3.1. Characterization of the Oligodiol Products
The oligodiol obtained from the reaction of DEG and PET catalyzed by ZnSO4·7H2O (after removal of the catalyst) was analyzed by FTIR, NMR, and LC-MS.
FTIR-ATR spectra of the oligodiols prepared by using ZnSO
4·7H
2O, shortened as DZS (
Figure 1), and Zn(OAc)
2·2H
2O, shortened as DZA, showed almost the same features. Compared to the FTIR spectrum of PET, the presence of the –OH end groups of the oligodiol was confirmed by a broad band at 3392 cm
−1, while the ethylene ether moiety appeared as bands at 2945 cm
−1 for the CH
2 asymmetric stretching, 2875 cm
−1 for the CH
2 symmetric stretching, and 1065 cm
−1 for the C–O stretching.
The DMSO-d6 solution containing oligodiol was treated with D2O to transform all the O–H end groups of oligodiol to O–D so that it would not appear in the spectrum and it would not interfere with the quantitative integral measurement. The strong signal at 3.64 ppm was due to HOD.
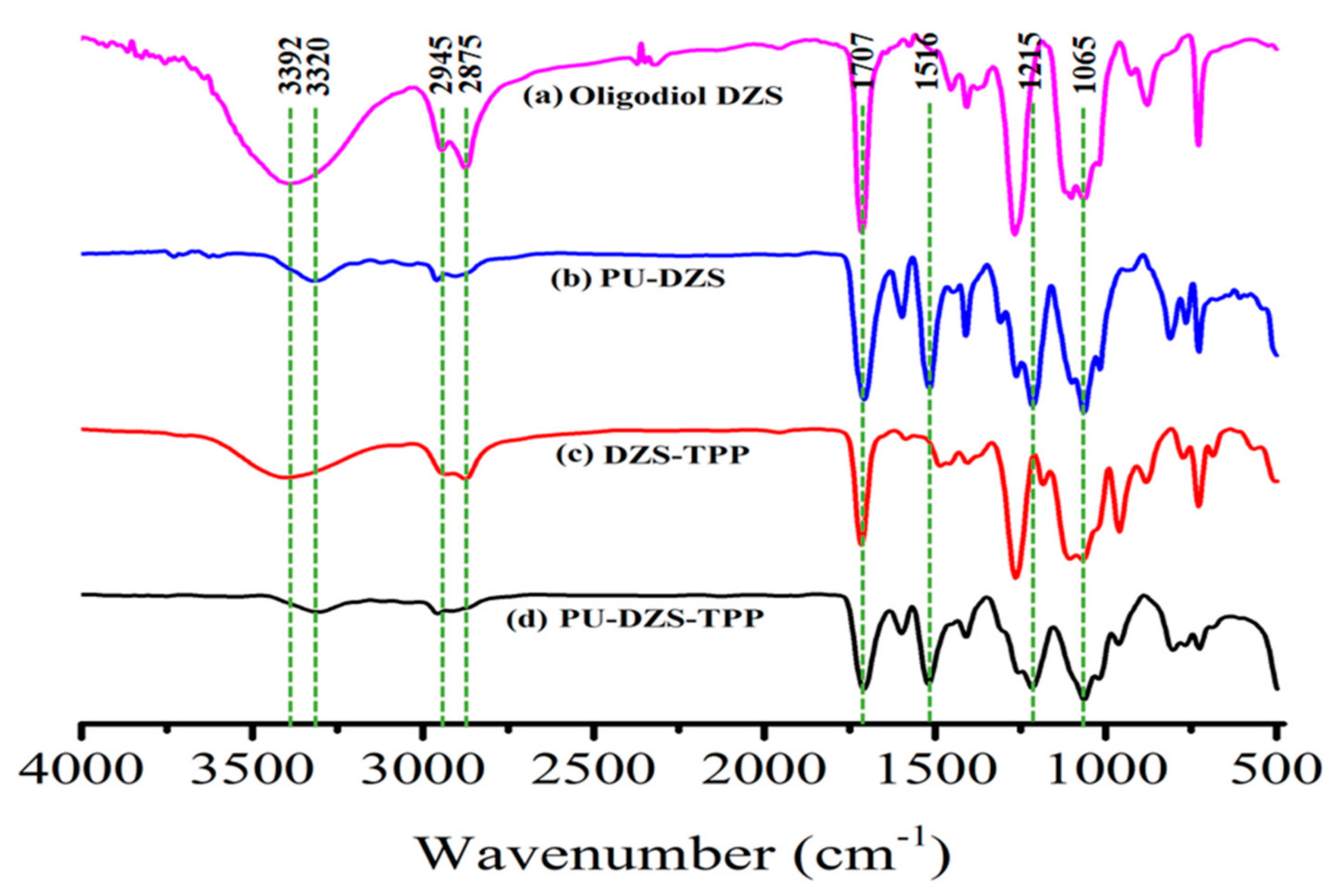
The
1H-NMR spectrum (
Figure 2) shows aromatic protons from 7.75 to 8.15 ppm, and aliphatic CH
2 appears as multiplet signals from 3.75 to 4.50 ppm. Weak signals at 3.58, 4.75 and 5.10 ppm are due to impurities. Resonance signals at 4.3 and 4.4 ppm are typical for –C
H2OCOAr with an integral of 3.97 protons, almost equal to 4.00 protons of aromatic CH. The
1H-NMR integrals are normalized with respect to one terephthalate unit or 4 aromatic protons. Resonances at 3.74 and 3.80 ppm are due to –C
H2CH
2OCOAr with 4.12 protons that also close to 4.00 protons. The –C
H2OC
H2– of ethers or –C
H2OH of end groups appearing at 3.41 and 3.49 ppm contributes to 15.31 protons.
The total number of aliphatic protons is 23.41. One monoethylene glycol unit, abbreviated as M, has 4 protons; therefore, the number of the M unit must be 5.85. The total aromatic protons are 4.00 (from the spectrum). One terephthalate unit, abbreviated as T, has 4 protons; therefore, the number of T units must be 1.00. Consequently, from the
1H-NMR spectrum, the molar ratio M/T must be 5.85/1.00. The initial D/(TM) molar ratio equals 2.5 (D stands for a DEG unit, which is equivalent to 2 M units, and PET is represented by TM) or the initial M/T molar ratio equals 6/1. A small difference of 0.148 M between the initial and final ratios could be due to the decomposition of M or D to form volatile materials during the reaction at high temperature under microwave irradiation. The volatile materials were isolated and characterized by GC-MS as a mixture of water and the decomposition products of M and D, such as 1,4-dioxane, acetaldehyde, 2-methyl-1,3-dioxolane, etc. [
18].
From the 1H-NMR spectrum, we could also determine the molar ratio of M (ether -CH2OCH2)/M (ester –CH2OCO) = 19.44/3.97 = 4.90/1. The newly formed ether bond must be [(4.90/2) − 1]/1 = 1.45/1, i.e., 1.45 mol ether bonds were formed per 1 ester unit. In theory, for 1 mol of PET or (TM) units completely reacted with 2.5 mol of D via the transesterification and ether formation, the following chemical equations were proposed:
- (1)
Transesterification: (TM) + D → DTM
- (2)
Ether formation: DTM + 1.5D → D2.5TM or equivalent to TM6.
The experimental value of 1.45 mol new ether bonds was quite close to the 1.5 mol proposed by the above calculation and theoretical equations.
The mass reduction during the reaction came not only from the decomposition of M or D but also the ether formation (2), when water was lost. The percent of mass reduction due to water removal was 1.45 × 18 × 100%/(2.5 × 106 + 192) = 5.70% (only 1.45 mol of new ether units was formed, and the other ether units came from the D structure). The percent of M units lost during the reaction was 0.148 × 62 × 100%/(2.5 × 106 + 192) = 2.01%. The total mass loss calculated from the 1H-NMR spectrum was 7.71%. The percent of experimental loss was 6.64%. The two values were very close, therefore, the characterization by 1H-NMR was correct.
Finally, the 1H-NMR spectrum demonstrated that both the transesterification of the diol with terephthalate ester and the ether formation from the alcohol end groups of DEG occurred during the glycolysis of PET by DEG. Consequently, the obtained product was a complex mixture of novel oligo-ester-ether-diols, and the quantity of excess DEG reactant was negligible.
The oligodiol sample was separated at first by liquid chromatography (LC), and each fraction at a different retention time was further analyzed by mass spectrometry (MS). From the mass spectrum, the molecular weight of each component was determined, and the molecular formula/structure was proposed.
The complex oligodiol structures were described by using abbreviated characters, such as T for the terephthalate unit or [COC6H4COO], M for the monoethylene glycol unit or [CH2CH2O], and D for the diethylene glycol unit that equals M2. Combining different T, M and D units and one molecule of water resulted in various oligodiol structures and molecular weights. The proposed structure would be correct if the calculated molecular weight was almost equal to the experimental m/z value from MS.
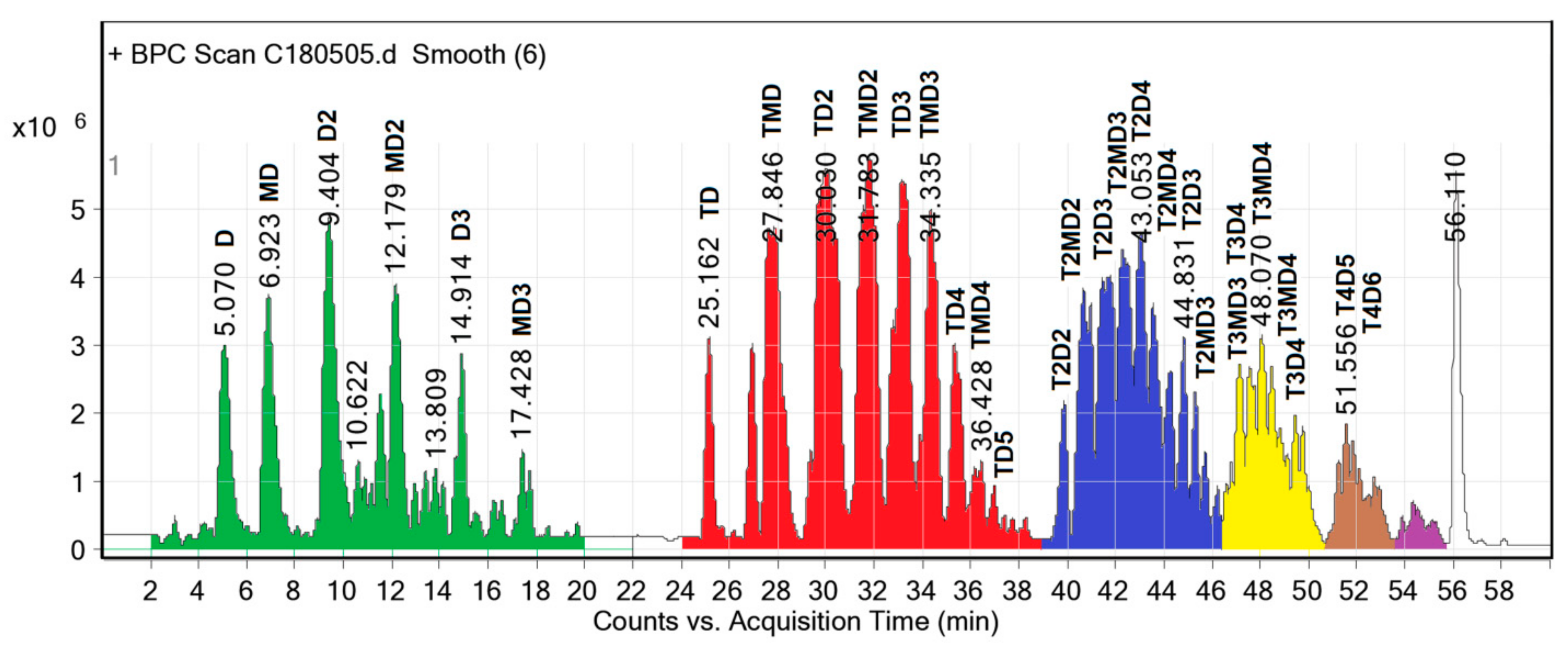
The chromatogram (
Figure 3) was separated into five groups, corresponding to five homologous series of structures or 67 different individual structures. Extremely weak signals were not considered.
(1) Retention time from 0 to 20 min.
Figure 3 shows a separation of the major components from 0 to 20 min, including D, MD, D
2, MD
2, D
3 and MD
3 as a homologous oligoether series of ethylene glycol (M) and diethylene glycol (D). The increment of homologous series was one M unit. The higher molecular weight molecules required longer retention time on the column. The most abundant component in this homologous oligoether series was the D
2 that was formed from two D molecules. The longest chain of this series was MD
3 that was created from at least three new ether bonds among one M and three D units, corresponding to an initial DEG/PET molar ratio of 2.5/1. This homologous oligoether series was not detected in the LC-MS result of the sample obtained after treatment with an aqueous 20% NaCl solution [
18], because the components had a high solubility, even in brine solution, and were removed from the final product mixture.
The reactant D not only contributes to the homologous oligoether series, but its peak height in LC (
Figure 3) also follows a good normal distribution curve. Here, we can conclude that the quantity of D is not in excess in the product mixture, but in an equilibrium quantity from the transesterification and ether formation reactions. For this reason, the removal of D from the reaction mixture is almost impossible and unnecessary.
(2) The retention time from 22 to 39 min in the chromatogram displayed peaks of a homologous monoterephthalate series with structures of increasing molecular, namely, TD, TMD, TD2, TMD2, TD3, TMD3, TD4, TMD4 and TD5, with the most abundant molecules being TD2 and TMD2. The increment of this homologous series was also one M unit. The smallest monoterephthalate structure was TD, which should be rewritten as MTM due to the probability of a carboxylic acid end group in TD being very low when diol was used in excess. The highest molecular weight component of the monoterephthalate series was TD5, arranged in the order of DTDDD or DDTDD, and formed by two ester and two ether groups.
Analogously, the diterephthalate series appeared from 39–46 min, the triterephthalate series eluted from 46–51 min, and the retention time of the tetraterephthalate series was from 51–53 min. All individual structures were also confirmed by MS.
Based on the above LC analysis, we can conclude that the oligodiols obtained without the aqueous 20% NaCl solution treatment are a complex mixture due to the presence of oligomers with longer ether chains, namely, TD4, TMD4, TD5, T2D4, etc., or that this is a mixture of various oligo-ester-ether-diols. Once again, the transesterification and ether formation reactions during PET glycolysis by DEG are confirmed by LC analysis. This is the first time that the structures of oligo-ester-ether-diols are experimentally confirmed. Here, we refer to the glycolyzed product as an oligo-ester-ether-diol and not as the commonly used polyol because the term polyol could be misunderstood as a polymer or a monomer that has multiple alcohol groups. Our product is an oligo-ester-ether-diol that has a low molecular weight with two alcohol end groups and contains terephthalate ester and ethylene glycol ether moieties in the linear chain. This novel diol is expected to offer the advantages of both ester and ether diols in the polyurethane formulation.
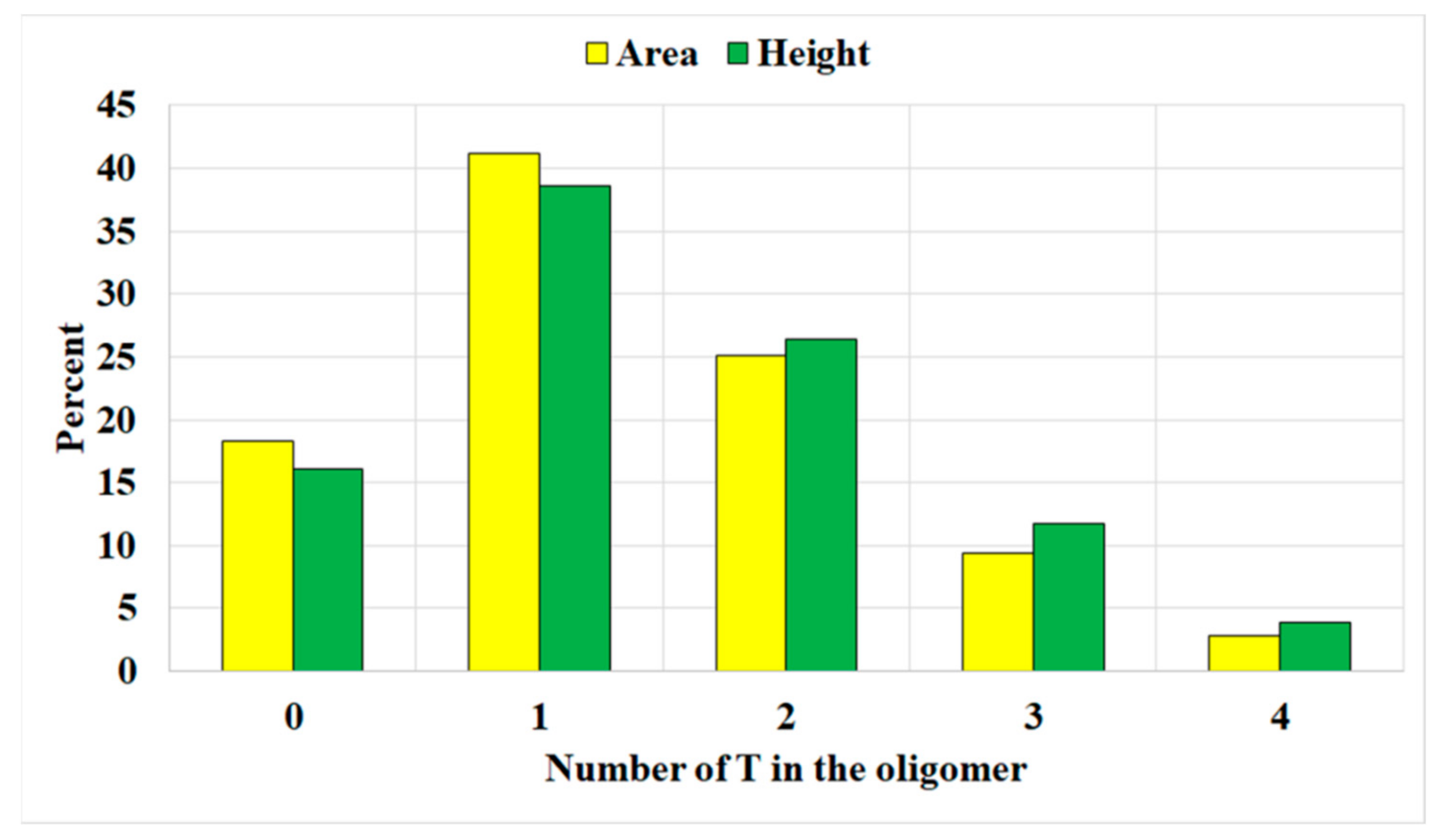
The distribution of terephthalate unit numbers in the molecule is depicted in
Figure 4. The major component of the oligodiol is monoterephthalate. The oligoethers without terephthalate units contribute to approximately 17%. The components containing larger terephthalate unit numbers have lower abundances. From the distribution, the average terephthalate unit in one oligomer chain is calculated as 1.367 by peak area or 1.492 by peak height.
The quantitative analysis of the LC-MS also shows different responses of peak areas and heights. At the lower terephthalate unit number or lower molecular weight side (
Figure 4), the intensities based on the area are stronger than those based on height. In contrast, at higher molecular weight regions, the relationship is reversed. For most LC analyses, the peak areas are used for quantitative calculations.
G.P. Karayannidis et al. [
8] used gel permeation chromatography to characterize the PET glycolysis product with DEG by using a 2.2/1 molar ratio with conventional heating. Three peaks in the GPC chromatogram were identified as three oligoester diols with mono-, di- and tri-terephthalate units. In our research project, oligodiol was characterized by the LC-MS method, and up to 67 different structures of 5 homologous oligo-ester-ether-diol series containing from zero to four terephthalate units were confirmed.
The calculations based on the area ratios of peaks in the chromatogram resulted in Mn = 475 g/mol, Mw = 581 g/mol, PDI = 1.22, the average molecular formula T1.37M5.79, and a molar ratio of M/T = 4.23. When the height ratios of peaks in the chromatogram were considered, it resulted in Mn = 507 g/mol, Mw = 615 g/mol, PDI = 1.21, the average molecular formula T1.49M6.08, and a molar ratio of M/T = 4.08.
The number average molecular weight (Mn) and weight average molecular weight (Mw) are calculated by using the formulas and , where Ni is the relative area or relative height of the peak in the chromatogram, and Mi is the molecular weight, as determined by mass spectrometry.
The M/T ratio calculated from LC was substantially smaller than the value obtained from 1H-NMR (5.825). We found that the area ratios of LC peaks matched the 1H-NMR spectrum better, and we chose Mn = 475 g/mol as the molecular weight of the oligodiol.
In comparison, even though the PET glycolysis was conducted in a microwave with a higher DEG/PET molar ratio of 4/1 and in the presence of Zn(OAc)
2, a larger M
n of 965 was obtained [
13].
Even though DEG was used in excess, the LC analysis showed that the quantity of unreacted DEG reactant in the oligodiol product was small due to the transesterification and ether formation occurring simultaneously and reaching an equilibrium state to form the oligo-ester-ether-diol. For this reason, purification by treatment with a cool 20% NaCl solution [
18] was unnecessary. In this research project, we use the glycolyzed product mixture directly for PU synthesis without purification steps, therefore, no additional waste is created, and a larger quantity of diol from waste PET is used.
The viscosity of the liquid oligodiols was determined by a Brookfield DVEELVTJO viscometer at 31 °C using an LV2-62 spindle. The viscosity value of the DZA sample was 219 cp and that of the DZS sample was 268 cp. The DZS oligodiol prepared by using the ZnSO4·7H2O catalyst showed a higher viscosity and thus a higher molecular weight than that of the DZA oligodiol. Both oligodiol samples remained in the liquid state at a temperature as low as −15 °C.
3.3. Polyurethane Characterization
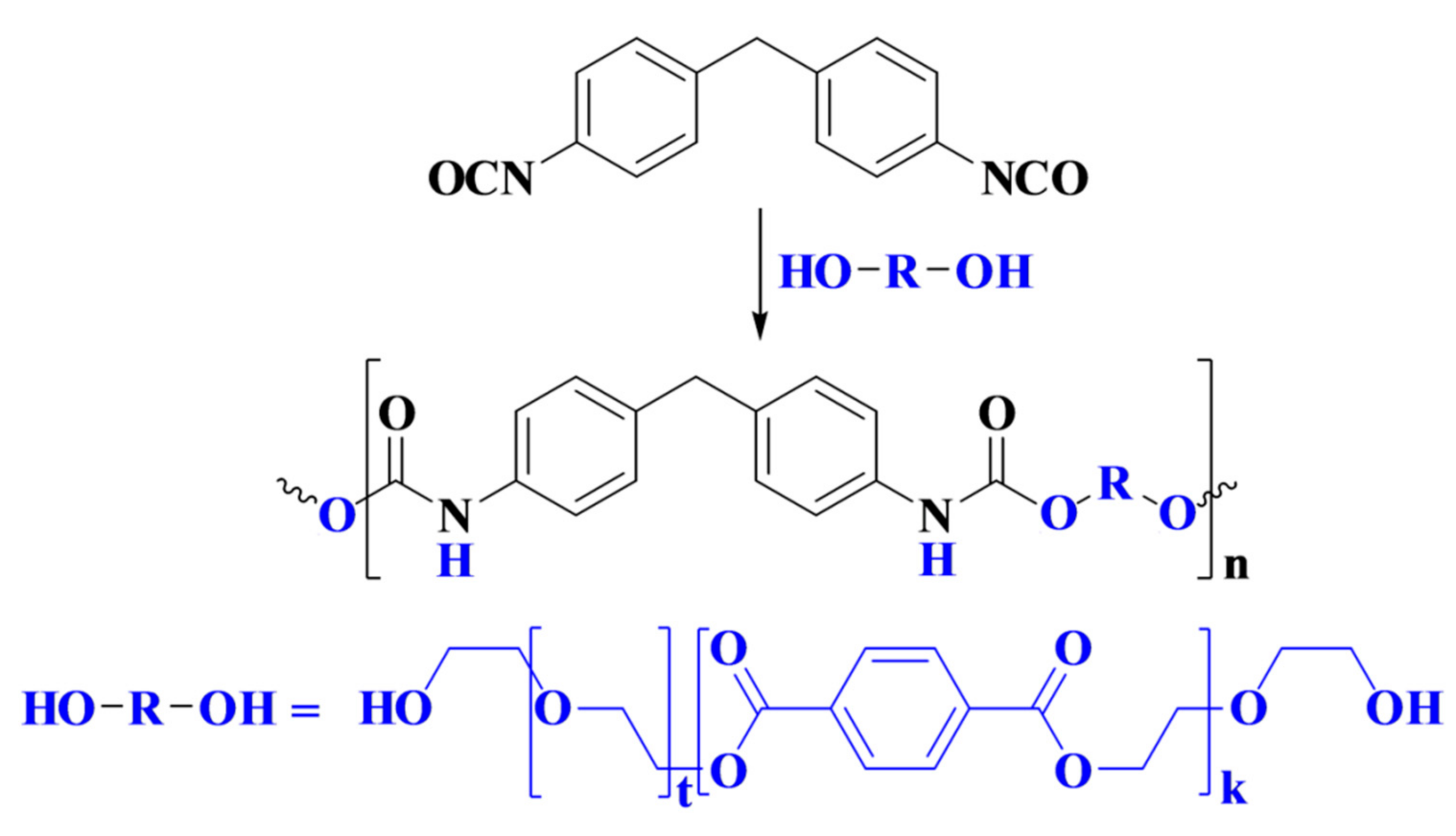
Rigid polyurethane foam (PUF) was prepared by the reaction of oligo-ester-ether-diol with methylene diphenyl diisocyanate (MDI), as depicted in
Scheme 1.
Foam was formed by the reaction of isocyanate with water to form carbamic acid, which is an unstable intermediate that is readily decomposed to evolve carbon dioxide and generate an amine. Consequently, this amine further reacted with isocyanate to form urea. For this reason, MDI was used in excess, and NCO/OH ratio was normally larger than 1.
At first, PU samples were prepared without adding the flame retardant so that the best foaming formulation and conditions were discovered. After that, by using the same formulation and procedure, the oligodiol samples were treated with triphenyl phosphate (TPP), as a flame retardant, before the foaming process.
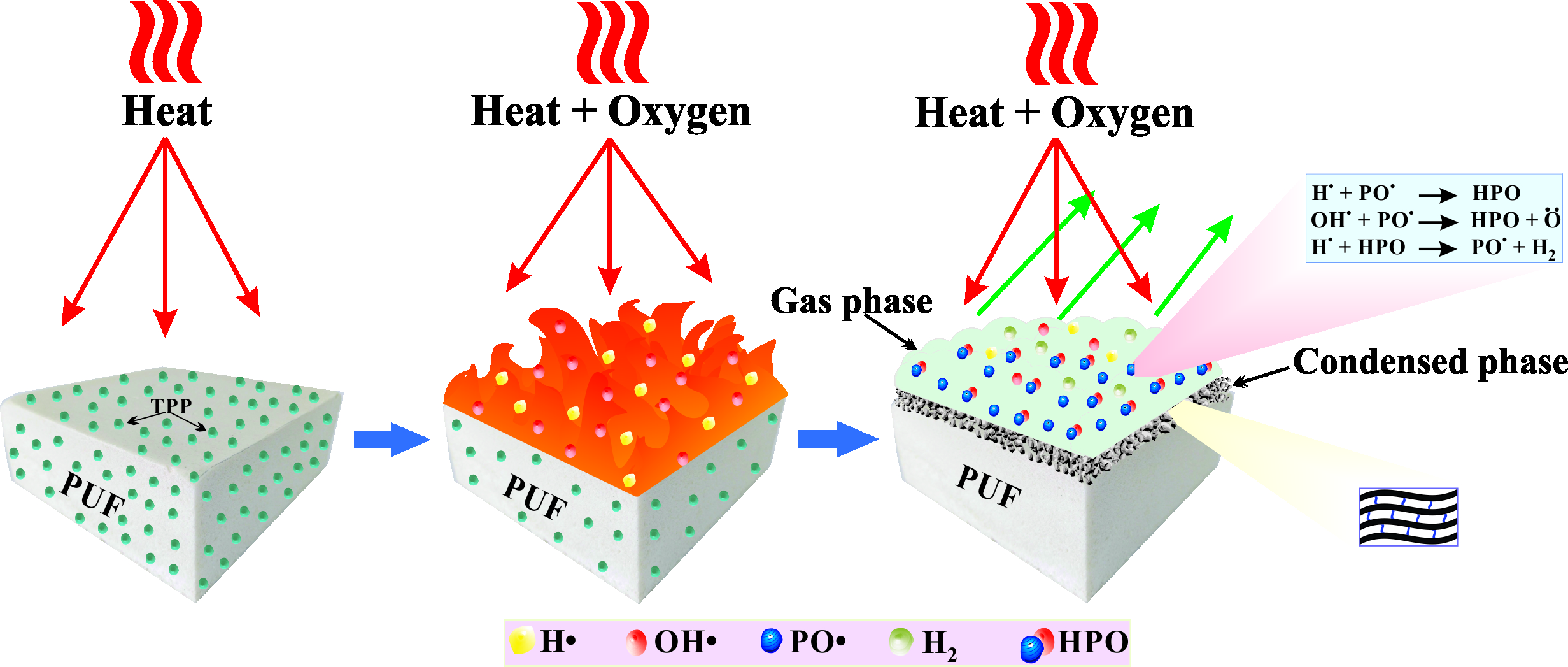
The ATR-FTIR spectra of PUs prepared from oligodiols DZS and DZA resulted in almost the same features. The spectra (
Figure 1b,d) showed typical peaks of the urethane group (–NHCOO–) at 3320 cm
−1 for NH stretching, 1707 cm
−1 for C=O stretching, 1516 cm
−1 for N–H bending and 1215 cm
−1 for C–O stretching. The C=O stretching at 1720 cm
−1 for the ester in the diol DZS reactant was shifted to a lower wavenumber for the C=O of urethane. The band at 1597 cm
−1 was caused by the C=C stretching of benzene rings. Additionally, the ether C–O stretching resulted in strong bands at 1099, 1065, and 1018 cm
−1.
After mixing TPP with DZS, the FTIR spectrum (
Figure 1c,d) showed a weak absorption at 1184 cm
−1 and a medium peak at 960 cm
−1 for P–O stretching vibrations.
3.3.1. Density, Pore Size, and Compression Strength
The measured densities of PU-DZS and PU-DZA were almost the same (102 ± 10 kg/m
3). Their values depended only on the quantity of water used as a foaming agent, and not that of the oligodiol. An attempt to reduce the density of the foam by increasing the amount of water in the formulation was not successful. The foam formed quickly, and collapsed. The PUF prepared from copolymer or polyester polyols usually has a density in the range of 25 kg/m
3 to 100 kg/m
3 [
19].
When TPP was added, the density of the PUF became lower (90 kg/m3) or the foaming process was more successful. TPP could play roles of a good solvent and a compatibilizer for oligodiol and MDI.
Both samples showed quite uniform cells (
Figure 5). The average cell size of PU-DZS (left) is larger than that of PU-DZS-TPP
25 (right), with the average diameters being 619 ± 65 μm and 408 ± 39 μm.
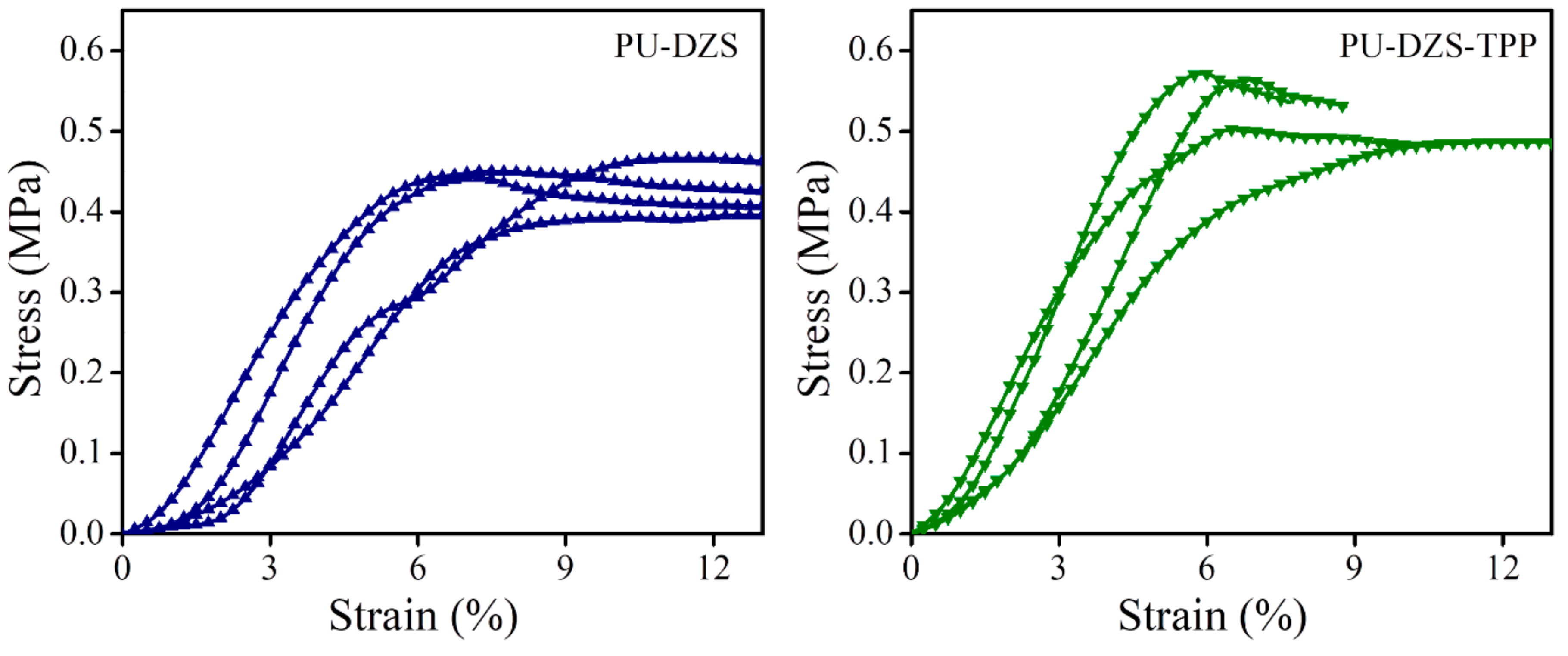
Compression strength was evaluated according to ASTM D1621 with a speed of 2.5 mm/m (
Figure 6). The smaller pore size of the PU-DZS-TPP
25 sample enhanced its yield strength (530 ± 45 kPa) compared to that of the PU-DZS sample without TPP (433 ± 31 kPa). The strain at the yield point showed a reverse relationship with corresponding values of 7.21 ± 1.90 and 9.4 ± 2.1%. When TPP was added, the cell size of the PUF decreased; therefore, it can withstand a stronger force, i.e., a higher yield stress and lower yield strain. Even though TPP reduced the T
g of the PU-DZS sample, compression tests were carried out at room temperature, which is below the T
g; therefore, the compression strength did not depend on T
g.
The density of our PUF samples (90–102 kg/m
3) is lower than the similar PUF prepared from PET glycolized by EG (from 150 to 510 kg/m
3 [
16]) and the compression strength of our PUF samples is also higher (433–530 kPa compared with 140–185 kPa [
16]). These better results could be explained by the effective role of flexible liquid oligo-ester-ether-diol prepared by PET glycolysis with DEG in foam formation and in mechanical strength enhancement.
3.3.2. Thermal Properties
The thermal transitions of the PUF samples were detected by DSC. The samples were heated (a) from 25 to 120 °C, (b) cooled from 120 to −50 °C and (c) reheated from −50 to 200 °C with heating/cooling rate ±10 °C/min.
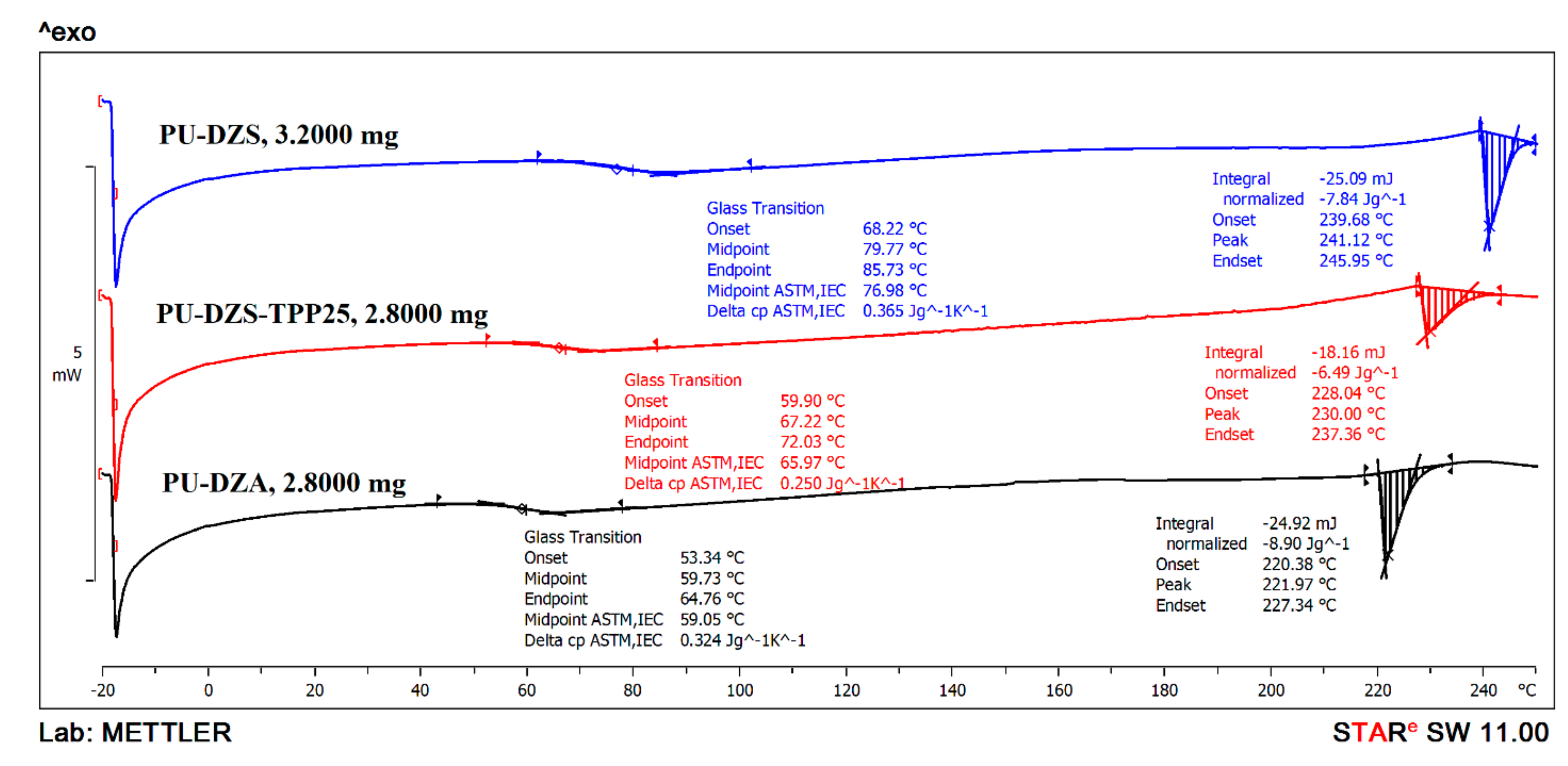
The glass transition temperature of the PUF is typical for ester or ether oligodiol soft segments in the chain. The DSC curves (
Figure 7) show that PU-DZS (T
g = 79.77 °C) has a higher glass transition temperature than that of PU-DZA (T
g = 59.73 °C). This result could come from the fact that PU-DZS was prepared from an oligodiol that has a higher molecular weight than that of PU-DZA, as confirmed by the higher viscosity. As TPP was added to PU-DZS, the T
g reduced to 67.22 °C (PU-DZS-TPP
25 curve). This is good evidence for the role of TPP as a plasticizer. The small TPP molecule was very compatible with the oligodiol soft segments in the PUF, and TPP also enhanced the flexibility of the soft segments; therefore, it reduced the T
g of the PUF.
At the high temperature range, the endothermic peaks that are typical for a melting process have the same increasing trend as that of the Tg values. The PUF has a high Tg and a high Tm. These can be the melting points of the soft segments of the terephthalate crystalline structure. The contribution of ethylene terephthalate segments to the total mass of PU in the PU-DZS sample is calculated as 48g/(48 + 66.3 − 7.62) × 100% = 45.0% of the oligodiol. The oligodiol was used as 66/145 × 100% = 45.5% of the PUF. The mass percent of ethylene terephthalate units in the PUF is 45.0% × 45.5% = 20.5%. This is also the mass percentage of the PET waste contribution in the PUF. Therefore, the % crystallinity of ethylene terephthalate units is (7.84(J/g)/140(j/g)/0.205) × 100% = 27.3%.
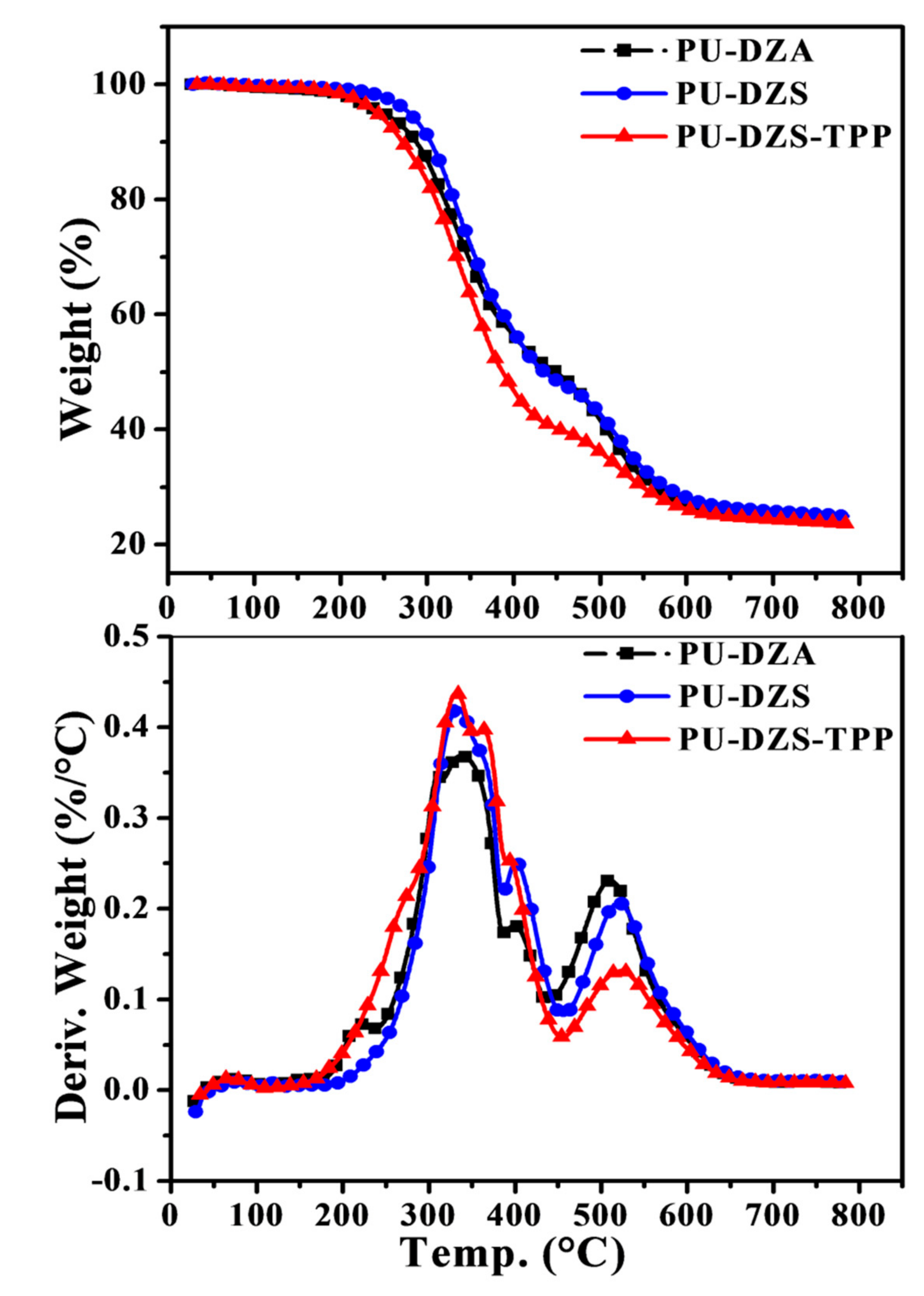
Two main degradation stages were distinguished following the TG and DTG curves (
Figure 8). When heating the PU-DZS sample from room temperature to 200 °C, there is a small weight loss due to volatile material (0.45%). A maximum weight loss is observed at 334 °C for the first stage from 200 °C to 460 °C, with an overall weight loss of 52.1%. In this temperature range, B.H. Kim, K. Yoon, and D.C. Moon [
20] suggested that the sequence of thermal degradation of rigid and soft polyurethanes based on methylene diphenyl diisocyanate was from hard to soft segments.
The second stage of decomposition covers 460 °C to 650 °C, with a maximum peak at 520 °C. The onset of the first decomposition at 284 °C is higher than the reported value of 260 °C [
13].
The low thermal decomposition of PU-DZA at 220 °C in the DTG curve, compared to that of PU-DZS, could be explained by the presence of Zn2+ catalyst residue and impurities in DZA, as evidenced by the dark color of the oligodiol. From room temperature to 438 °C, the PU-DZA sample lost 41.50%.
TPP is a flame retardant, and it has simultaneously worked as an ecologically friendly plasticizer for triacetyl cellulose [
21] and poly(vinyl alcohol) [
22]. When TPP was added to the PUF, the decomposition trends were almost unchanged. The char content at 800 °C of PU-DZS-TPP
25 (23.65%) was not quite different from that of the PU-DZS sample (24.92%). The calculated residue at 800 °C of PU-DZS-TPP
25 was 0.90 × 24.92% + 0.10 × 0.00% = 22.43%. Assuming that the char was only formed from TPP, we had equation 0.90 × 24.92% + 0.10 × X% = 23.65%. The solution to this equation is X = 12.2%. Consequently, at least 87.8% of the TPP converted into the gaseous phase and only 12.2% converted into char, or TPP mainly played the role of flame retardant in the gaseous phase. This conclusion again confirmed our previous statements [
22].
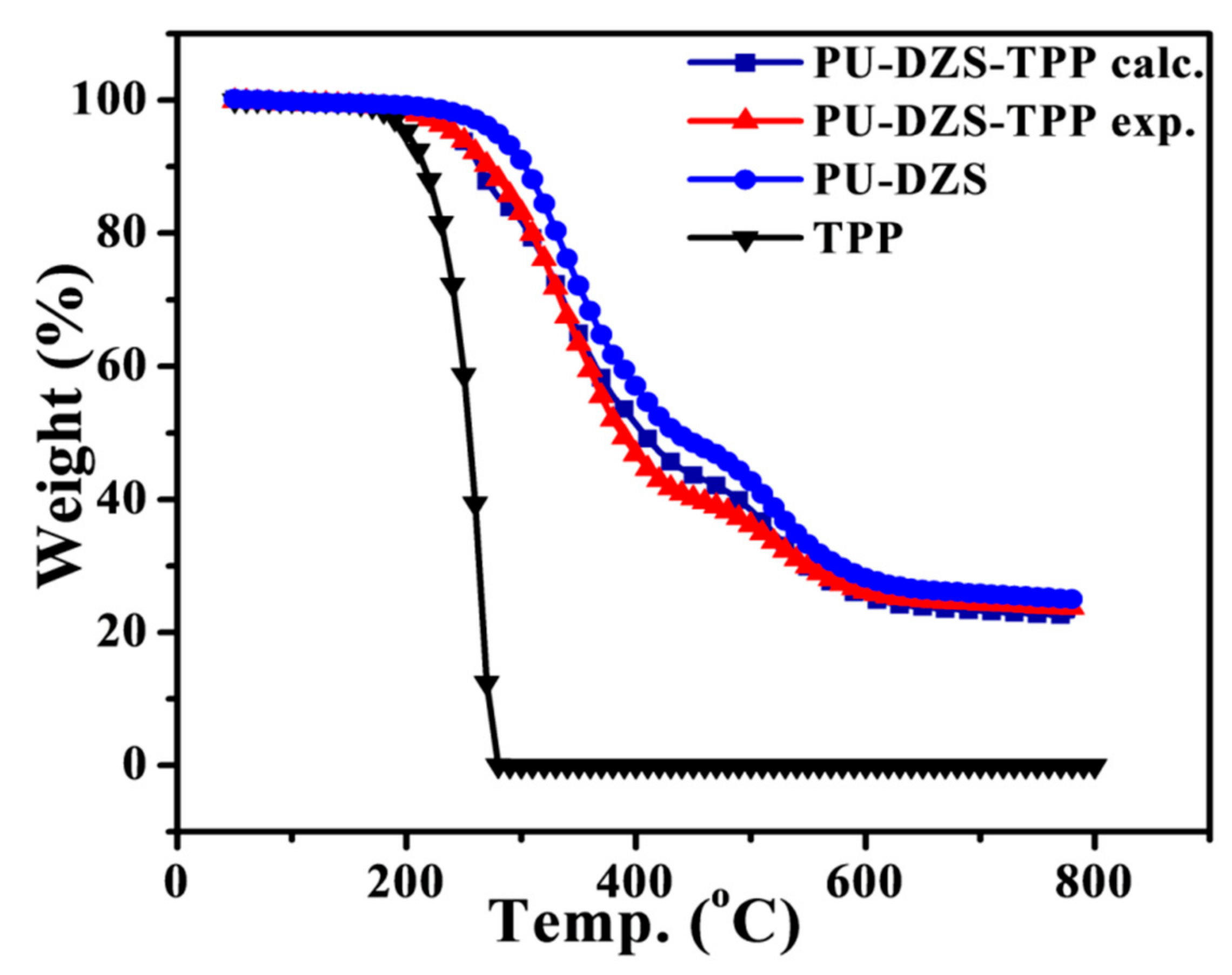
To provide proof of the TPP effect on the thermal degradation behaviors of the PUF, experimental TGA data were compared with TGA values calculated by using the additive rule (
Figure 9). The obtained results show that the addition of TPP slightly accelerated the thermal decomposition of PU-DZS in the temperature range of 320–530 °C. There was an interaction between TPP and PU-DZS, but it did not significantly contribute to the fire retardant property. The main flame retardant mechanism for TPP occurred entirely via the gas phase, and the char layer of PU-DZS, acting in the condensed phase, was partially responsible for the flame retardancy of the mixture.
3.3.3. Flammability and the Mode of Action of PUF
Rigid PUF is a highly flammable material, and it is not easy to find an effective single-component halogen-free flame retardant (FR). To achieve good flame retardancy, the addition of a char-forming agent along with a phosphorus FR is required. However, in this study, the rigid PUF prepared from recycled PET oligodiol contains aromatic moieties and is more thermally stable. Therefore, a single phosphorus FR (as TPP) was added in 5–25 pph loadings to find the UL94 V-0 ranking. The designated compositions and test results are shown in
Table 1. The PU-DZS sample without TPP was highly combustible, having a low LOI value of 17%; the flame propagated along the test specimen to the holding clamp with a horizontal burning rate of 120 mm/min, so no rating was recorded for the UL94 HB test. When 5 pph TPP was added, the PUF sample also did not satisfy the UL-94 HB test; however, the horizontal burning rate decreased to 75 mm/min. No UL-94 V rating was recorded for the PU-DZS-TPP
5 sample. The sample with the 15 pph TPP loading obtained the V-1 rating, while the V-0 ranking was obtained for the PUF sample containing 25 pph TPP; furthermore, the LOI value was found to be significantly increased from 17% to 21%.
The UL-94 and LOI results are quite good and surprising when considering the rather low content of phosphorous in TPP. Commonly, additions of more than 30 wt % TPP to polymers can achieve good flame retardancy. The results observed in this study—that only 25 pph (10 wt %) TPP loading could give a UL-94 V-0—clearly indicate that the chemical structure of the FR or the polymer itself and the fireproof mode of action of the FR and/or the polymer matrix need to be considered.
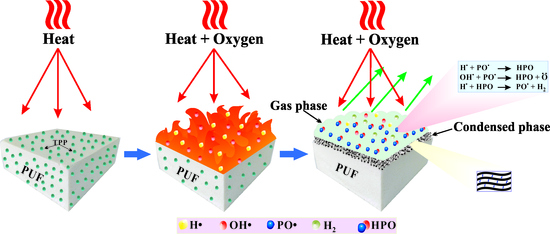
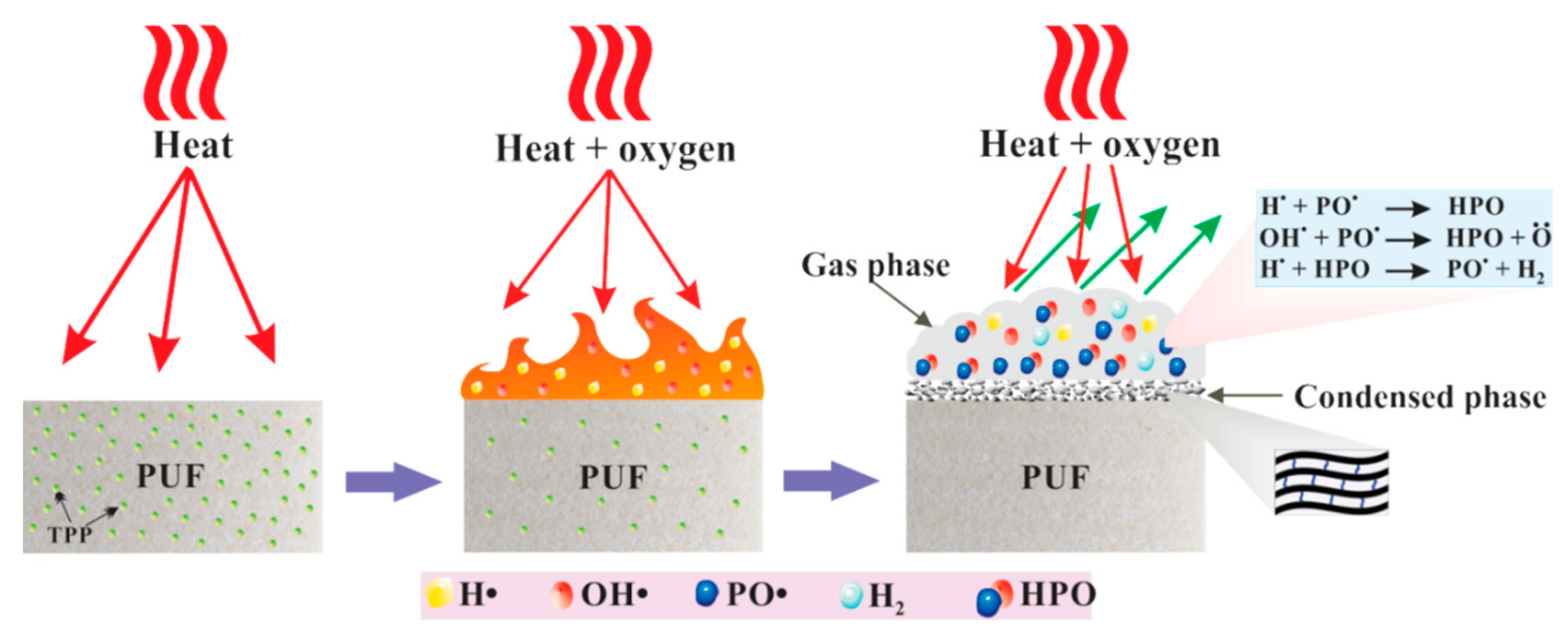
It is well known that TPP vaporizes and yields active radicals such as PO
2●, PO
●, and HPO
● during combustion. These radicals act as scavengers for H
● and OH
● radicals and thus extinguish the flame of the combustion process [
23,
24]. As mentioned above, the PU-DZS prepared from recycled PET oligodiol and MDI has aromatic moieties and becomes more thermally stable. According to the thermal decomposition in
Figure 8, the weight loss of PU-DZS is 74% in the temperature range of 250–650 °C, leaving 26% residual char. This means that the flame retardancy mechanism of PUF mainly depends on the gas phase, but the condensed phase also plays an important role. TPP evaporated in an early step and acted as a radical scavenger or crosslinker for the pyrolytic products of PU-DZS.
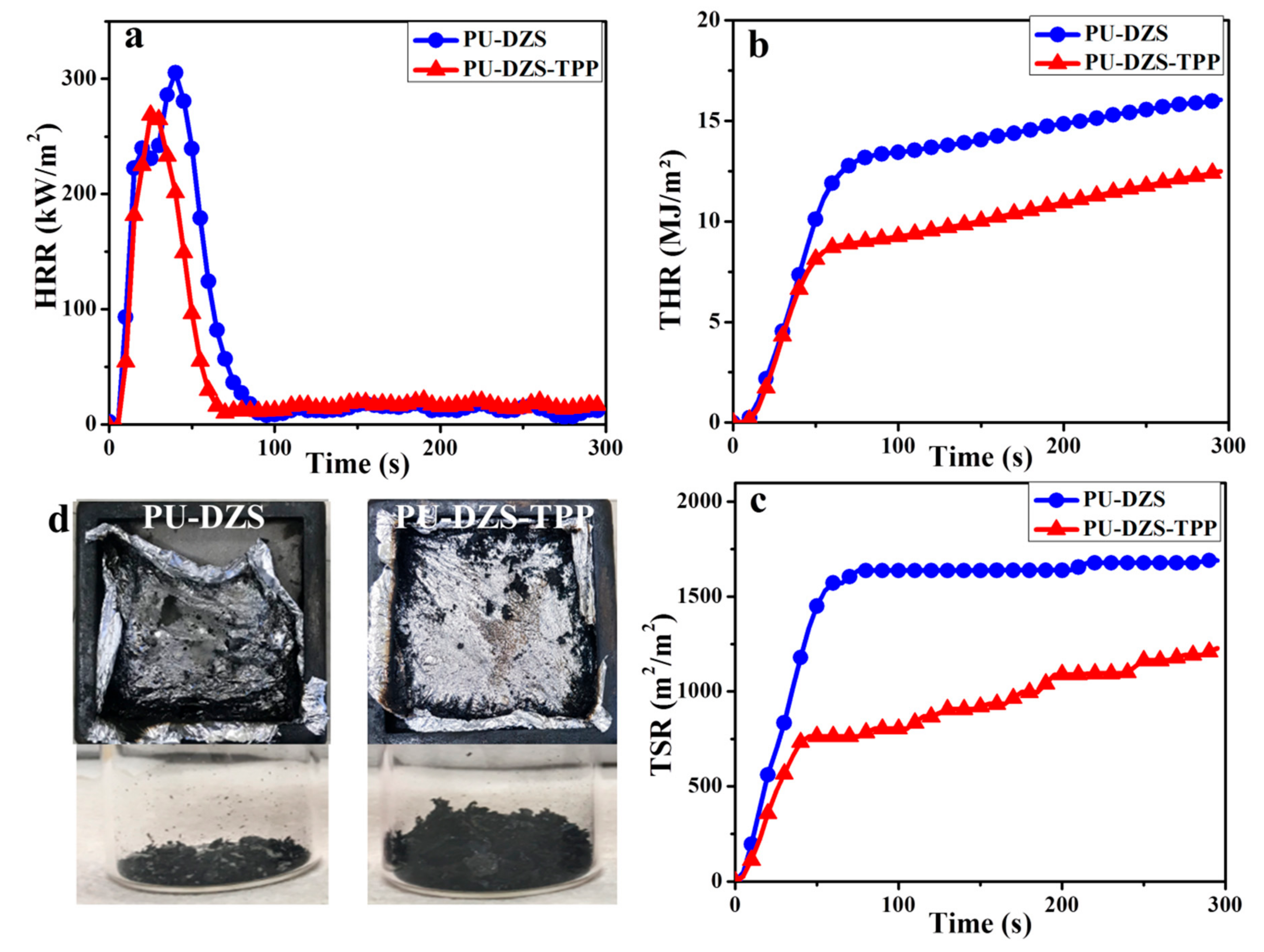
The cone calorimeter is a bench-scale instrument used to determine the flammability parameters of materials in laboratory. It is one of the most useful tests to compare with real-world fire conditions, and there were correlations between the cone calorimeter data and UL-94 V rankings [
25,
26,
27]. The flammable performance of the neat PU-DZS and PU-DZS-TPP
25 was evaluated by the cone calorimeter, and the testing results including the heat release rate (HRR), the peak HRR (PHRR), the total heat release (THR) and the total smoke release (TSR) are shown in
Figure 10. The UL94 and LOI results are in good agreement with the cone calorimeter data. One might expect to observe lower PHRR and THR values for PU-DZS-TPP with a better UL-94 V ranking. However, PU-DZS-TPP
25, classified as UL94 V-0, had lower PHRR, THR, and TSR values than the neat PU-DZS (no UL94 rating).
The PHRR, THR, and TSR of PU-DZS-TPP
25 were reduced by 12.9%, 22.8%, and 26.7%, respectively. As mentioned above, we know that the mode of action of TPP mainly inhibits gas-phase combustion, which effectively lowers the amount of oxygen consumed and the heat generated. In other words, TPP mostly acted in the gas phase and left very little residual char from combustion. Therefore, the PHRR is reduced, but not much, and it was assumed that the significant decreases in TSR and THR may be due to the prominent gas-phase mechanism of TPP and the partly condensed phase action of TPP and/or PUF (
Figure 11). The data also allowed further assumptions that there was a crosslinker between the pyrolytic products of PU-DZS and TPP to form residual char. However, these char layers are not stable enough to undergo further thermo-oxidative degradation, and therefore, very little residual char can be obtained after the cone calorimeter test.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}