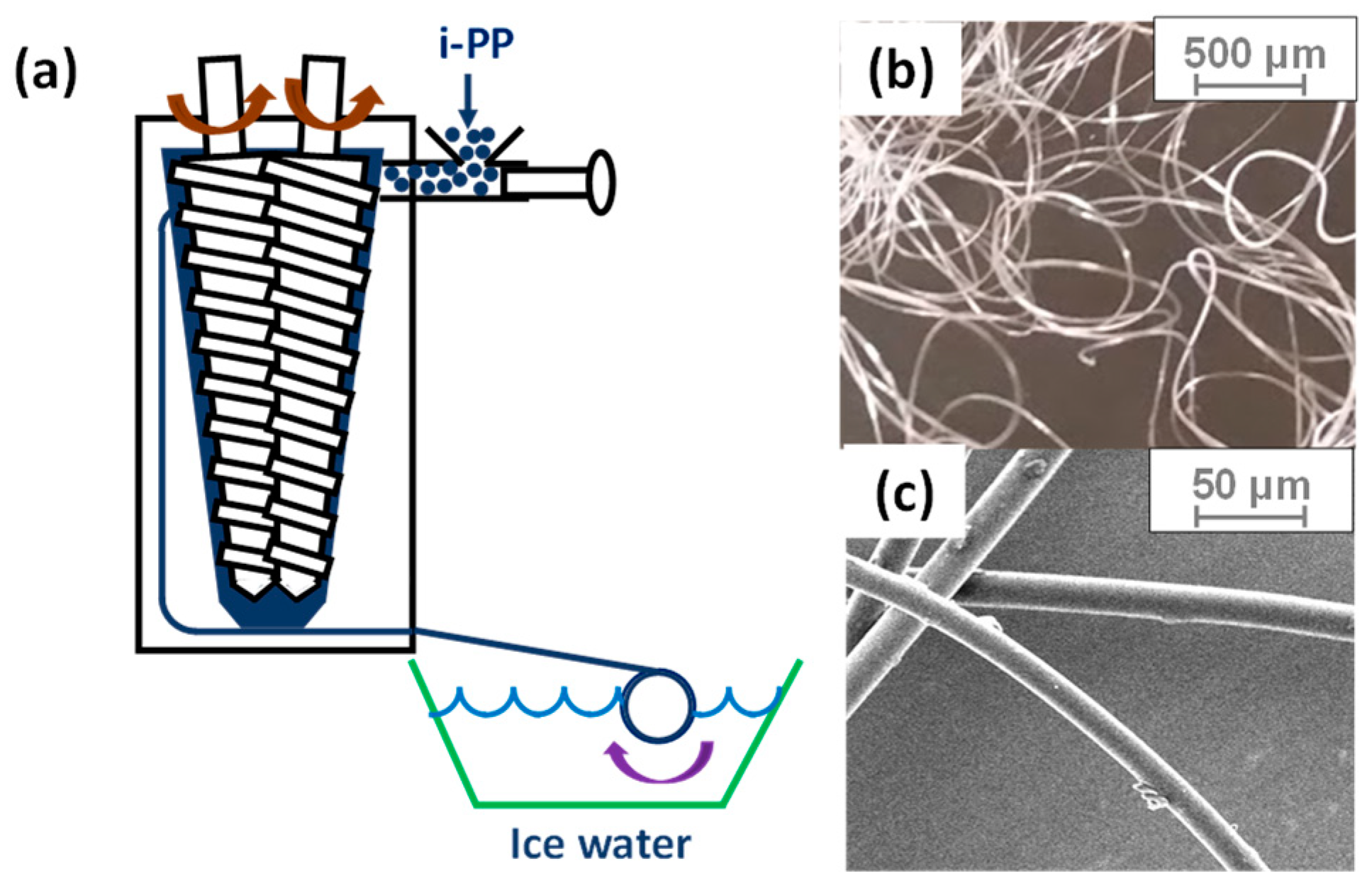
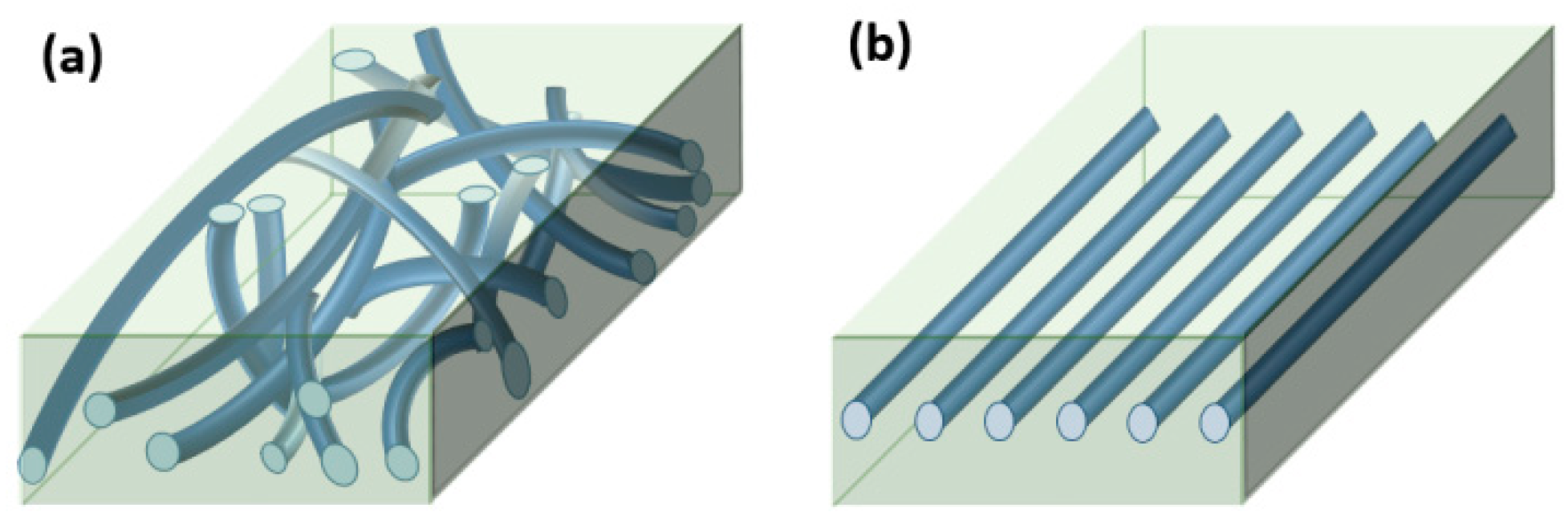
2.2. Sample Preparation
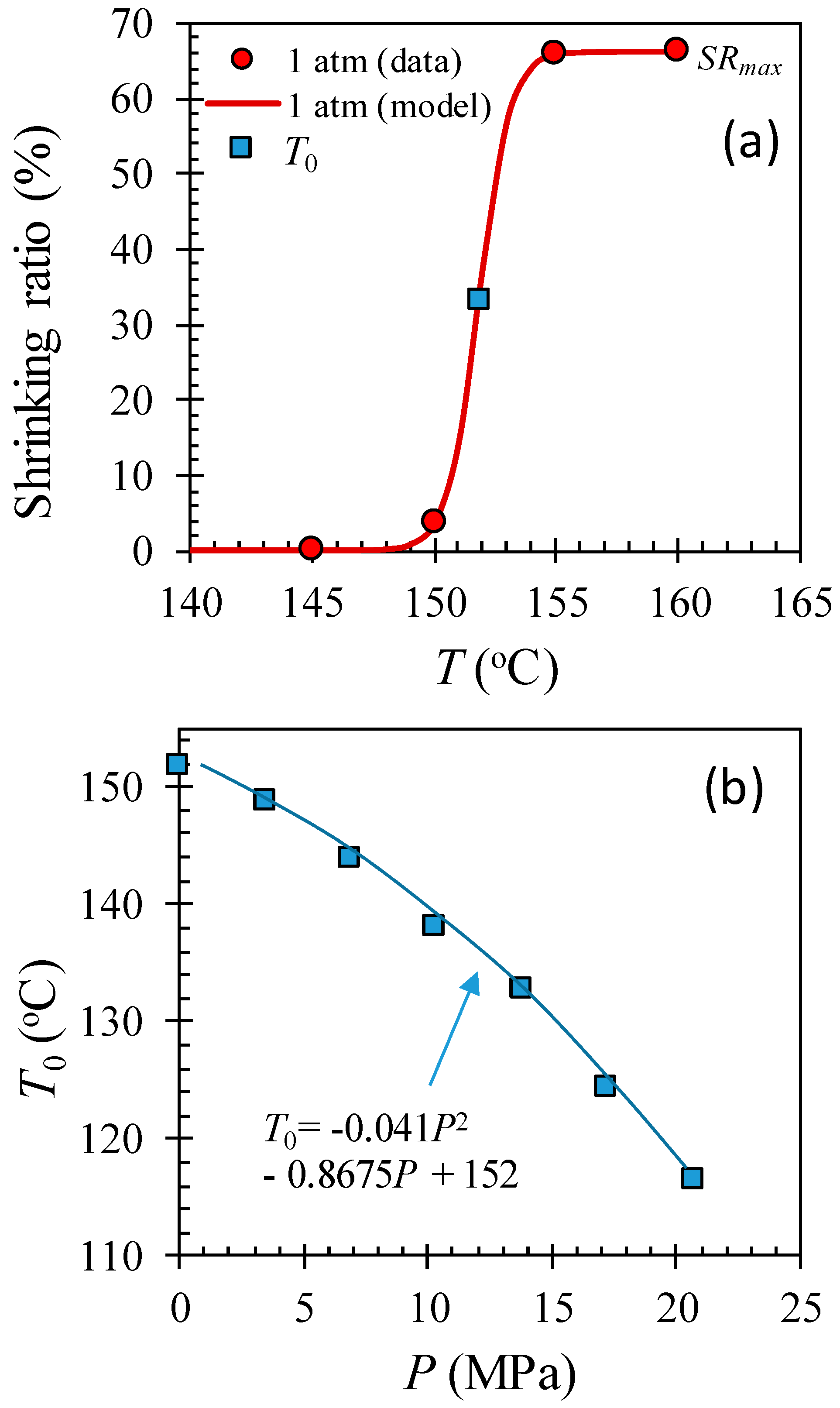
2.3. Microfiber Shrinkage Ratio
2.4. Shrinking Behavior of HAS Microfibers
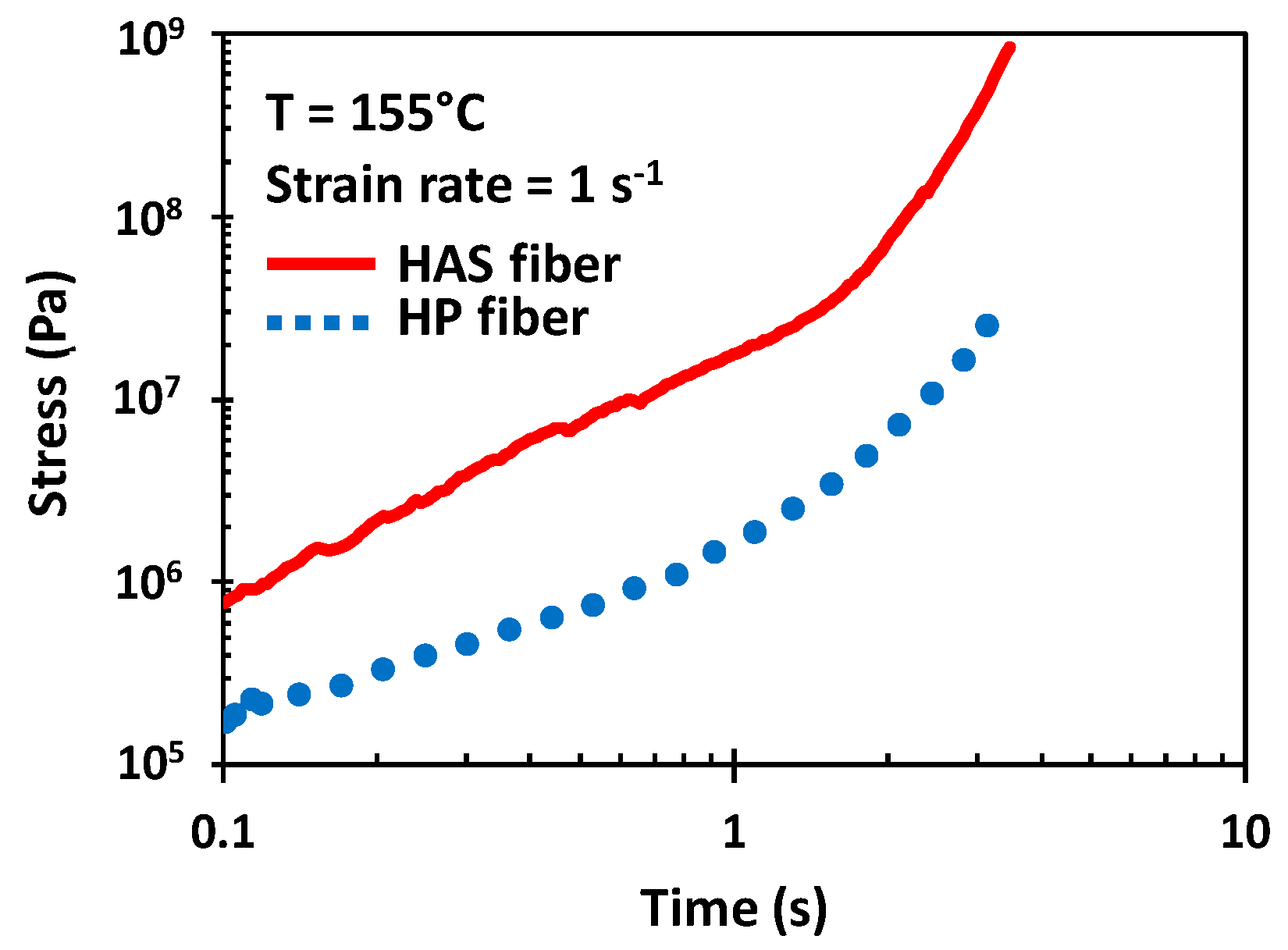
2.5. Tensile Properties of Microfibers
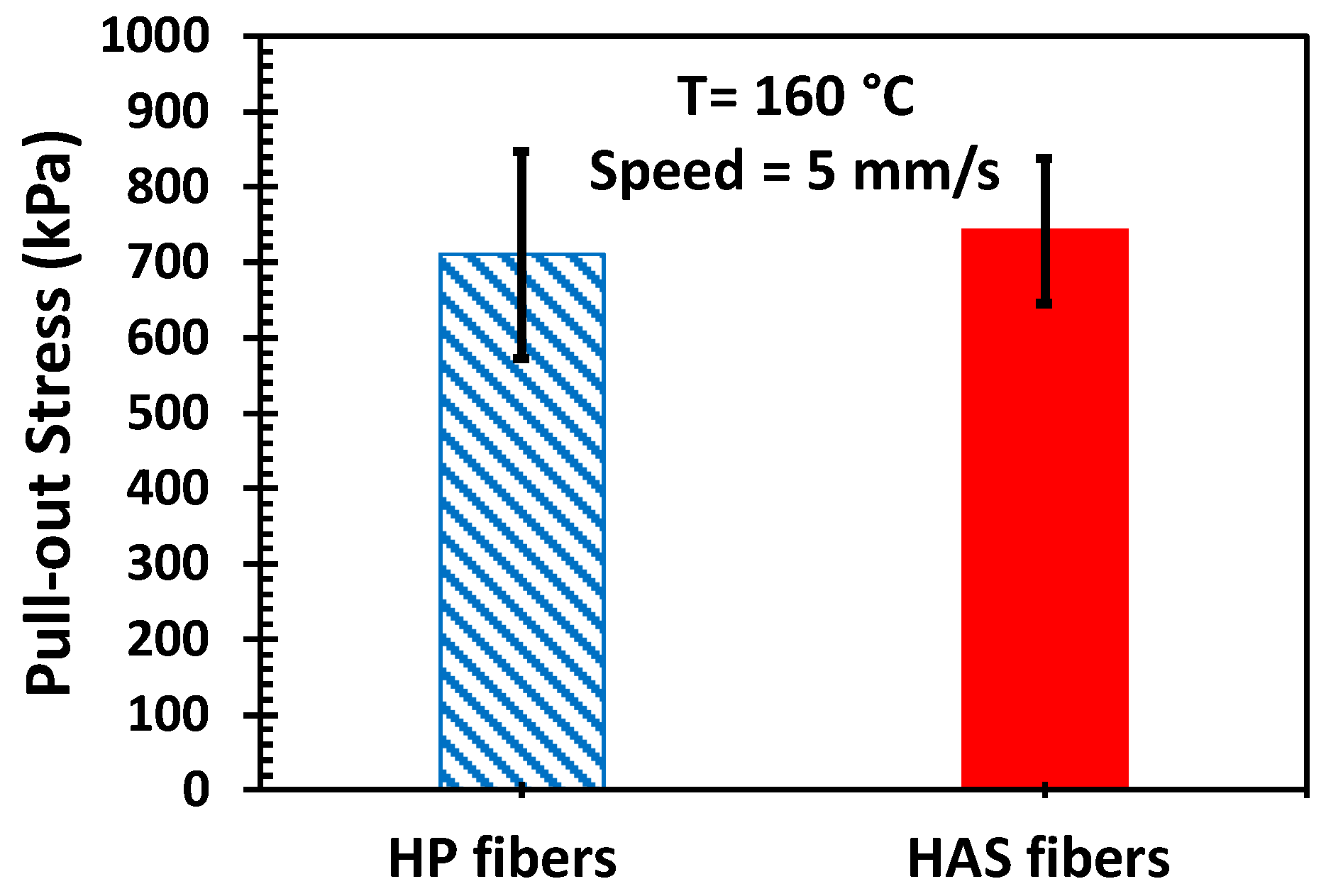
2.6. Single Microfiber Pull-Out Test
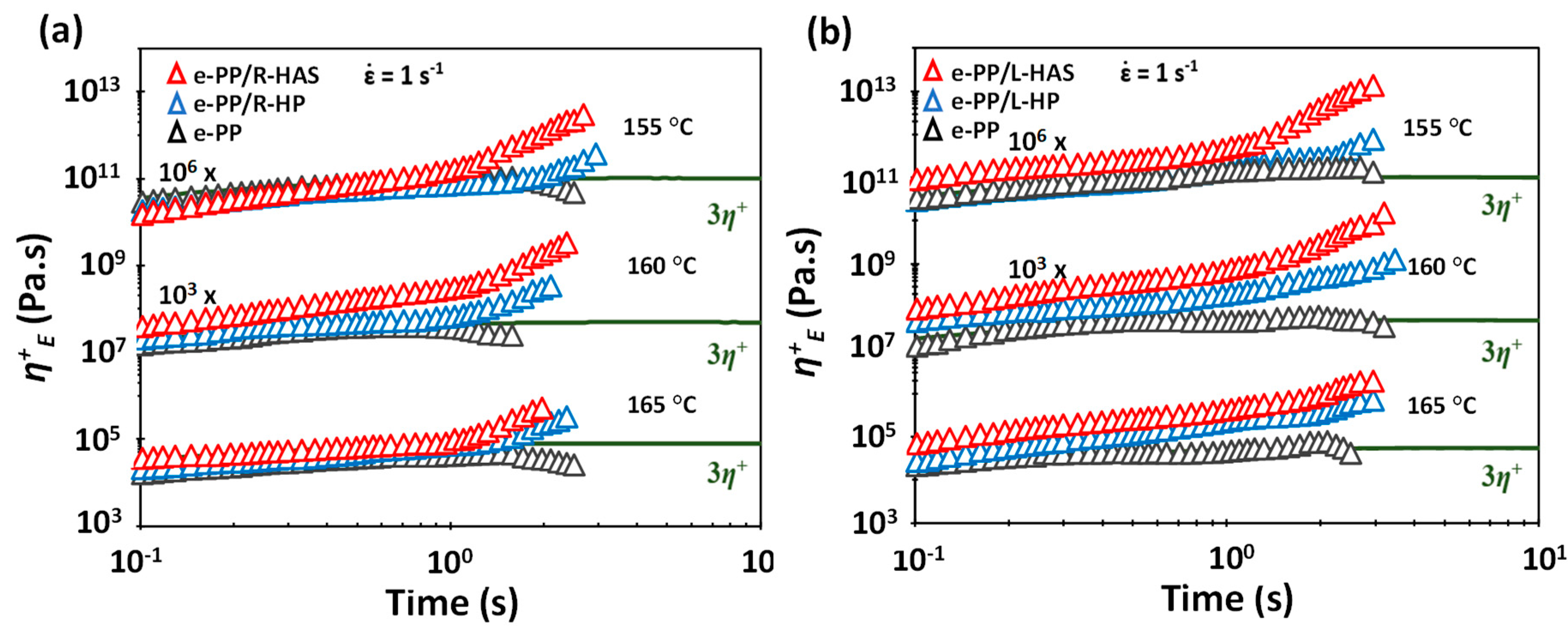
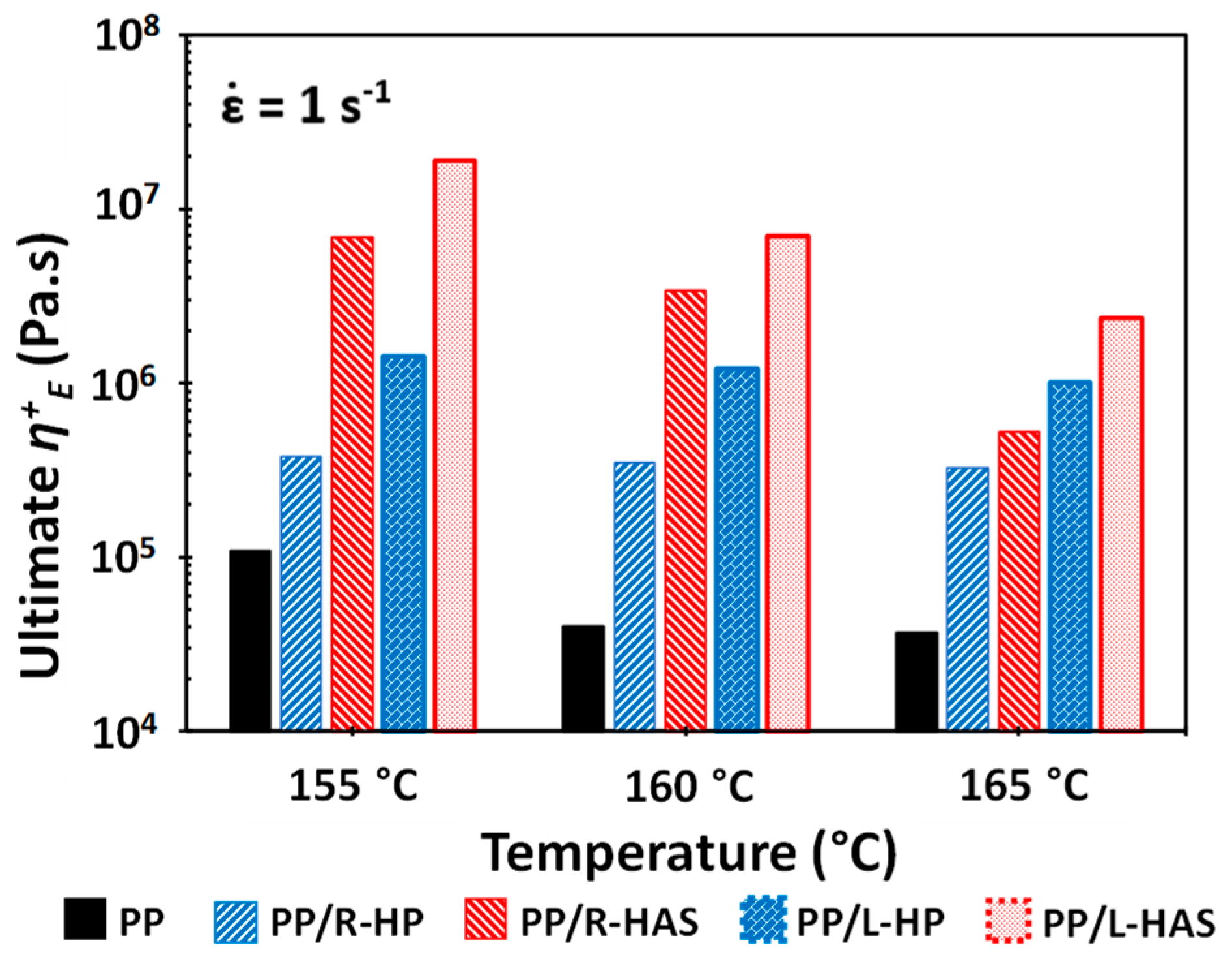
2.7. Extensional Rheometry
2.8. Shear Rheometry
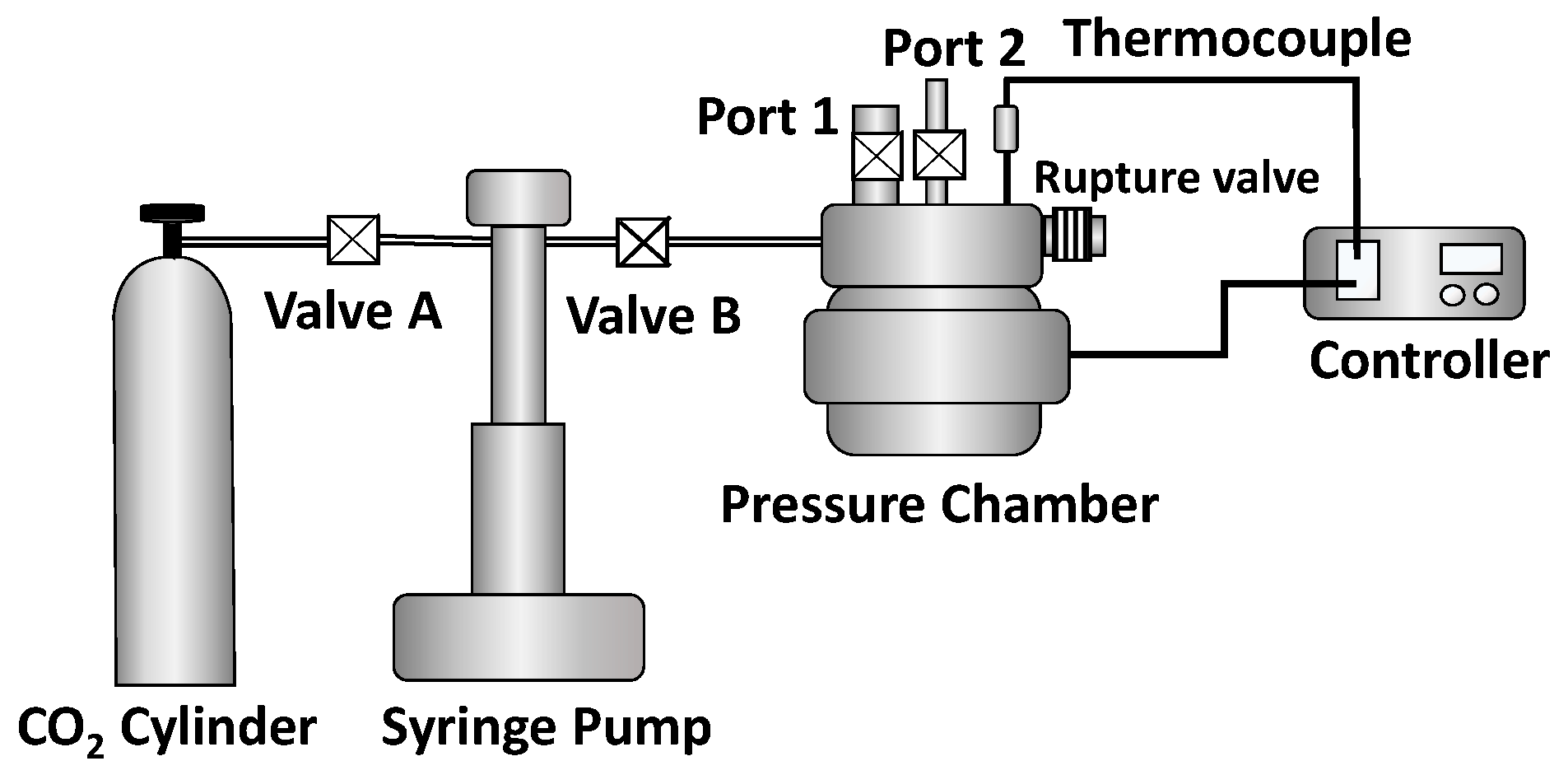
2.9. Batch Physical Foaming
3. Results
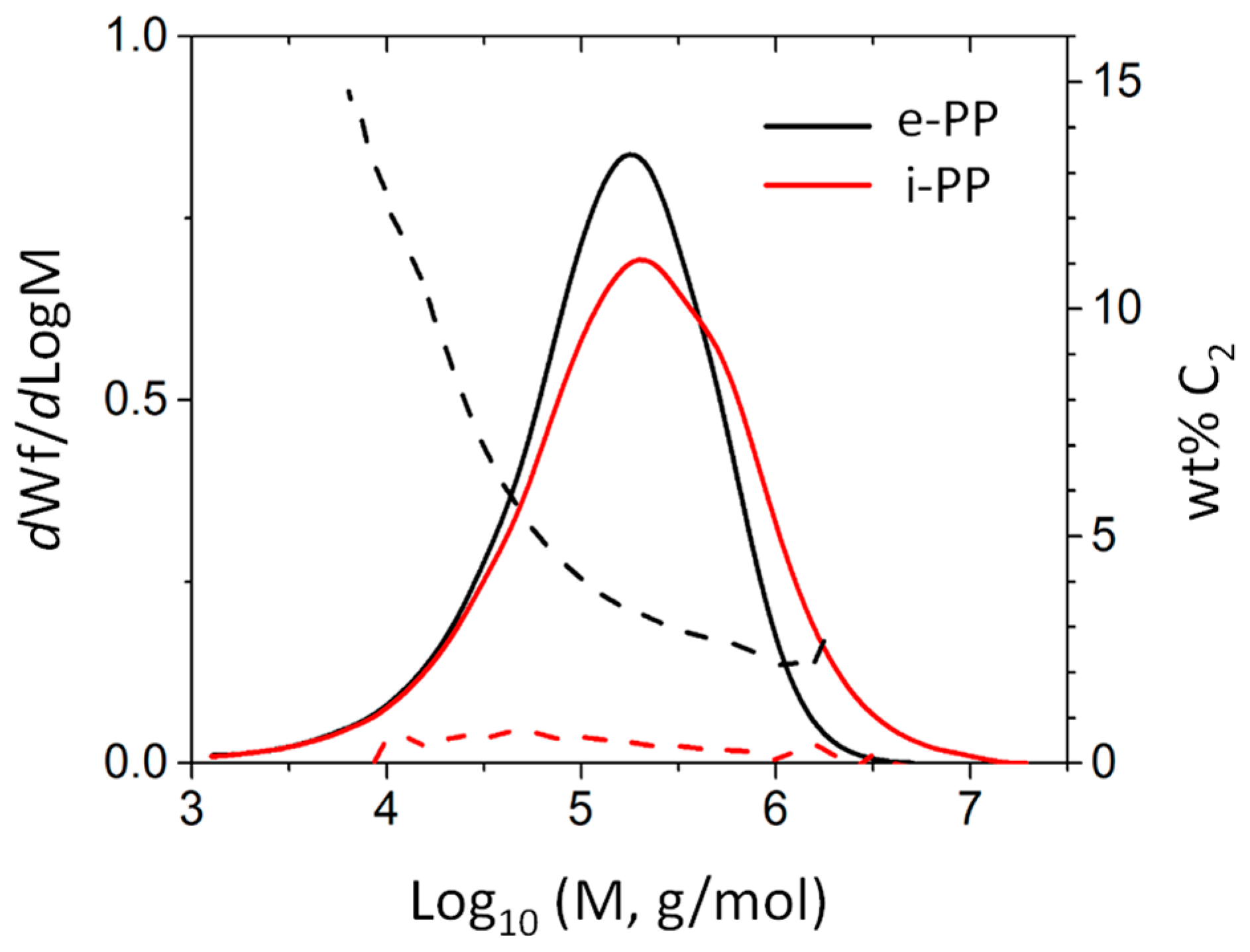
3.1. Polymer Characteristic Analysis
3.2. Tensile Test Properties of HAS and HP Microfibers
3.3. Microfiber Pull-Out Test
3.4. Extensional Rheometry
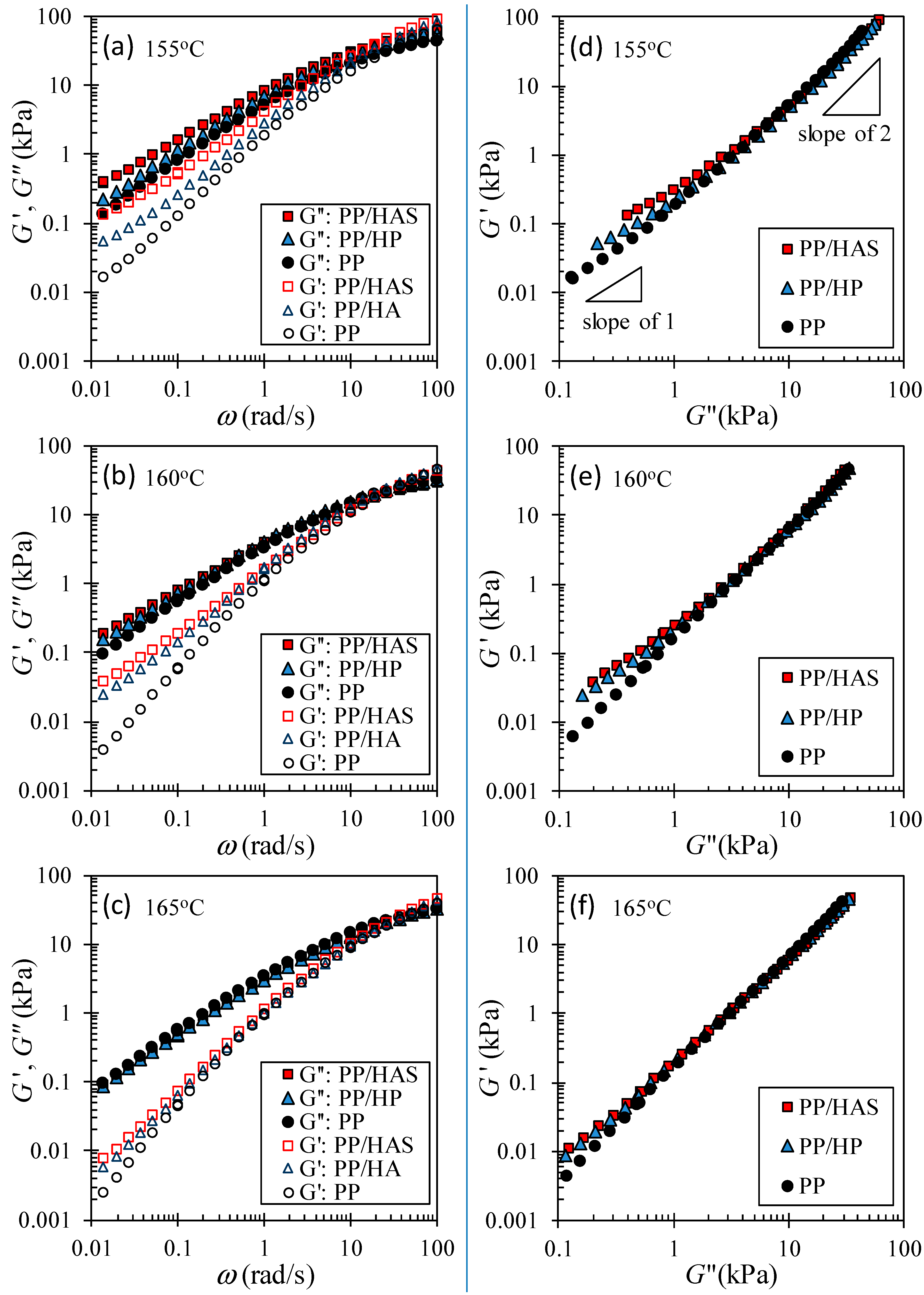
3.5. Linear Viscoelastic Shear Behavior
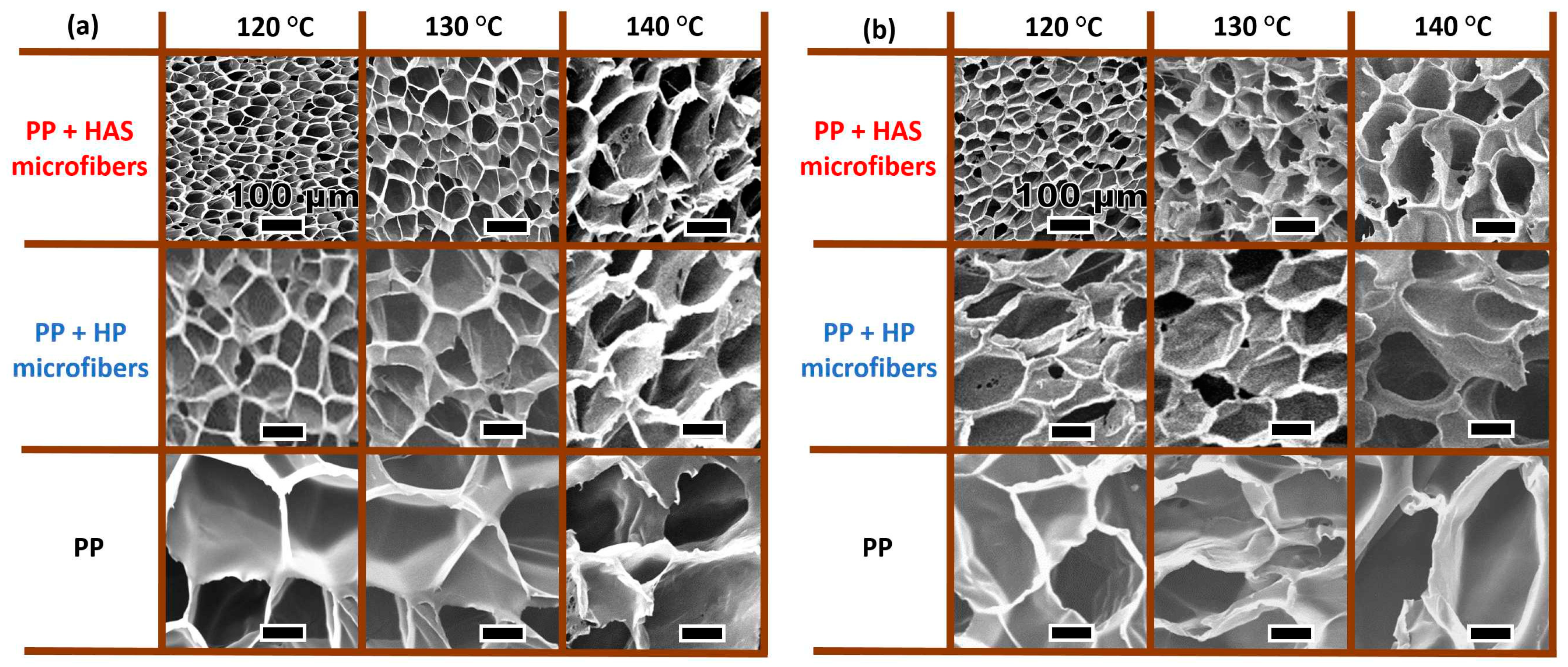
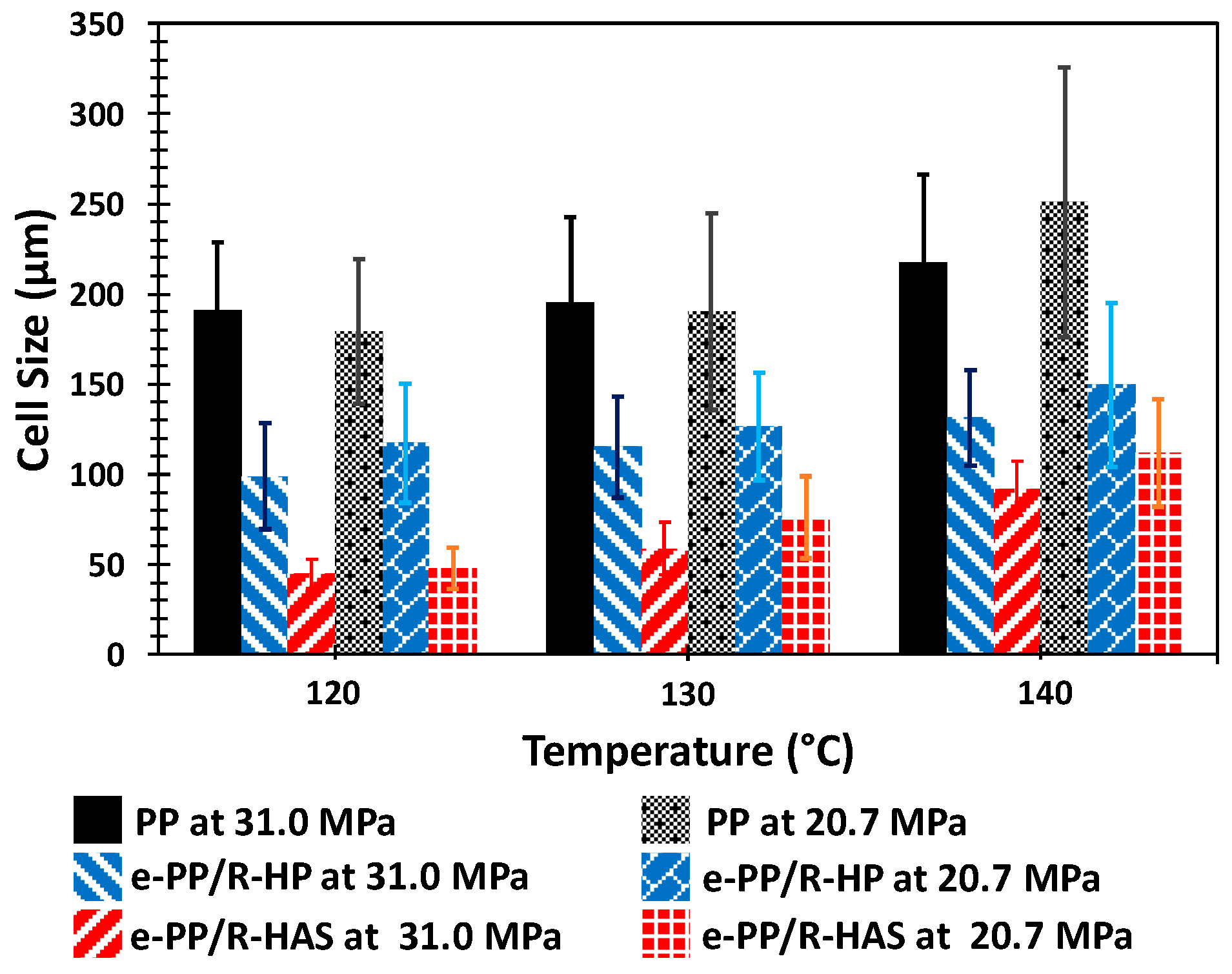
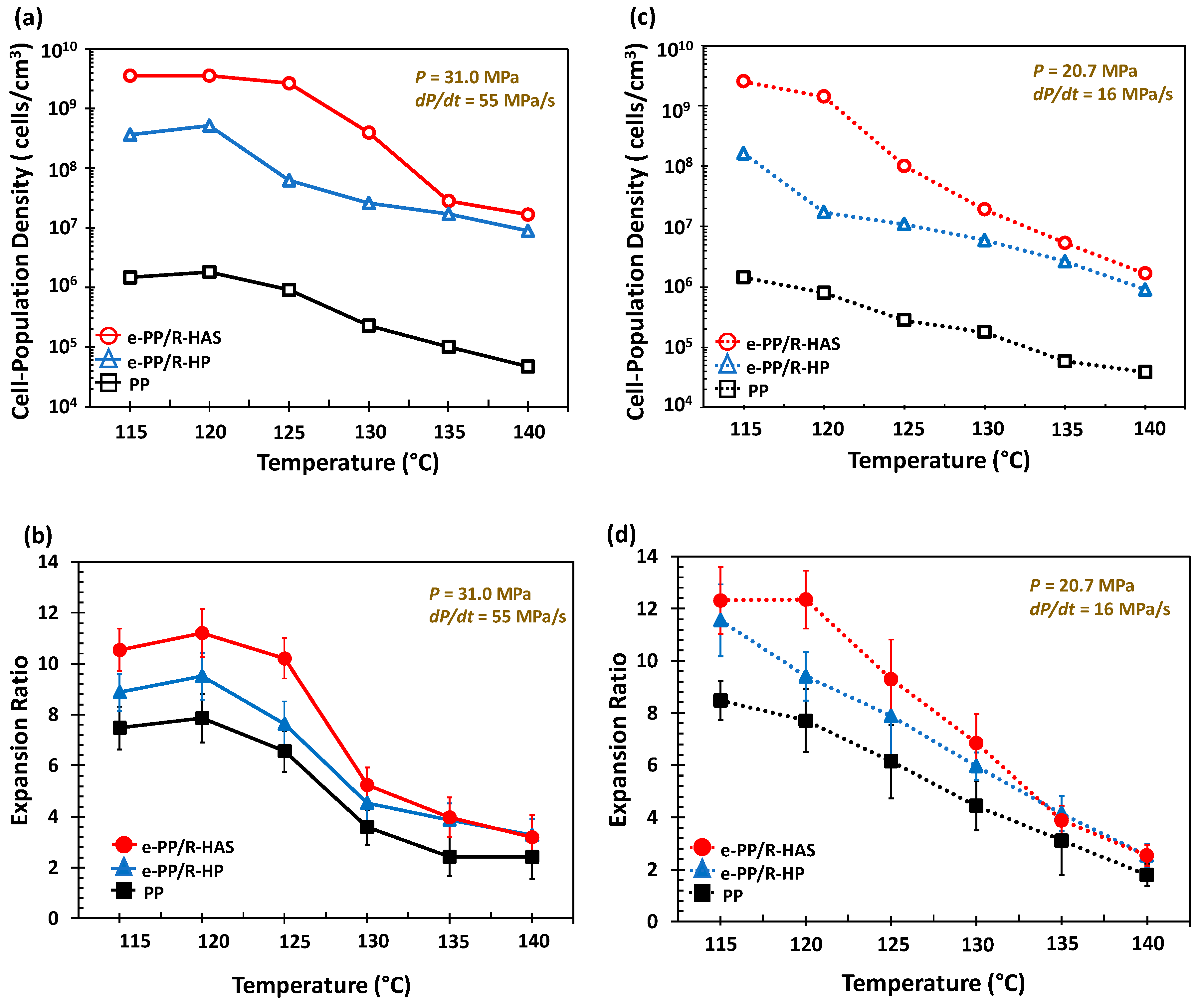
3.6. Batch Foaming
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gabriel, C.; Münstedt, H. Strain Hardening of Various Polyolefins in Uniaxial Elongational Flow. J. Rheol. 2003, 47, 619–630. [Google Scholar] [CrossRef]
- Rizivi, A.; Andalib, Z.K.M.; Park, C.B. Fiber-spun Polypropylene/Polyethylene Terephthalate Microfibrillar Composites with Enhanced Tensile and Rheological Properties and Foaming Ability. Polymer 2017, 110, 139–148. [Google Scholar] [CrossRef]
- Spitael, P.; Macosko, C.W. Strain Hardening in Polypropylenes and Its Role in Extrusion Foaming. Polym. Eng. Sci. 2004, 44, 2090–2100. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, Z.; Guan, Y.; Wei, D.; Zheng, A. Investigation of Extensional Rheological Behaviors of Polypropylene for Foaming. J. Cell. Plast. 2013, 49, 317–334. [Google Scholar] [CrossRef]
- Zhang, Y.; Parent, J.S.; Kontopoulou, M.; Park, C.B. Foaming of Reactively Modified Polypropylene: Effects of Rheology and Coagent Type. J. Cell. Plast. 2015, 51, 505–522. [Google Scholar] [CrossRef]
- Stange, J.; Münstedt, H. Effect of Long-chain Branching on the Foaming of Polypropylene with Azodicarbonamide. J. Cell. Plast. 2006, 42, 445–467. [Google Scholar] [CrossRef] [Green Version]
- Torres, E.; Li, S.; Costeux, S.; Dealy, J.M. Branching Structure and Strain Hardening of Branched Metallocene Polyethylenes. J. Rheol. 2015, 59, 1151–1172. [Google Scholar] [CrossRef]
- Tabatabaei, S.H.; Carreau, P.J.; Ajji, A. Rheological Properties of Blends of Linear and Long-chain Branched Polypropylenes. Polym. Eng. Sci. 2010, 50, 191–199. [Google Scholar] [CrossRef]
- Jahani, Y.; Ghetmiri, M.; Vaseghi, M.R. The Effects of Long Chain Branching of Polypropylene and Chain Extension of Poly(ethylene terephthalate) on the Thermal Behavior, Rheology and Morphology of Their Blends. RSC Adv. 2015, 5, 21620–21628. [Google Scholar] [CrossRef]
- Ameli, A.; Jung, P.U.; Park, C.B. Electrical Properties and Electromagnetic Interference Shielding Effectiveness of Polypropylene/carbon Fiber Composite Foams. Carbon 2013, 60, 379–391. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, W.; Zou, J.; Turng, L. Dual-scale Modeling and Simulation of Film Casting of Isotactic Polypropylene. J. Plast. Film. Sheet. 2016, 32, 239–271. [Google Scholar] [CrossRef]
- Kim, E.S.; Park, H.E.; Lee, P.C. In situ Shrinking Fibers Enhance Strain Hardening and Foamability of Linear Polymers. Polymer 2018, 136, 1–5. [Google Scholar] [CrossRef]
- Maddah, H.A. Polypropylene as a Promising Plastic: A review. Am. J. Polym. Sci. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Drabek, J.; Zatloukal, M. Evaluation of Thermally Induced Degradation of Branched Polypropylene by Using Rheology and Different Constitive Equations. Polymers 2016, 8, 317. [Google Scholar] [CrossRef]
- Mohebbi, A.; Mighri, F.; Ajji, A.; Rodrigue, D. Current Issues and Challenges in Polypropylene Foaming: A Review. Cell. Polym. 2015, 34, 229–338. [Google Scholar] [CrossRef]
- Yang, Y.; Boom, R.; Heerden, D.V.; Kuiper, P.; Wit, H.D. Recycling of Composite Materials. Chem. Eng. Process. 2012, 51, 53–68. [Google Scholar] [CrossRef]
- Münstedt, H. New Univeral Extensional Rheometer for Polymer Melts. Measurements on a Polystyrene Sample. J. Rheol. 1979, 23, 421–436. [Google Scholar] [CrossRef]
- Bach, A.; Rasmussen, H.K.; Hassager, O. Extensional Viscosity for Polymer Melts Measured in the Filament Stretching Rheometer. J. Rheol. 2003, 47, 429–441. [Google Scholar] [CrossRef]
- Meissner, J.; Hostettler, J. A New Elongational Rheometer for Polymer Melts and Other Highly Viscoelastic Liquids. Rheol. Acta. 1994, 33, 1–21. [Google Scholar] [CrossRef]
- Kim, J.; Mai, Y. Engineered Interfaces in Fiber Reinforced Composites, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1998; ISBN 9780080426952. [Google Scholar]
- Baired, D.G.; Collias, D.I. Polymer Processing: Principles and Design, 2nd ed.; Wiley: Hoboken, NJ, USA, 1993; ISBN 9780470930588. [Google Scholar]
- Dealy, J.M.; Morris, J.; Morrison, F.; Vlassopoulos, D. Official symbols and nomenclature of The Society of Rheology. J. Rheol. 2013, 57, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Fujiwara, K.; Takikawa, T.; Takishima, S.; Masuoka, H. Solubilities and Diffusion Coefficients of Carbon Dioxide and Nitrogen in Polypropylene, High-density Polyethylene, and Polystyrene under Higher Pressures and Temperatures. Fluid Phase Equilibr. 1999, 162, 261–276. [Google Scholar] [CrossRef]
- Park, H.E. Effects of Pressure and Dissolved Carbon Dioxide on the Rheological Properties of Molten Polymers. Ph.D. Thesis, McGill University, Montreal, QC, Canada, 2005. [Google Scholar]
- Wang, L.; Lee, R.; Wang, G.; Chu, R.K.M.; Zhao, J.; Park, C.B. Use of Stereocomplex Crystallites for Fully-biobased Microcellular Low-density Poly(lactic acid) Foams for Green Packaging. Chem. Eng. J. 2017, 327, 1151–1162. [Google Scholar] [CrossRef]
- Tran, M.; Gong, P.; Detrembleur, C.; Thomassin, J.; Buahom, P.; Saniei, M.; Kenig, S.; Park, C.B.; Lee, S. Reducing Thermal Conductivity of Polymeric Foams with High Volume Expansion Made From Polystyrene/expanded Graphite. SPE ANTEC Indianap. 2016, 2016, 1870–1881. [Google Scholar]
- Carslaw, H.S.; Jaeger, J.C. Conduction of Heat in Solids, 2nd ed.; Clarendon Press: Oxford, UK, 1986; ISBN 9780198533689. [Google Scholar]
- Wunderlich, B. Macromolecular Physics. Vol. 3: Crystal Melting; Academic Press: New York, NY, USA, 1980; pp. 61–64. [Google Scholar]
- Han, C.D.; Baek, D.M.; Kim, J.K.; Ogawa, T.; Sakamoto, N.; Hashimoto, T. Effect of Volume Fraction on Order-Disorder Transition in Low Molecular Weight Polystyrene-block-Polyisoprene Copolymers. Macromolecules 1995, 28, 5043–5062. [Google Scholar] [CrossRef]
- Kossuth, M.B.; Morse, D.C.; Bates, F.S. Viscoelastic Behavior of Cubic Phases in Block Copolymer Melts. J. Rheol. 1999, 43, 167–196. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
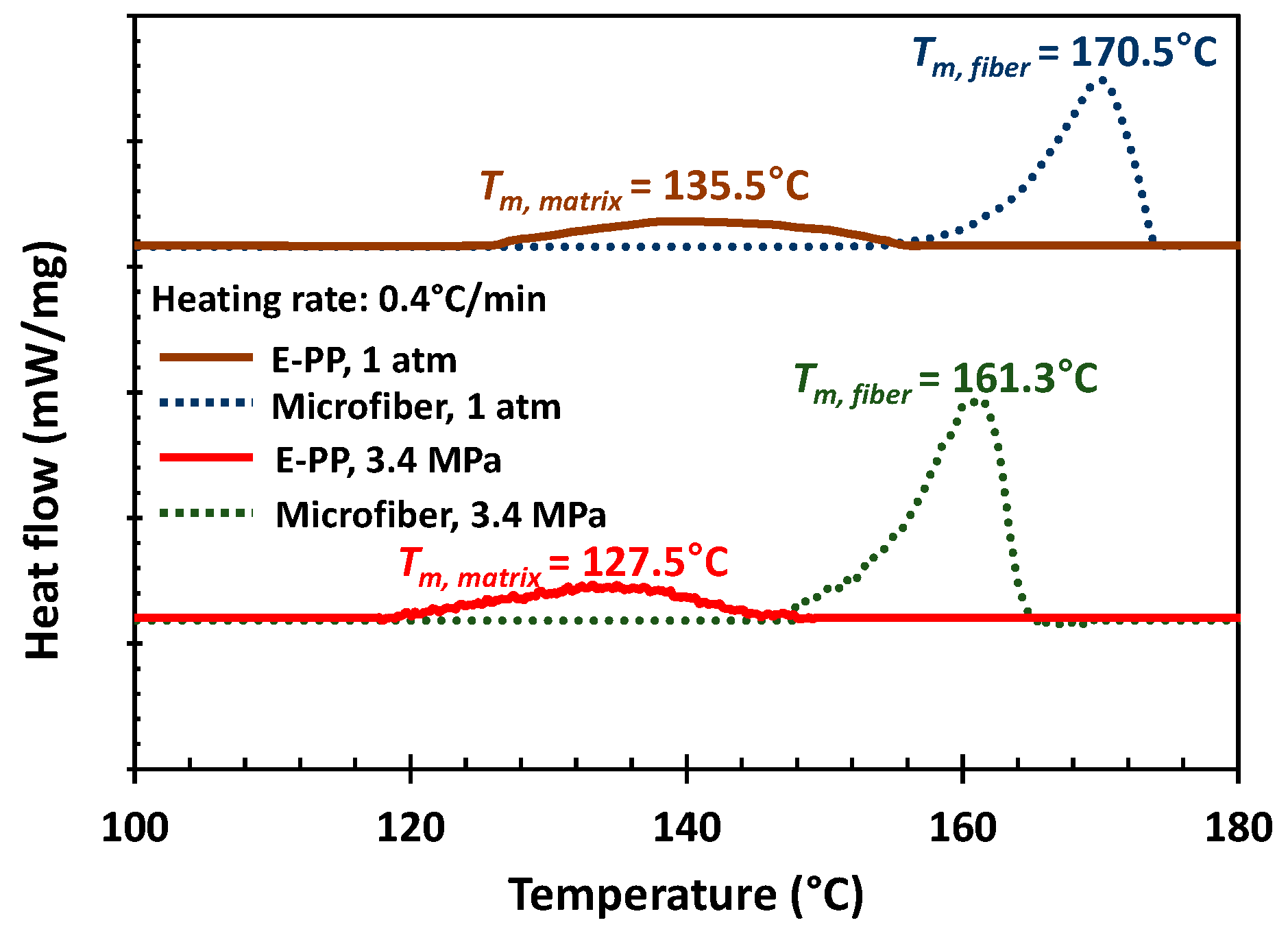
| Code | Function | Molecular Weights (kg/mol) | Mw/Mn | Tacticity (Triad mol %) | Comonomer Content | Peak Tm (°C) | Crystallinity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mn | Mw | Mz | rr | mm | mr | mol % | mass % | 1 atm | 3.4 MPa with CO2 | ||||
| e-PP | matrix | 55 | 252 | 621 | 4.58 | a 84.7 | propylene (92) ethylene (8) | propylene (94.5) ethylene (5.5) | 135.5 | 127.5 | 14.5 | ||
| i-PP | micro- fiber | 60 | 451 | 2080 | 7.55 | 3 | 93 | 4 | b N.A. | b N.A. | 170.5 | 161.3 | c 62.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.S.; Park, H.E.; Lopez-Barron, C.R.; Lee, P.C. Enhanced Foamability with Shrinking Microfibers in Linear Polymer. Polymers 2019, 11, 211. https://doi.org/10.3390/polym11020211
Kim ES, Park HE, Lopez-Barron CR, Lee PC. Enhanced Foamability with Shrinking Microfibers in Linear Polymer. Polymers. 2019; 11(2):211. https://doi.org/10.3390/polym11020211
Chicago/Turabian StyleKim, Eric S., Heon E. Park, Carlos R. Lopez-Barron, and Patrick C. Lee. 2019. "Enhanced Foamability with Shrinking Microfibers in Linear Polymer" Polymers 11, no. 2: 211. https://doi.org/10.3390/polym11020211
APA StyleKim, E. S., Park, H. E., Lopez-Barron, C. R., & Lee, P. C. (2019). Enhanced Foamability with Shrinking Microfibers in Linear Polymer. Polymers, 11(2), 211. https://doi.org/10.3390/polym11020211