3.1. Heck Coupling for Obtaining Silylalkenyl Units Anchored at the Si–O–Si Core
Heck coupling (HC), also known as the Mizoroki–Heck reaction, is generally a palladium catalyzed coupling process between unsaturated C=C bonds (here, Si–HC=CH
2) and an aryl halide (usually –I, –Br) resulting in the formation of alkenyl-substituted arenes. It occurs in the presence of additional phosphine ligand and equimolar amount of a base, although there are many variations of this process, a wide scope and tolerance to many organic functional groups is observed [
40].
Mono-functional silsesquioxanes are generally less frequently applied in obtaining compounds with interesting photophysical properties. Nevertheless, there are a few interesting examples to present that reflect the application of Heck coupling as a method for their synthesis. It is worth mentioning that the compounds are characterized by better thermo- and photostability. Liras et al. presented the synthesis of boron dipyrromethane (BODIPY) derivatives of POSS and their photophysical characterization (
Figure 5) [
41].
The elaborated synthetic procedure was not so effective, and enabled the respective mono- and disilsesquioxane substituted BODIPY derivatives (BODIPY–POSS) to be obtained with low (9%) to high yields (88%). Their photophysical properties were determined in EtOAc solution and poly(methyl methacrylate) (PMMA) film. Maxima absorption wavelengths were similar in both systems, with λab in a range of 521 to 536 nm, and maxima fluorescence emission wavelengths λem ranging from 532 to 575 nm. Thermal stability was improved for both systems, i.e. mono- and disilsesquioxane substituted BODIPY derivatives. The most photostable hybrid dyes were the systems with a POSS unit linked to the BODIPY by one or two ethylene bonds (π-conjugation). The results obtained indicate a possibility for these systems to be applied where high thermo- and photostability are required.
Monovinylhepta(cyclohexyl)silsesquioxane was reported as an efficient reagent towards Heck coupling (mediated by Pd(P
tBu
3)
2]) with p-iodophenyl pyrylene diimide by Lucenti et al. [
25]. This compound synthesized with a moderate 37% yield when compared with an analogous one, obtained via stoichiometric hydrolytic condensation (mediated by MeC
6H
4SO
3H) of hepta(cyclohexyl)silsesquioxane trisilanol with respective tri(methoxy)silylpropyl pyrylene diimide (23% yield). These two systems, where POSS unit and pyrylene diimide moiety were separated either via an aliphatic (propyl) or styryl spacer, were analyzed in terms of their absorption/emission spectra and compared to their pyrylene diimide counterparts. The type of the linker (i.e., aliphatic or arene) did not bear any influence on the photophysics of these. The analyses as expected showed slight red-shift emissions of up to 10 nm in solution (CHCl
3), but with very good photoluminescence quantum yields (up to 100%). The analogous analysis for solids resulted in higher red-shifted emission spectra due to the expected aggregation and π–π intermolecular interactions. However, the photoluminescence quantum yields for the POSS-based systems were twice as high as for the respected organic analogue that suggests suppressed aggregation. This favors its possible use in the fabrication of efficient emissive devices. This part is presented in this section for the ease of product comparison, despite the fact that it also concerns condensation reactions.
Heck coupling was also applied by Liu et al. in the synthesis of thermally stable mono- and octa- azobenzene-functionalized T
8-based dyes (
Figure 6) [
42]. The procedure enabled formation of the designed compound with yields of 67% and 80%; however, the reaction conditions were more severe in comparison with the previous example. Both systems were proved to be thermally stable with
Td5% = 273 and 383 °C for the mono and octa products, respectively. Introduction of these azobenzene moieties reduced the crystallinity of the compound withthe increasing amount of these groups in the molecule. The trans–cis photoisomerisation was examined and supported by DFT (Density Functional Theory) calculations. These systems were fluorescent with a maximum emission wavelength λ
em at about 400 nm, which enables their potential application as blue light emission materials.
Octa-functional POSS-based OLEDs have attracted far more attention mainly due to the possibility to anchor chromophores moieties (i.e., eight groups) onto the Si–O–Si core. Heck coupling may be conducted in a few combinations and towards the formation of fully substituted products as well as without full substitution.
Pioneering work was presented by Sellinger et al. when they reported on the use octavinyl silsesquioxane (OVS) in a Heck coupling process with a mono-brominated version of a highly π-conjugated arene system in the presence of [Pd(P
tBu
3)
2], obtaining hexa-substituted OVS with a 75% yield (
Figure 7) [
43]. Incomplete substitution was found probably due to a steric hindrance of the chromophore used and steric availability of vinyl groups. The obtained compound was verified in terms of its thermal stability, showing very high
Td5% = 465 °C. Its film and solution (toluene) photoluminescence spectra revealed λ
em = 430 and 434 nm respectively. The resulting electroluminescent device showed 18% improvement in external quantum efficiency over their small organic chromophore analogue.
Analogously, not-fully-substituted pyrene-based T
8 cages were obtained by Ervithayasuporn et al. in Heck coupling using a [Pd(OAc)
2]/PPh
3/NEt
3-mediated reaction that, after two days, resulted in the formation of a product with four pyrene substituents (as the most abundant species) with a 41% yield (
Figure 7) [
44]. The photophysical properties of the product were determined and broad emission bands λ
em = 406, 426 and 494 nm of low intensity in DMSO and sharp λ
em = 398, 421 and 445 nm of high intensity in THF were revealed. The compound revealed the solvent’s polarity dependant fluorescence, the π–π* emission of sole pyrene moieties was observed in THF, while in DMSO the formation of a pyrene–pyrene excimer through space has been postulated (the authors suggest its confirmation due to a large Stokes shift of Δλ = 143 nm). This compound was also tested as a sensor for several types of anions and, interestingly, was proved to encapsulate fluoride that results in a π–π* fluorescence enhancement in DMSO. Recent research by Ervithayasuporn et al. is related, with the synthesis of an analogous compound, but with four anthracene substituents (
Figure 7) [
45]. The synthetic procedure was based on Heck coupling reaction with the same catalytic system to obtain the desired product with 86% yield. Its photophysical parameters disclosed similar solvent’s polarity dependant fluorescence. It was also used to perform additional tests for possibility to encapsulate a variety of anions (e.g., F
−, OH
−, CN
− and PO
43−) by observation of fluorescence quenching due to from charge-transfer complex formation. This resulted in the possibility to use this compound as a sensor of the abovementioned anions.
Heck coupling to yield pyrene derivatives was also performed by Sellinger et al. They first reported on octavinyl T
8 cage functionalization, performing a [Pd(P
tBu
3)
2]-mediated reaction with bromopyrene [
46]. This was performed to obtain complete substitution (8–substituted as most abundant–P8) in comparison to the double Heck reaction product (14–substituted as most abundant–P14) with a >80% yield that was thermally stable up to 495 °C. These two systems were also analyzed in terms of their photoluminescence properties in solution (toluene), λ
em = 425 nm (P8) and 431 nm (P14), as well as in thin films, λ
em = 494 nm (P8) and 506 nm (P14). This meant a 70–75 nm red-shift effect when compared results in solution vs. in a solid state that suggested possible pyrene unit aggregation. The problem of two high “density” pyrene units was resolved by performing an efficient functionalization of vinyl-substituted T
8, T
10 and T
12 cages with bromopyrene and a mixture of bromopyrene and a smaller steric reagent (i.e., 4-heptylbenzene) using the same catalytic Pd system (
Figure 7) [
47].
Obtained products (i.e., monopyrene(heptaisobutyl)silsesquioxane, along with the pyrene/heptylstyrene-substituted T8, T10 and T12 cage product mixture) were analyzed with TGA (ThermoGravimetric Analysis), showing their high thermal stability up to 359–469 °C. The films prepared (using solution-processed deposition) from the obtained materials exhibited a sky-blue emission (λem in the range of 459–486 nm for thin films, and λem in the range of 400–449 nm for the toluene solution). These results may also suggest chromophore aggregation in a solid state. The obtained compounds were tested in solution-processed OLEDs prepared to obtain materials showing current efficiencies of 3.64% and 9.56 cd A−1, respectively.
Xu et al. presented a Heck coupling-based strategy to obtain anthracene-substituted T
8 cage via a [Pd(OAc)
2]/PPh
3-catalyzed reaction at room temperature in various reagent stoichiometry resulting in differently substituted products (
Figure 8) [
48]. The substitution was also statistical, but with the majority of the presented forms of products that depended on the possibility to form a complex between palladium with bromoanthracene substrate.
Their
Td5% in the range of 407 °C (H3), 468 °C (H1) and 480 °C (H2), respectively, suggest high thermal stability in comparison with bromoantracene
Td = 246 °C. The photophysics were verified and the UV-vis spectra revealed the presence of three absorption maxima wavelengths λ
ab = 356, 373 and 390 nm for solution in toluene. The emission spectra were recorded in toluene, showing emission maxima wavelengths λ
em = 440 (H1), 461 (H3) and 458 (H2) nm, which were compared to the spectra for solids that disclosed red-shifts Δλ = 42–51 nm, suggesting known phenomena of possible anthracene unit aggregation (the smallest effect for H1). The same authors reported on the further modification of dianthracene(hexavinyl)octasilsesquioxane (H2), and using Heck coupling to introduce one diazophenylnaphtalenediamine moiety followed by an amidation process to introduce a complex structure of a chromophore that possess broad-band absorbing properties [
49]. The presence of different chromophores allows the absorbing bands of the dyes to be combined, reduces the possibility of their aggregation and possesses a broad red-emission band at λ
em = 638 nm.
Completely octa-substituted T
8 cage obtained via Heck coupling reaction was reported by Liu et al. and used for the synthesis of a water-soluble hybrid nanodot based on POSS and conjugated electrolytes for the Two-Photon Excited Fluorescence (TPEF) analysis that imaginesthe cellular and cancer nuclei (
Figure 9) [
50,
51].
The Two-Photon Excited Fluorescence is a microscopic method used to reduce photodamage to a cell, improve resolution and minimize cellular autofluorescence, but it has rarely been exploited, due to the lack of efficient absorption materials that can enter nuclei and in turn selectively stain them. The desired fluorophore was anchored to the POSS core via a Heck coupling procedure of octavinyl silsesquioxane (OVS) and respective monobromo-substituted chromophore in the presence of [Pd(OAc)2/P(o-Tol)3] with a 45% yield. The quaternized silsesquioxane derivative was soluble in water and its average diameter of 3.6 ± 0.3 nm enabled easy penetration of cellular nuclei. The photophysical properties were verified and absorption maxima λab bands at 320 and 465 nm corresponded to the fluorene and benzothiazole units, but they red-shifted by 34 and 75 nm, respectively, due to the –HC=CH–POSS moiety extending the conjugation system. On the other hand, the λem was located at 617 nm and exhibited a large Stokes effect at 152 nm.
An interesting scientific concept was proposed and performed by Liu et al., who used octavinyl silsesquioxane (OVS) and multibromo-substituted arenes in a Heck Coupling process to yield crosslinked 3D networks of highly hyperbranched structures with a >80% yield (
Figure 10) [
52,
53].
The reaction procedure was also modified by using mixtures of different chromophores (e.g., BP and Py) in various stoichiometry, which resulted in the formation of macromolecular 3D systems with different chromophore content. The purification procedures of obtained polymeric materials, due to their 3D structure, was necessary due to the presence of non-reacted comonomers, and post-reaction inorganic salts and other residues within the matrix. The isolation was mostly based on filtration and washing with different solvents. For this reason, a Soxhlet apparatus was used with THF and MeOH to obtain pure products. All of the obtained compounds were analyzed in terms of their proven thermal resistance and high
Td5% at ~390 °C, but the high porosity of these materials was revealed by during BET (Brunauer–Emmett–Teller) analysis. Depending on the type (and amount, if mixture) of respective arene, respective hyperbranched products exhibited various surface areas from 358 to even 848 m
2g
−1 (when M3 was used as a reagent). The most interesting part of the research was disclosed in defining the photophysical properties of both substrates (M1–M3, BP, Py). In the case of M1, M2 and M3, which showed high blue fluorescence (λ
em at ca. 435 nm) in solid state and in solution (DCM = dichloromethane), the respectively obtained hyperbranched products showed strong red shifts (observed at λ
ex = 365 nm), and green (λ
em = 509 nm for prod.1, derived from M1; λ
em = 518 nm for prod.2, derived from M2) or yellow (λ
em = 540 nm for prod.3, derived from M3) fluorescence. This may be derived due to the extended π–π conjugation in the 3D network by additional –HC=CH– linkage. The ethanol dispersion of these materials was applied to obtain sensors for various nitroaromatic explosives by fluorescence quenching [
53]. On the other hand, modulation of the stoichiometry of BP and Py in Heck coupling with OVS resulted in the formation of nine types of materials that altered the amount of BP and Py units in the frameworks. As a result, they exhibited continuous color change from bluish-green at λ
em = 485 nm, to green at λ
em = 528 nm, yellow at λ
em = 555 nm and finally to red at λ
em = 605 nm. High fluorescence quantum yields in the solid state were calculated and ranged from 2.46% to 22.42% [
52]. These systems were used along with the thiol-containing polysiloxane matrix to produce a UV-LED device that shows color-transformable properties by simply turning the irradiation on and off.
Heck coupling using the silsesquioxanes may be performed also in the reverse functional group placement (i.e., to applied octa(halogenophenyl)silsesquioxane and styrene derivatives as a source of –HC=CH
2 moiety). Respective research on this issue was pioneered by Laine et al. [
54]. The primary reagent for the study was cubic octa(phenyl)silsesquioxane (OPS) that was subjected to bromination via a Fe-catalyzed reaction that resulted in [octa(polybromophenyl)]silsesquioxane. The resultant compounds had a different substitution of bromine at the Ph ring, but this could be controlled by reaction stoichiometry to obtain a para-brominated 2,5-dibrominated derivatives. A more-efficient and selective procedure for the synthesis of analogously octa(p-iodophenyl)silsesquioxanes using ICl was also reported [
55,
56]. These two systems are perfect halogenophenylPOSS reagents for the Heck and other coupling processes (e.g., Suzuki-type) [
57]. In 2010, Laine et al. reported on synthesis of a series of octa(stilbene)silsesquioxane derivatives using octa(p-iodophenyl)silsesquioxane and octa(bromophenyl)silsesquioxanes (on average 67% p-substituted, 24% meta, 9% ortho and 3% disubstituted) with respective p-substituted styrenes in Heck coupling reaction conditions (i.e., [Pd
2(dba)
3] (1 mol %)/[Pd(P
tBu
3)
2] (2 mol %) that resulted in octa(stilbene)silsesquioxanes with diverse p-phenyl substituents with high 62–86% yields (
Figure 11) [
58].
The desired octa-substituted products were also accompanied by compounds with not-completely substituted phenyl rings, which was a result of not-fully halogenated octa(phenyl)silsesquioxane, especially in the case of Br [
54]. The corresponding products were characterized in terms of their thermal resistance (
Td5% in the range of 318–429 °C). The greatest emphasis was placed on their photophysics assessment and its comparison with trans-stilbene and Me-stilbene-Si(OEt)
3. The UV-vis spectra of products (in THF) were similar in shape and red-shifted 5–10 nm (λ
ab 298–320 nm) to their molecular counterparts, but their large red shift of 60–100 nm was visible for the emission spectra of these systems when compared with trans-stilbene and Me-stilbene-Si(OEt)
3 (λ
em 334–436 nm)
. The greatest difference was noted for stilbene and p-Me-stilbene silsesquioxane derivatives that were also collated with UV-vis and PL spectra for octa(p-Me-stilbene)silsesquioxane and its hepta(p-Me-stilbene)phenylsilsesquioxane. Altogether, the absorption–emission changes that were observed along with the theoretical calculations and additional analysis of stilbene–siloxane and cyclosiloxane, respectively, suggest that the large red-shift may have resulted from interactions of the stilbene π* orbitals with a LUMO located inside the cage and involving all Si and O atoms, as well as the organic substituents that interact in the excited state. Interestingly, for the NMe
2 derivative the solvatochromism phenomenon was observed mainly due to the charge–transfer interaction when the polarity of solvent was changed.
Studies on stilbene derivatives of silsesquioxanes were also conducted. One of the aspects to be studied was the usage of octa(halogenephenyl)silsesquioxane reagents substituted with bromine at phenyl ring in different places (i.e., octa(o-bromophenyl)silsesquioxane, but also octa(2,5-dibromophenyl)silsesquioxane and octa(2,4,5-dibromophenyl)silsesquioxane) [
59]. This provided more functionalities per unit volume, exceeding the density when compared to POSS-based dendrimers. These compounds were used for analogous Heck coupling procedure (including catalytic systems, etc.), leading to corresponding octaphenyl silsesquioxane (OPS)-based styrenes with 8, 16 and 24 styryl substituents, with R = 4-Me, R = 4-NBoc and R = 4-Ac groups, respectively [
60]. Interestingly, the thermal parameter for these systems is similar to their octa-substituted derivatives. The blue shift of the absorption and the red shift of the emission spectra for the (R-o-styryl)
8OPS suggest the interactions of the Si–O–Si cage with the ortho-substituted organic groups. On the other hand, the (Rstyryl)
16OPS exhibited a longer conjugation length, resulting in higher values of Stoke’s shift and (Rstyryl)
24OPS behavior that revealed the existence of both a regular π–π* transition and a charge transfer, which could be explained by two excited states involving the LUMO inside the Si–O–Si cage.
The synthetic procedure of Heck coupling was also conducted in a controlled copolymerization of octa(p-iodophenyl)silsesequioxane with 1,4-divinylbenzene (DVB) and 1,4-diethynylbenzene (DEB). Laine et al. reported on a specific formation of copolymeric systems derived from octa(p-iodophenyl)silsesequioxane embedded between DVB or DEB fragments (
Figure 12) [
61].
The synthetic routes that were tested were based on changing the reaction sequence of octa(p-iodophenyl)silsesequioxane Heck Coupling functionalization (carried out with specific stoichiometry) and linking each silsesquioxane via Heck or Sonogashira cross-coupling with DVB or DEB. The procedure resulted in final copolymeric systems with a moderate 39%–51% yield and respected average molecular weights in the range of 9–30 kDa, as well as suggesting that the formation of di(p-iodostyryl)hexastyrylsilsesquioxanes as the first reaction sequence results in higher molecular weights of the resulting copolymeric systems. The photophysics of the two types of copolymers significantly depended on the organic linker. The DVB copolymers exhibited very little conjugation due to high 1,4-DVB fragments, whereas the DEB showed a strong red-shift at ~40 nm emission. This suggests electron delocalization through the Si–O–Si cage. Again, the p-aminostilbene copolymer derivative showed charge-transfer stabilization.
Laine and co-workers expanded the studies conducted on T
10 and T
12 cages with reactive Si–HC=CH, p-iodophenyl and bromophenyl substituents. They previously reported on the possibility of fluoride ion (TBAF = tetra-n-butylammonium fluoride) catalyzed cage rearrangements between polyvinyl- and polyphenylsilsesquioxane, resulting T
10 and T
12 with mixed reactive group placement (depending on the reaction condition and stoichometry) [
30,
31]. The research concerned using vinylT
10/12 cage mixture and its functionalization using Heck and double Heck coupling with bromostyrene, 1-bromonaphthalene and 9-bromoanthracene, as well as Grubbs first generation catalyzed cross-metathesis of the vinylT
10/12 with p-substituted styrenes (
Figure 13) [
62].
In parallel, the reverse Heck Coupling (i.e., reaction) of (Br-styrenyl)T
10/12 cage mixture with p-substituted styrenes was conducted (
Figure 14).
The results for both processes are presented here for the ease of their comparison (CM is described in
Section 3.3). The described catalytic processes are parallel routes for analogical T
10 and T
12 structure formations, and also similarly efficient. The products were compared, their thermal resistance was verified and they exhibited
Td5% up to 445 °C. The UV-vis and PL spectra in THF were analyzed and, in general, were similar to those obtained for T
8 analogues. Interestingly, the UV-vis absorption maxima of products obtained via Heck and double Heck coupling were almost the same (in the range 254–374 nm), while the emission of double-functionalized compounds was a bit red-shifted (310–465 nm), as should be expected for the slightly extended, excited state of conjugation. The p-substituted stilbene-based T
10 and T
12 exhibited emission maxima dependent on the electronic properties on the substituents (e.g., the largest red shift of λ
em for the = –NH
2 group, that results from a charge transfer). For this derivative, a solvatochromism was also observed for MeCN (acetonitrile), THF and cyclohexane solvents used. Additionally, the dependence of emission spectra shapes (maxima) and their intensity on the solvent used (MeCN:THF, hexane:THF) and the spectra collected for various compound concentration suggested their aggregation. This possible aggregation may be responsible for the exciplex formation and seems to differ significantly from the one known for traditional organic molecules. These findings might suggest smaller band gaps, meaning a possible use of these systems in 3D hole/electron transport devices.
The research connected with the use of T
10 and T
12 cages was carried on for copolymeric systems. Again, Laine et al. performed experiments using octa(vinyl)silsesquioxane T
8 (OVS) and octa(p-iodopheyl)silsesquioxane T
8 to obtain a mixture of T
10 and T
12 with mixed vinyl- and p-iodophenyl-substituent placement (with a 90% yield) along with an analogous reaction for vinyl and phenyl-substituted T
10 and T
12 cages [
63]. The vinyl/phenyl-mixed T
10 and T
12 systems were substrates for the following:
Heck Coupling reaction (analogous catalytic conditions described in [
61]) with 1,4-dibromobenzene and 4,4′-dibromostilbene, resulting in lightly branched non-linear copolymeric systems with a 83–86% yield. This is the fastest way to obtain the desired copolymeric compounds.Cross-metathesis (described in detail in
Section 3.3) reaction, using Grubbs first generation catalyst with 1,4-dibromobenzene to obtain (again) slightly branched non-linear copolymeric system with a 89% yield.The vinyl/(p-iodo)phenyl-mixed T
10 and T
12 cages were substrates for the reverse synthetic path (i.e., cross-metathesis reaction, using Grubbs first generation catalyst with 1,4-dibromobenzene to obtain a slightly branched non-linear copolymeric system with a 95% yield).The abovementioned copolymer (i.e., its p-iodophenyl substituents) was post-modified via a Heck coupling reaction using R-styrenes (H-styrene, OMe-styrene) to obtain R-styrene functionalized copolymer with a 67% yield.
All these six types of copolymers were tested in terms of their photophysical properties, with results collated for those with bis(vinyltriethoxysilyl)benzene and 4,4′-bis(vinyltriethoxysilyl)stilbene used as models. All copolymers exhibited a red shifted emission spectra that were strongest for the copolymers obtained via Heck Coupling (described in the first point) at ca. 20–60 nm for the DVB derivative when compared with the model compounds. This is attributed to shorter conjugated linkers, enabling Si–O–Si cage participation in the 3D excited states. The compounds were obtained in particular because of their 3D interaction in the excited state that corresponds with their application in OLEDs devices.
3.3. Cross-Metathesis and Related Modfications for Silylalkenyl Fragment Formations
Cross-Metathesis (CM) is one of the fundamental catalytic transformations in the chemistry of organosilicon compounds [
67]. In this manner, it is a TM-carbene-mediated (TM = Ru, Mo, etc.) process of C=C bond cleavage and formation of unsaturated organosilicon derivatives. Silsesquioxanes, which are promising precursors with Si–HC=CH
2 reactive moiety may be placed in this group of reagents [
68,
69,
70,
71]. CM may be applied as a single synthetic route leading to desired POSS-based compounds, or used as a valuable tool for obtaining precursors of silsesquioxanes with reactive groups for consecutive reactions (e.g., Heck or Sonogashira coupling). In this matter, the described process was used to obtain a series of compounds exhibiting interesting photophysical properties.
Cole-Hamilton et al. contributed to this issue with the use of octavinyl silsesquioxane (OVS) grafted with styrene derivatives using Grubbs first generation catalyst (4 mol %) and 2–3 equiv. of styrene per one Si–HC=CH
2 group and resulting in good 67%–78% product yields (
Figure 16) [
72,
73]. The modifications of styryl rings resulted in changes in absorption and emission spectra, especially when aldehyde moiety was introduced and resulted in quenching emission. The POSS derivatives were reported to exhibit red shifts and broader spectral emissions due to the involvement of a silsesquioxane core in electronic delocalisation. It should be noted that CM reaction conditions may be improved so that not as much styrene excess is required to complete OVS conversion (24 h) [
74].
Cross-metathesis was employed as precursor synthetic process for obtaining mono(p-bromostyryl)heptaisobutylsilsesquioxane (M1-Br) by Naka et al. [
75]. This compound was applied as a reagent in two subsequent Sonogashira coupling reactions, resulting in dumbbell-shaped π-conjugated systems with POSS as pendant groups (
Figure 17).
Unfortunately, the reaction conditions were based on the use of 3 equiv. of para-bromo-styrene, and 3 mol % of Grubbs first generation catalyst (added to the reaction in two portions) enabled the formation of precursor M1-Br with only a 68% yield, contrary to the procedure developed in our group [
76]. The next steps of the process were performed with moderate 37%–58% yields of respective final dumbbell products (DA13). These were characterized in terms of their photophysical and thermal properties in comparison with their organic counterparts. Their
Td1% was respectively high (268–282 °C). The interesting part of the research concerns absorption and emission spectra performed in a solution vs. in a solid. The λ
em maxima in the solid state were red-sifted in a range of 34–50 nm and equaled 401, 542 and 520 nm for DA13, respectively. This was noticed after UV irradiation (352 nm) that revealed blue, yellow and greenish-yellow emissions in POSS-based systems contrary to their organic analogues. These results also indicate the participation of the Si–O–Si cage in the delocalization of electrons, and prevention of the π-conjugated parts from interaction.
Laine et al. have profoundly contributed to the development of cross-metathetic transformation using the most useful reagent in this process (i.e., octa(vinyl)silsesquioxane T
8 (OVS), but also decavinylT
10 and dodecavinylT
12 cages). The selective introduction of substituted (E)-styrenyl moiety at Si–O–Si cores and for the bromo-derivatives enabled their consecutive modification via Heck coupling (
Figure 18) [
71,
77]. Interestingly, the cross-metathesis for OVS proceeded with 0.5 mol % of the catalyst and at room temperature only in the cases of T
10 and T
12 at 40 °C. The purification of these compounds concerned filtration over Celite, precipitation and column chromatography, obtaining final products with 74%–82%yields. Additionally, the p-NH
2-vinylStilbene product was transformed into respective benzamide derivatives. All of the compounds were characterized in terms of photophysical properties. For T
8 compounds, the absorption (λ
ab 260–358 nm) and emission (λ
em 304–482 nm) spectra showed red shifts in comparison with their organic counterparts (styrene and p-vinylstilbene).
The octa(XStyryl)silsesquioxane T
8 showed smaller red-shifts than octa(R′Stilbenevinyl) silsesquioxane T
8compounds (10–40 nm shifts vs. 30–80 nm shifts—i.e., Stokes shifts at ca. 50 nm) as might be expected due to a larger conjugation for the R′StilbenevinylT
8 compounds. The red shift is also larger in the presence of more electron-donating groups (–OMe). The NH
2StilbeneVinylT
8 exhibited effects of solvent polarity on its emission behavior (DCM and MeCN) due to charge-transfer (CT) interactions that were also proved by DFT calculations, as noted previously [
58]. The StilbeneVinylT
10 and T
12 derivatives were separated effectively due to the proper solvent choice. Their photophysical properties were compared along with the StilbeneVinylT
8 to observe the impact of the cage size and symmetry measured in solution (THF) and films. In general, the red-shift emission was observed for all three compounds, due to the π-conjugation, and the additional red-shifted spectra for films revealed the aggregation of chromophores, as expected. Additionally, two-photon spectroscopy analysis was performed to compare the polarization and non-linear absorption properties of the separated T
8, T
10 and T
12 cages along with the p-triethoxylsilylvinylstilbene and p-vinylstilbene. Due to the differences in symmetry, especially between T
10 and T
12 cages, these compounds displayed changes in the dipole moment of excitation and exhibited different emission maxima wavelengths, with excitation at λ
ex = 333 nm (T
10 − λ
em = 386 nm, high intensity), λ
ex = 400 nm (T
10 − λ
em = 453 nm) and λ
ex = 800 nm (T
10 − λ
em = 450 nm, the highest intensity when compared with T
8 and T
12). Indeed, the observed impact on fluorescence decreased for bigger T
12 cages, which may have been derived from the cage symmetry and/or the chromophores’ proximity. Moreover, a red-edge effect in these 3D molecular structures was observed (i.e., the red-shifted emission wavelengths upon shifting of the excitation wavelength to the red end of the abs. spectra. The highest effect was observed in the case of deca(Stilbene)silsesquioxane T
10, which suggests a possible application of these compounds as a new class of hybrid materials offering switchable emissions.
The cross-metathesis process, leading to polymeric POSS systems (e.g., ADMET reaction), was described in
Section 3.1 due to the ease of comparison of the products, and is analogous in structure to a Heck coupling reaction [
63].
The Núñez research group presented an interesting combination of analogous cross-metathesis and Heck coupling sequences leading to octa(carborane-Stilbenevinyl)silsesquioxane T
8 cages (
Figure 19) [
78]. The cross-metathesis of OVS with p-bromostyrene was perfumed using the synthetic procedure reported by Laine [
71]. As a result, octa(Br-styrene)T
8 was applied as a reagent in efficient Heck coupling with four types of carboranyl-substituted vinylstyrene, producing a good isolation yield of 1–4 products (43–65%). The importance of this research lies in the combination of carboranes known from their 3D electron delocalization and the fact that they were additionally coupled with π-conjugated arene fragments along with the T
8 silsesquioxane unit, enhancing thermal stability and also affecting the electronic properties of these systems. All compounds were characterized in terms of their absorption–emission properties (analysis performed in DCM) and supported by DFT calculations. The absorption spectra shapes were similar with λ
ab near 338 nm and all emission maxima wavelengths red-shifted to ~392 nm (λ
ex = 340 nm). Interestingly, the highest symmetry of product 1 in comparison with products 2–4 (
Figure 19) was visible in the broadening of fluorescence in the red part of the spectra that indicates intermolecular interactions of the o- and m-substituted carborane units. This also caused the fluorescence quantum yield to be the highest for 1 (~60%). Additionally, the emission spectra of drop-casted films revealed its strong bathochromic effect (λ
em 450–484 nm) due to the aggregation and intermolecular interactions of substituents. However, these systems exhibited remarkable thermal resistance, with up to 87% residue on the initial weight at 1000 °C, making them excellent candidates for luminescent materials.
3.5. Hydrosilylation of Alkenes and Alkynes as Effcient Procedures for the Introduction of Silylalkanyl and Silylalkenyl Moieties tothe Si–O–Si Core
In the chemistry of organosilicon compounds, the hydrosilylation process is of key importance and, in this case, it has a predominant role as a fundamental industrial tool for the synthesis of the aforementioned systems [
87,
88]. A catalytic insertion of Si–H into multiple bonds, in particular into carbon–carbon unsaturated bonds (C=C and C≡C) including heteroatom, occurs in the presence of transition metal complexes (TM complexes), especially platinum Karstedt’s catalyst = [Pt
2(dvds)
3], and nickel, rhodium and iridium based systems in homo- and heterogeneous forms, but also with Lewis acids (e.g., AlCl
3). The reaction conditions, catalytic systems and reagents’ structures influence the stereo-or even regioselectivity of hydrosilylation leading towards saturated and unsaturated molecular, but also macromolecular organosilicon systems [
82]. Hydrosilylation is also a very popular catalytic procedure to anchor organic dyes onto a Si–O–Si core of a cubic T
8 cage (POSS) or a double-decker (DDSQ) type.
Imae and Kawakami’s group reported a new type of POSS derivative containing carbazole as a photo- and electroactive chromophore [
89,
90]. The carbazole unit was introduced via hydrosilylation between octa(dimethylsiloxy)silsesquixane (Q
8M
8) and 9-vinylcarbazole in the presence of commercially available Karstedt’s ([Pt
2(dvds)
3]) as well as Speier’s (H
2PtCl
6) catalyst (
Figure 22).
The reaction was performed in dry toluene using a small excess of olefin to ensure complete conversion of the reactants, and it led to the formation of the expected product with a 70% yield. Its TGA showed that the product was stable up to ~400 °C in air, and 450 °C in N2 (i.e., it exhibited higher stability than its organic analogoue, poly(9-vinylcarbazole) (PVCz), which was also proved by DSC anaysis and was expected by POSS addition). The Tg = 37 °C was much lower than that of the PVCz (Tg = 190 °C) due to the flexible spacer between POSS and carbazole. The photophysical parameters were analyzed in solution (THF) and in a solid state, and were compared to their organic couterparts (9-ethylcarbazole (EtCz) and poly(9-vinylcarbazole) (PVCz)). The emmision spectrum of the product (λex = 250 nm) showed two peaks, λem = 353 and 370 nm, which is almost the same as that of the EtCz. Similar compatibility was obtained in terms of quantum yields in air (Φp = 0.27 and ΦEtCz = 0.30). On the other hand, the PVCz showed weak and broad peaks with a lower quantum yield (ΦPVCz = 0.10) resulting from the formation of excimer. The λem for solids was more red-shifted towards the wavelength region 350–400 nm and 450 nm, indcating a greater aggregation in crystalline product than that in amorphous material, but the values of the photoluminescence quantum yields were the same. The result suggests that the introduction of the rigid POSS core isolated each of the carbazole units, preventing their aggregation and the formation of excimer, which resulted in a useful concept to apply this new material in a solid-state optelectronic device.
Interesting research on the synthesis of carbazole-substituted double-decker-shaped silsesquioxane (DDSQ-4Cz) also obtained via a hydrosilylation reaction (mediated by Karstedt’s catalyst) was presented by Miyashita et al. (
Figure 23) [
91].
The obtained material’s thermal stability was higher than 355 °C. Photophysical properties of DDSQ-4Cz were disclosed via UV-vis and photoluminescence analysis and compared with its organic analogue (N-vinylcarbazole). The absorption spectrum (in CHCl3) revealed of two peaks (λab between 325–350 nm) atributted to the π–π* absorption of carbazole, which closely resembled that of N-vinylcarbazole. Interestingly, the photoluminescene spectra of DDSQ-4Cz and N-vinylcarbazole were practically the same (λem = 350–300 nm), indicating that the four carbazole units were isolated by the rigid core, thereby preventing excimer formation. The authors also demonstrate the obtained material’s potential application in organic electronic diodes. They fabricated an electroluminescent device that showed a maximum brightness of 320 cd m−2 at 20 V drive voltage with 40 mA cm−2 current density. This proved that DDSQ are promising compounds useful for OLEDs.
Dudziec and Marciniec’s groups reported on dihydro-substituted double-decker silsesquioxane (DDSQ-2SiH) efficient modification using Pt-Karstedt’s mediated hydrosilylation reaction protocol with precise reaction time control (FTIR in situ). This facilitated selective formation of a series of molecular as well as macromolecular DDSQ-based systems with ethyl-bridged π-conjugated arenes (
Figure 24) [
92]. Obtained compounds were verified in terms of their thermal stability (higher than organic couterparts) as well as their photophysics.
A UV-vis analysis of the resultng molecular and macromolecular compounds in comparison to the respective arenes was performed. For the DDSQ-based products, the absorption spectra combined the shape of the DDSQ unit as well as the organic counterpart. For the 1-(4-vinylphenyl)naphthalene derivative, the absorption spectrum showed a 21 nm blue-shifted torn peak at λ
ab = 257 and 330 nm, which was different than the basic arene (λ
ab = 278 nm). This result may suggest the involvement of Si–O–Si core in the excited state, as has already been reported by Laine and Sellinger for mono- and octa-functionalized silsesquioxanes [
47,
58]. The photophysical and thermal features could enable the application of these compounds in OLEDs devices. Our group studied the hydrosilylation of olefins and alkynes with silsesquioxanes with one, four and eight reactive Si–H moieties, which constitute a valuable group of precursors used in the formation of photoactive materials [
93,
94,
95,
96,
97].
In 2014 Wang, He and Chin demonstrated a practical route to the synthesis of a series of three hybrid polymers containing a periodically interconnected DDSQ unit and oligofluorenes in the main chain [
98]. Hydrosilylation was employed to prepare these materials by sequential addition of dihydro-substituted DDSQ (DDSQ-2SiH) and fluorenes: monomer (
n = 1), dimer (
n = 2) and trimer (
n = 3) in the presence of the Karstedt’s catalyst (
Figure 25).
The obtained copolymers (P1–P3) had molecular weights of Mn = 9700, (Polydispersity Index) PDI = 2.55; Mn = 18,700, PDI = 1.45; and Mn = 39,300, PDI = 1.34, respectively, and were soluble in common oragnic solvent. TG analyses indicated a high level of thermal resistance in the obtained systems, up to 446 °C. This means that the block copolymer containing DDSQ units displayed an improvement in their thermal stability when compared to neat poly(fluorene) (PF, 417 °C). The authors also demonstrated that the thermal stability of these products was affected by the ratio of DDSQ in the polymer chain, since the DDSQ can lower thermal conductivity and work as a thermal barrier to isolate heat. All polymers were thermoplastic, with significantly higher Tg (up to 125 °C) than the corresponding PF (Tg = 55 °C). The optical properties of the hybrid nanocomposites were determined and compared with the chromophores used. The absorption and emission spectra (in solution and solid) were different. According to the literature, conjugated polymers exhibited significant changes in their solid-state spectra because of interchain interaction (e.g., for PF, λem changed from 459 to 539 nm in the solid state). Surprisingly, in obtained products, no obvious λem peak shift was observed for solid- and solution-state spectra, while the λab of polymer films (solid) was slightly blue-shifted in comparison to those in a solution. These slight shifts were probably due to the aggregation-induced disorder. The authors of the study observed small 4–13 nm red shifts in the PL λem for solution compared to the solid state, mainly because of the restriction in conformational motion. It is worth mentioning that λab and λem gradually red-shift along with the increase of chain length (i.e., from P1 to P3). It was demonstrated that all the hybrid polymers exhibited a quantum yield higher than 95%, while for the PF it was only 79%. The reason for such high values of P1–P3 quantum yields could be the confinement of excitons within each oligofluorene unit due to the interconnecting DDSQ cage. Thus, the oligofluorene fragments behaved as isolated, not conjugated chromophores. According to the authors, these results suggested that the optical properties of oligofluorenes were faithfully maintained in the proposed final materials. To demonstrate the potential of synthesized materials in organic electronic applications, the authors successfully prepared two OLED devices that maintained color stability and purity when compared to PF.
In 2006, Shim et al. demonstrated that a hydrosilylation reaction may be used for the efficient synthesis of POSS-based blue-light electroluminescent (EL) nanoparticles containing terfluorene moieties on each of their eight arms [
99]. This material was obtained via the Pt-catalyzed hydrosilylation between commercially available octa(dimethylsiloxy)silsesquioxane T
8 (Q
8M
8) and an allyl-functionalized terfluorene (
Figure 26).
The material displayed high thermal stability (Td5% = 379 °C) and good film-forming properties (tests on a quartz or an indium tin oxide plate). The photophysical analyses were performed in solution (THF) and in solid state. The maximum λab in solution was 352 nm (353 nm in a solid state) with PL emission maxima λab at 394 and 415 nm (401 and 420 nm in a solid state). These results were compared with those for the organic counterpart poly(dihexylfluorene) and were coincided both in solution and in solid state. This good spectral overlap suggests that POSS–FL can be used as a dopant of blue-light-emissive conjugated polymers, such as polyfluorenes, to increase their quantum efficiencies through energy transference and the isolation of chromophores, as well as in applications requiring electroluminescent nanoparticles. It is worth mentioning that the external quantum efficiency in EL devices of POSS-derivative doped poly(dihexylfluorene) blends were found to be four to eight times higher than that of their organic analogue.
Other examples based on the same procedure for anchoring the chromophore part onto the T
8 core were presented by Lee, Mochizuki and Jabbour group [
100]. They used a hydrosilylation process for the synthesis of a series of macromolecular materials composed of an inorganic Q
8M
8 core and organic fluorescent emitters covalently attached either monochromatically or in a combination of mixed emitters (POSS with different chromophore systems) (
Figure 27).
Hydrosilylation was performed using Karstedt’s catalyst with fixed and controlled reagents’ stoichiometry; however. the final product yields may be not satisfying and may suggest further optimization of the procedure. Monochromatic POSS-emitter products (1–3) were obtained with a 60%, 47% and 61% yield, respectively, while isolated yields of POSS containing a combination of two different emitters (4–7) were not higher than 26%. All compounds were proved to have higher thermal stability than their organic emitters. The most interesting part of the research was disclosed in defining the photophysical properties of obtained compounds. Solution and thin-film photoluminescence spectra were measured and compared for both the free emitters and the modified POSS. It was demonstrated that monochromatic products (1–3) have similar absorbance and emission spectra to their free emitter counterparts both in solution and in a solid state. Thus, using POSS core as a scaffold does not influence the color of the emitters applied. Surprising results were obtained for products bearing more than one type of emitter (4–7). It was found that lower-energy emissions (orange or yellow) dominated in the photoluminescence spectrum for materials 4–7 due to a strong intramolecular energy transference from the neighboring higher-energy blue emitter on the POSS. This high degree of energy transference derives from two major factors: the overlapping of the blue emitter’s emission band with its lower-energy absorption band, and the blue and orange emitters’ short intramolecular distance. Overall, the authors indicated that these materials were easily processable and could be spin-coated from solution for the fabrication of OLED devices. They were also prepared and tested with dopants of selected POSS-based systems.
In 2009, Hwang et al. described the synthesis of a similar type of material, i.e., POSS-based electroluminescent nanoparticles, containing anthracenenaphthyl fragments at each of their eight arms [
101]. The procedure involves the hydrosilylation of Q
8M
8 with vinyl-functionalized 9-naphtalene-2-yl-10-phenyl anthracene in the presence of 2 mol % of Karstedt’s catalyst (
Figure 28).
POSS–NPA was analyzed to investigate its thermal and photophysical properties. It was found to be thermally stable up 450 °C, with high Tg = 154 °C. The UV-vis absorption and PL emission maxima of the final product in solution (chlorobenzene) were found to be 378 and 433 nm, while those characteristics for the product in a solid state were 379 and 464 nm, respectively. These results indicate the aggregation of POSS–NPA molecules and their intermolecular interactions. The authors fabricated an electroluminescent (EL) device that shows blue light emissions.
Xu and Su reported on the synthesis of a new star-type POSS-based molecular hybrid containing stilbene chromophore [
102]. These materials were prepared by the hydrosilylation of a series of alkynylstilbenes with octahydridosilsesquioxane (OHS) (
Figure 29).
The reaction was performed in refluxing 1,2-dichloroethane (DCE) in the presence of platinum dicyclopentadiene complex [Pt(dcp)], and led to the formation of expected products with isolated yields ranging from 42 to 51%. However, this catalyst seems to have lower selectivity, as in all cases the formation of α and β isomer mixture was detected (α:β = 39–43:57–61). All products exhibited high thermal stability (Td5% up to 320 °C, Tg up to 250 °C). The THF solutions of the products were easily cast into films, whereas films of the chromophore could not be obtained in the same conditions. The photophysics were verified and the UV-is spectra (in THF) revealed the presence of strong absorption peaks at λab = 336 (H1), 339 (H2), 335 (H3) and 360 (H4) nm. It was also found that alkoxy chain length exerts little influence on the absorption of products H1–3. In the case of H4 the absorption peak was red-shifted, which may be attributable to a stronger intramolecular interaction and formation of the excited state. It was demonstrated that the products (H1–4) and their free emitter counterparts showed nearly the same λab and broader absorption bands for the hybrids (H), that may have originated from the σ–π conjugation effect of Si–C=C– in obtained materials. The authors proved that the combination of an inorganic POSS core with stilbene chromophores endowed the resulting materials with novel optical limiting properties and high thermal stability. However, there wano information on how the presence of α and β isomers in H affected these properties.
In 2016, Le et al. developed a new spherosilicate-based compound containing carbazole and pyrene units [
103]. This new organic–inorganic amorphous hybrid material was obtained in a two-step reaction involving: (1) synthesis of chromophore using alkylation of
N-dipyrenylcarbazole with allyl bromide, and (2) hydrosilylation of obtained olefin with Q
8M
8 in the presence of Karstedt’s catalyst (
Figure 30).
The authors demonstrated that this newly-developed POSS derivative exhibits excellent thermal stability, efficient control of POSS dispersion behavior and good film-forming properties. The photophysical properties of the product were determined and compared with chromophore used in solution (CHCl3) and in solid state. The UV-vis spectra of both compounds showed a strong absorption band at λab = 281 nm, which can be attributed to the π–π* local electron transition of the carbazole fragment, and a longer-wavelength absorption band at λab ca. 349 nm, which corresponds to the π–π* electronic transition of the chromophore backbones. The shape and position of the PL emission spectra was different for the product and chromophore, which can be attributed to the fact that bulky POSS cages reduce the extent of substrate aggregation. This conclusion also supports measurement of PL quantum efficiency (PLQE) of the product (90%), which was much higher than that of the separate chromophore (66%). The results of emission in solid state were interesting, and showed ca. 20 nm red-shift in λem compared to the solution-phase spectra. This suggests the presence of weak van der Waals forces and intermolecular π–π interactions of adjacent chromophore fragments. Although significantly red-shifted behavior was observed in the film state, the final product exhibited an emission band centered at 454 nm, which was substantially blue-shifted compared to that of the substrate film (463 nm). These observations indicate that the POSS cage inhibits the aggregation of chromophore units to improve their color purity and stability both in solution and the solid state. This also promotes its possible applications in solution-processed optoelectronic devices.
Li and Xu et al. reported on the synthesis of two organic–inorganic nanohybrid materials obtained by decorating spherosilicate POSS cores with 2,3,4,5-tetraphenylsilole units through a one-step hydrosilylation process (
Figure 31) [
104]. As presented in
Figure 31, Q
8M
8 and Q
8V
8 were used as synthetic scaffolds and two different silole derivatives were chosen as their reagents. Expected products (
1,
2) were isolated with 57% and 47% yields, respectively. Their thermal and photophysical properties were determined along with their utility as potential sensors for the selective detection of nitroaromatic explosives in aqueous media. As expected, thermal stability of these systems was improved (up to 390 °C). Photophysical properties of both products were solvent-dependent. It was observed that product
1 emits weakly at 486 nm in THF and at 496 nm in a mixture of THF/water (1:9), while in 90% water content the photoluminescence intensity is 68-fold higher than in pure THF.
These results suggest that the PL of 1 clearly demonstrates the aggregation-enhanced emission (AEE) of this molecule. Similar emissive behavior was also observed for 2. For a quantitative comparison, measurements of fluorescence quantum were performed. The absolute fluorescence quantum values of the thin solid films of both products reached as high as 51% and 45%, respectively, meaning they were higher than those of their corresponding parent silole molecules in the solid state. Obtained results proved that the POSS core improved the fluorescence quantum yield of the AEE materials in both the solution and in the aggregated state.
Further examples of the use of silsesquioxane derivatives in hydrosilylation with azo-chromophores in order to form functionalizable POSS nanoparticles have been reported by the Xu, Tsukruk and Miniewicz groups independently [
105,
106,
107,
108,
109]. The procedures, allowing the synthesis of POSS derivatives with azobenzene mesogenes, have been proposed via the hydrosilylation of respective octahydro-substituted T
8 silsesquioxanes. Selected structures of final materials (
1–
10) are presented in
Figure 32.
Xu presented a group of azobenzene-containing POSS-based star-like hybrid functional materials obtained via hydrosilylation octahydridosilsesquioxane with three different azobenzene chromophores containing terminal alkynyl moiety as the reactive fragments [
109]. The reactions were carried out in the presence of platinum dicyclopentadiene complex [Pt(dcp)] at 80 °C, using an eight-fold molar excess of olefin relative to silsesquioxane. However, the process was not selective, and expected products were obtained as a mixture of α- and β-adducts with yields ranging from 48% to 54%. Analyzed thermal stability was proved to be higher than of the respective chromophores and suggests its enhancement with anchoring chropomhores onto POSS core. Interestingly, the authors did not observe any sign of melting or glass transition of the products up to the decomposition temperatures, though all substrates were crystalline solids. These amorphous characters of compounds may result from a star-like structure with rigid azobenzene chromophore units protruding from a spherical POSS core in 3D thus minimizing any π–π interactions. The photophysical properties of all products were disclosed using UV-vis and PL analyses, and were compared with their organic analogues. Product 10 and its corresponding azobenzene chromophore displayed nearly the same maximum absorption wavelengths and spectral pattern, indicating that Si–O–Si core has no significant effect on the electronic structure of the final material. The UV-vis spectra of products (8–10) in THF showed strong absorption bands at λ
ab = 356, 347 and 365 nm, respectively, which can be assigned to the π–π* local electron transition of the azobenzene fragments. It was also proven that azobenzene moieties influence the λ
ab (e.g., the maximum absorption peak of compound
9 shows a 9 nm blue shift compared to that of
8, which may have resulted from the rigid spacer –PhCOO– unit twisting the planar azobenzene fragment. Analogous results were achieved for spectra recorded in a solid state (λ
ab = 360, 347 and 366 nm). This suggests the star-type structure in products enables avoiding the aggregation for azobenzene units in solid state. The authors showed that obtained materials had good film-forming properties, high thermal stability and novel optical limiting properties that could be potentially applied as opto- and electroluminescent materials.
Miniewicz et al. reported on two types of novel nanoparticles based on a POSS surrounded by eight covalently attached units of azobenzene mesogens differing in the length of their aliphatic chains (
1,
2) (
Figure 32) [
105,
106]. The compounds were selectively obtained in β isomers using Karsted’s catalyst and obtained with 68.5% and 71% yields, respectively. The UV-vis spectra (in CHCl
3) of the stable forms of
1 and
2 exhibited features that were characteristic of parent azobenzene molecules in the wavelength range 250–550 nm, as well as a strong π–π * band at λ
ab ca. 338 nm (for
1) and 335 nm (for
2) and much weaker broad bands with maxima at λ
ab ca. 445 nm (for
1) and 440 nm (for
2), respectively. These results suggest that, in equilibrium and at room temperature, the azobenzene units are predominantly in their trans configuration. Different absorption spectra were recorded after homogeneously being irradiated by intense broad UV light (360 nm) for 2 min. The disappearance of a band at 335 nm and the increase of intensity of the 440 nm band was observed. This transformation is characteristic of the reversible trans–cis isomerization process in azobenzene units. In further studies, the authors proved that the addition of the POSS compound with mesogenic branches containing azo dye undergo photoisomerization enhanced by UV light.
The Tsukruk group presented a similar type of nanoparticles, but have reported two different approaches to fabricating azo-functionalized POSS compounds via Karstedt’s mediated hydrosilylation. These were carried out in 48 and 72 h, with high 83–94% yields (
Figure 32) [
107,
108]. Firstly, they obtained branched azo–POSS structures based on azo dyes possessing flexible spacers of different chemical natures and lengths between the POSS core and the azobenzene units (
3–
5). Secondly, they synthesized the final material by isolating the azobenzene moieties with a constant short spacer between the azo dyes and POSS core (
6,
7). The product
7 was obtained as mixture of α and β isomer, and the distribution of these isomers at molecular level was statistical. The higher thermal stability of all systems was proved (for products
3 and
5 the melting point temperatures were detected). The photophysical properties of all products were determined and compared with azo dyes (in solution and solid state). The UV-vis spectra of prepared azobenzene-modified POSS compounds showed two characteristic bands: λ
ab at 334–347 nm, which was related to the π–π* transition of the trans-azobenzene form, and λ
ab at 427–443 nm, originating from a typical n–π* transition. It was also observed that λ
ab peak positions (π–π*) of the initial substrates were identical to their respective POSS products. However, the absorption bands corresponding to π–π* transition in the trans isomers of all products were broader. This may have resulted from partial chromophore–chromophore aggregation of the azobenzene moieties. The UV-vis absorption spectra of the prepared films closely resembled solution spectra. Moreover, the products with the isolated azobenzene fragments (
3–
5) revealed a significant decrease in the trans–cis photoisomerization rate in solution as compared to other systems (
6,
7). In the solid state, the photoisomerization rates of all products were almost the same. These results indicate that the presence of isolation groups effectively prevents aggregation of substituents attached to the POSS core in films.
A different part of POSS-based frameworks exhibiting interesting photophysical properties are TM complexes, mainly Ir and Pt, that are anchored by functional groups playing the role of a ligand to the silsesquioxane core. The phosphorescent TM complexes used as emitters in OLEDs have attracted much attention because they can utilize singlet as well as triplet excitons through spin-orbital coupling with metal ions [
110]. Platinum, and particularly iridium, are the most applicable metals, as they possess high quantum efficiency, brightness, a variety of colors and short excited-state lifetimes, and may be used as an alternative to classical OLEDs or in electrochemical devices, etc. On the other hand, silsesquioxane-based systems using conjugated arenes as substituents exhibit interesting photophysical and also thermal properties. Their incorporation into organic-light-emitting materials prevents aggregation of the organic moieties that lead to emission quenching and/or the lowering of quantum efficiency. The POSS core is suggested to act as a hybrid hole-transport material when compared to traditional hole-transport materials. Additionally, rigid Si–O–Si core improves the thermal stability of the resulting material. These silsesquioxane features have encouraged researchers to consider its application as a scaffold for organic substituents that play role of a ligand to anchor the metal ion and constitute a POSS-based complex. In this part, the proper choice of a coordination sphere for a desired complex (i.e., the presence of respective functional groups at the Si–O–Si core acting as donating groups in ligands, as well as modulating metal ancillary ligands), has evolved over the recent years, and interesting reports on TM complexes anchored onto silsesquioxane cores appeared. Their application in the fabrication of light-emitting devices has been observed previously [
110]. The literature review suggests a hydrosilylation reaction (mediated by Karstedt’s complex) as an easy methodology, since the bridge or spacer between the POSS core and the potential ligand play a minor role [
111]. This is why the aliphatic connection resulting from hydrosilylation of alkene moiety by Si–H groupis the predominant synthetic procedure that enables formation of POSS-based compounds and their use as potential ligands for TM-complex synthesis.
Jabbour et al. pioneered this issue and in 2009 reported on the efficient formation of a series of not only iridium-based, but also platinum-based complexes (
Figure 33) [
112,
113]. The synthetic procedure involved a few stages, which is a general approach for this kind of research. First, the proper design of an organic ligand structure is required. Usually the
N- and
O-based ligands are the most favorable, (e.g., based on acetylacetone and pyridine derivatives (2-phenylpyridine, 3-phenylisoquinoline), etc.).
Formation of a respective alkenyl-functionalized linker, placed on the ligand moiety, was used for combining this molecule with the POSS core. Next, the selection and introduction of a proper ancillary ligand to the TM coordination sphere is important. The final part of the research concerns performance of a hydrosilylation reaction to anchor the complex with its functionality onto the POSS core. In these examples, the mono- and octa-substituted POSS was used as a reagent, with the –OMe2Si-H moiety as reactive group. In the case of octa-functional systems, one Ir-based complex fragment and one and two Pt-based complex fragments were attached to the silsesquioxane. The rest of the Si–H moieties were reacted with allyl-carbazole. The authors obtained these systems with diverse yields, from very high (92%), to moderate (54–63%) and rather low (~20%), and their photophysical properties were analyzed (CHCl3, solid film). In general, the absorption properties of these molecules derive from the absorption of metal-to-ligand charge transfers (MLCT) at λab ca. 340–350 nm. The carbazole moieties, together with ligand-centered absorption units, result in shorter wavelengths. New emission peaks at λem = 465 and 610 nm (green and green-yellow) from the complex with POSS system were red-shifted, especially in solid state for a molecule with two complex moieties in a molecule due to the excimer/aggregate states. Nevertheless, these systems were used in the fabrication of electroluminescent devices that revealed quantum efficiency to be enhanced.
Yu et al. reported on the efficient formation of silsesquioxane-based phosphorescent materials consisting of an emissive Ir(III) complex and carbazole moieties (
Figure 34) [
114,
115].
These systems were based on 3-(pyridin-2-yl)coumarinato N,C4) as ancillary ligand and varied the additional ligand that was used to bind the POSS core with allyl group (i.e., 8-hydroxyquinolinolate and acetylacetone derivatives). The authors obtained compounds with one and two Ir moieties anchored to one POSS molecule with 12.5%, 23% and 55% yields, respectively. Their enhanced thermal stability was proven. The photoluminescence spectra of the POSS systems (in DCM and solid state) were obtained. Again, the absorption spectra were derived from carbazole units (λab = 360 nm) as well as spin-allowed and spin-forbidden metal-to-ligand charge transfers (MLCT) (λab = 400 nm). The emissions of these systems were similar and revealed the presence a weak green emission at λem = 535 nm, along with a dominant red emission at λem = 635 nm for 8-hydroxyquinolinolate, a strong green emission at λem = 530 nm and a dominant green emission λem = 567 nm. The quenching of the emission within the concentration rise was diminished due to the separation of the chromophores onto the POSS core. These compounds were also successfully applied to the fabrication of electroluminescence devices exhibiting high brightness and higher external quantum efficiencies.
In 2016, You and Su reported on a synthesis of POSS-based systems containing one, two and three moieties of Ir(III) complex that were also based on 3-(pyridin-2-yl)coumarinato N,C
4) as an ancillary ligand, and allyl acetylacetone modified with n-Bu-carbazole molecule (
Figure 35) [
116].
These systems were obtained using the abovementioned general synthetic approach via hydrosilylation, with 26.4%, 14.7% and 6.4% yields, respectively, that may suggest the optimization of reaction conditions for better yields. Obtained compounds were verified in terms of their thermal resistance, which was proven. The absorption and emission spectra were, in general, analogous to the previously reported ones (i.e., absorption derived from the carbazole unit and spin-allowed and -forbidden metal-to-ligand charge-transfer (MLCT) transitions). The emission (λex = 430 nm) spectra resembled each other and exhibited mainly green emission at λem = 530 nm and minor green-yellow at λem = 570 nm. The results also present a reduction of quenching from Ir-complex moiety interactions due to a bulky POSS core. The electroluminescent device fabricated using the abovementioned compounds revealed an enhanced quantum efficiency that proved its application utility.
3.6. Other Catalytic Reactions leading to the Formation of POSS-Based Systems of Interesting Photophysical Properties
A group of catalytic processes known as “click reactions” has gained a profound interest in (bio-) organic chemistry and also materials chemistry, a term that was successfully introduced into literature by Sharpless in 2001 [
117]. In general, these are perceived as mainly one-pot, modular processes, with a wide tolerance to other functional groups that generate minimal and inoffensive byproducts. They are supposed to be stereospecific and are characterized by a high thermodynamic driving force to favor a single product formation with a high yield. Additionally, the process would preferably have mild and simple reaction conditions, along with commercially available reagents to obtain products easily isolated, mainly via crystallization or distillation. Many of these criteria are subjective and depend on a certain type of a process, but several reactions fit the concept better than others, and have been applied in the chemistry of silsesquioxanes [
118]:
[3+2] cycloadditions, such as Huisgen 1,3-dipolar cycloaddition and particularly Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC)
thiol-ene coupling reaction (TEC)
strain-promoted azide-alkyne cycloaddition (SPAAC)
Diels–Alder reaction
thiol-Michael addition
oxime ligation
One of these processes, applied in the synthesis of silsesquioxanes, is the 1,3-dipolar Huisgen cycloaddition of azides to alkynes, to give form 1,2,3-triazoles (CuAAC) [
119]. This is a catalytic reaction, mediated mostly by copper(I) salts (but also ruthenium or silver) and reducing agents (e.g., sodium ascorbate) [
118,
120].
A combination of the 1,3-dipolar Huisgen process followed by palladium Sonogashira coupling was used in the synthesis of oligophenyl-ethynylenes with heptaisobutylsilsesquioxane used as pendant groups by Ervithayasuporn et al. (
Figure 36) [
121].
These two consecutive processes turned out to be efficient methods (84–99% yields) for the synthesis of interesting, highly π-conjugated organic systems, with silsesquioxane units at two independent molecule fragment, of high thermal stability (Td5% up to 369 °C and exhibited Tg in a range of 80–197 °C for R’ = Phe oligo-pPE POSS). Their photophysical properties were analyzed in solution (THF) and also in a solid state. While the maxima absorption in solution equaled λab = 373–403 nm, the emission spectra (excitation at 350 nm) shift from deep blue to sky blue, and red-shifted (λem = 409–443 nm) as the amount of aromatic fragments of end-capping molecules increased, as well as with the extended conjugation of in the main chain. The emission spectra of photoluminescence measured in the solid state were strongly shifted towards longer wavelengths (λem = 470–533), which resulted in interchain orbital interactions referred to aggregation bands. Nevertheless, the extended π-conjugation of oligo-pPEs POSS maintained relatively high photoluminescence quantum efficiencies when compared to mono-pPEs POSS. The authors reported on the preliminary and successful use of mono- and oligo-pPEs POSS (R’ = Py) as active dopants in the fabrication of dye-doped OLED devices.
An interesting example of octazido-functional POSS was reported by Xu et al. with two types of functionalities of different stoichiometry obtained via a 1,3-dipolar Huisgen reaction (
Figure 37) [
122]. The authors used octa(azidomethylene)spherosilicate obtained as an efficient azido-bering reagent [
123]. The synthetic procedure was based on a different kind of copper complex, along with the reducing agent (i.e., [Cu(PPh
3)
3Br] and KF) that resulted in very high yields of products bearing two types of functionalities B and Y: W
71, W
62 and W
53, which abbreviations derive from the stoichiometry of the acetylenes applied.
The obtained compounds were thermally more stable (Tg in a range of 192–273 °C) than their organic counterparts (B and Y). Their photophysical properties were analyzed in comparison to the acetylenes (B and Y) as reference molecules in solution and in a solid state. The absorption and emission spectra (shape, intensity and also λem) of the acetylenes were dependent on the solvent use, and were collected in THF and also THF/water (from a 0 to 90% water fraction). Additionally, the emission spectra of Y were strongly fixed on the amount of water content and were red-shifted ca. 60 nm. As a result, the emission spectra of W62 and W53 possessing two (W62) and three (W53) Y groups also exhibited the THF/water mixture emission dependence. The emission spectrum of W62 in THF presented two peaks at λem = 410 and 440 nm, but with the increasing content of water an enhancement of two new red-shifted peaks at ~530 and 570 nm were noted. As a result, the light band was fully covered from 400 to 700 nm, making the realization of a white light emission possible. Interestingly, this feature was preserved in the solid state and enabled its application as dopants in the synthesis of white-light-emitting materials.
López-Arbeloa, García-Moreno and Chiara et al. presented T
8 silsesquioxane-based dyes that were obtained by a copper(I)-catalyzed 1,3-dipolar cyclo-addition of octakis(3-azidopropyl)octasilsesquioxane with alkyne-substituted 4,4-di-fluoro-4-bora-3a,4a-diaza-
s-indacene (BDP) chromophore (
Figure 38) [
124].
The reaction conditions (also reagent stoichiometry) as well as the type of Cu catalyst affected the number of BDP substituents attached to the T8 core, and the process was carried out to obtain mono-BDP-substituted and fully (i.e., octa-BDP) substituted product. These two systems, along with their respective BDP analogues (as model compounds), were analyzed in terms of their photophysical properties. P1 and P8 exhibited similar absorption and fluorescence properties (P1: λab = 526 nm, λem = 540 nm; P8: λab = 523 nm, λem = 546 nm) but P8 revealed substantial fluorescence decay that was attributed to the strong π–π interaction of BDP units and formation of H-type or “sandwich” aggregates. Additionally, the fluorescence of P8 was strongly dependent on the type of solvent used (its poor solubility in non-polar media), and acetone was found to be the best. The compounds were analyzed for their lasing properties (in solution and in a solid state) in comparison to the BDP molecular analogues. Interestingly, compound P1 reached a laser efficiency of up to 56% (in the solid matrix) and up to 60% (in ethyl acetate solution) while maintaining a high photostability when compared to the parent BDP model (35%). This proves the high application potential of BDP–POSS-based systems in the formation of new photonic materials as alternative sources for optoelectronic devices.
An interesting work on the radical thiol–ene reaction (TEC) (mediated via DMPA = 2,2-Dimethoxy-2-phenylacetophenone) of propargyl thiol and octa(vinyl)POSS (OVS) functionalization, and its further ligation with rhodamine 6G, was presented by Mironenko et al. (
Figure 39.) [
125].
The authors reported on the photophysics of compound and related absorption (λab = 532 nm)—emission spectra (λem = 554 nm) that were pH dependent and increased in intensity with a decrease in pH value. Due to their problems with solubility, an increase of intensity was noted for higher-content ethanol in the water/ethanol solution. This compound was tested as a fluorescent chemosensor for a variety of metals, and the results indicated a positive response for Au3+ ion (50:50 water/ethanol, pH = 1 and molar concentration of rhodamine units of 10−5 M). Under these conditions, there was a strong fluorescence emission enhancement. As a result, POSS–Rh8 may be used as selective fluorescent chemosensor for the selective determination of Au(3+) ions in aqueous media.
One of the reactions applied in the chemistry of silsesquioxanes was free radical copolymerization leading to hybrid materials that are more amenable to solution techniques (e.g., spin-coating that results in large-area device fabrication). On the other hand, the phosphorescence of metal complexes, especially iridium or platinum, and their use in specific types of OLED devices was reported [
110]. However, these complexes may suffer from self-quenching in the solid due to interaction aggregation. This may be improved by anchoring them onto the Si–O–Si core (described in
Section 3.4). The idea of grafting of non-conjugated polymers (as a transparent matrix) with the metal complex covalently attached to the polymer backbone was studied. In this scenario, solubility would be additionally improved in comparison to the conjugated counterparts, thus facilitating application.
An interesting paper of Ling and Lin et al. reported on the efficient synthesis of poly(methylmethacrylate) via the AIBN (2,2′-Azobis(2-methylpropionitrile))-mediated radical copolymerization of two types of methylmethacrylates (bearing mono(propyl)heptaphenyl(silsesquioxne) = M3, carbazole unit = M1, with styryl-based iridium complex = M2) used in different molar ratio (POSS from 2 to 8 mol %) and resulting in copolymeric systems (
Figure 40) [
126]. Depending on the specific ratio of comonomers, the molecular weights varied (
Mn = 5100–19,400 g/mol, suggesting formation of other oligomeric systems), with PDI from 1.4 to 2.2.
The obtained materials were characterized in terms of their absorption emission properties, comparing the results with analyses for each monomer M2, M3 along with the copolymer without POSS (i.e., M2 + M3 and final ternary copolymers M1 + M2 + M3 with 2–8 mol % POSS contribution). The absorption spectra of copolymers in DCM showed peaks at λab = 237, 261, 294, 328 and 342 nm, which came from the carbazole segment and iridium complex moiety. The photoluminescence spectra (λex = 365 nm) in solution exhibited blue and green emission bands λem ca. 420 and 435 nm (derived from the host carbazole unit M1), with additional green emission bands at λem ca. 530 nm (derived from the guest iridium complex fragment M2). The band at 530 nm was enhanced with the increase of iridium complex content from 0.5 to 5.0 mol % while the bands around 420 and 435 nm decreased in intensity. There was energy transference suggested by the host (donor) carbazole moiety to the guest (acceptor) iridium complex moiety, which is also dependent on the content of Ir units along with the POSS moiety. However, the ternary copolymer with 5 mol % or iridium units and 2 mol % of POSS exhibited a significant enhancement of the green emission that may have indicated the separation of Ir complex units, preventing their aggregation, so that the concentration-quenching was restrained in the solution. In a solid state, the emission spectra presented only one little red-shifted green emission band (up to ~10 nm) but there was an interesting enhancement of the spectra intensity for the copolymer with 0.5 mol % of Ir complex content, meaning that the POSS unit prevented the aggregation of Ir units more effectively—a phenomenon that was more intensive with an increase in POSS content. The quantum efficiencies for these systems in a solid state were also higher, up to 52% (when compared to binary copolymers M2 + M3, Φ = 7–24%), which suggests that incorporation of POSS units enhances luminescence profoundly. Thermal resistance was also improved. These aspects make the obtained materials definite candidates for OLED devices.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
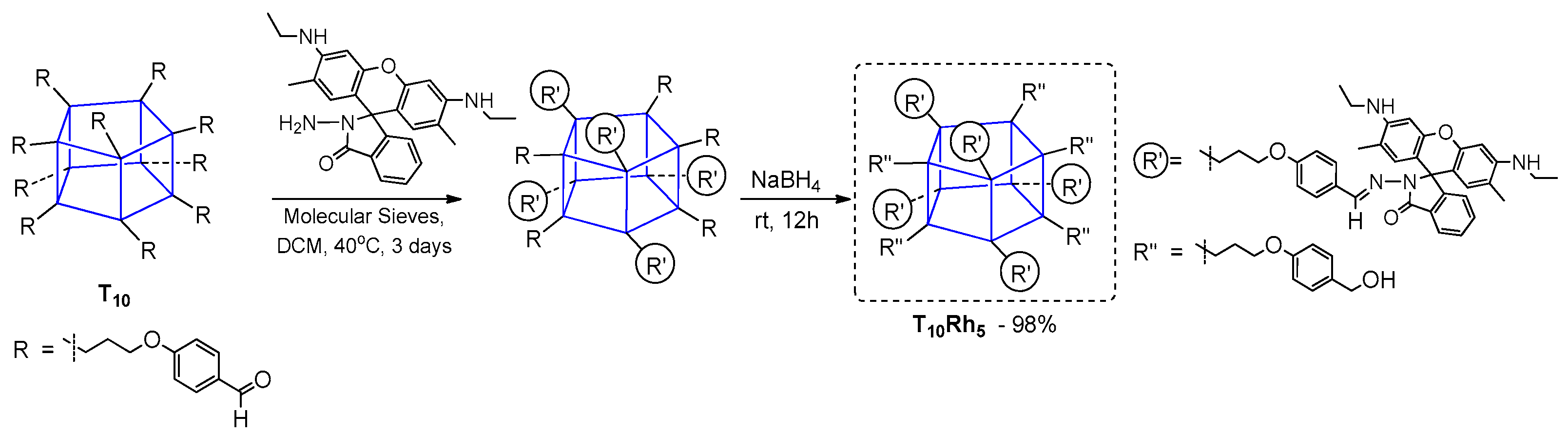{kind=link}
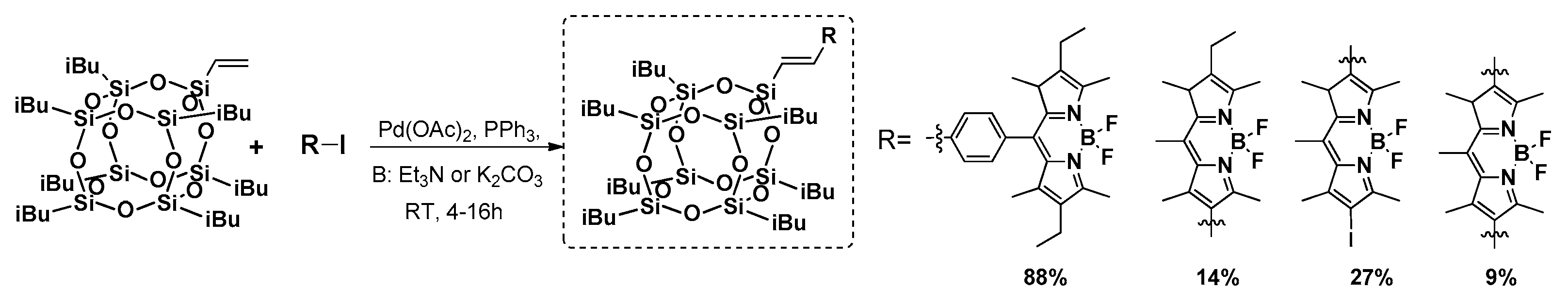{kind=link}
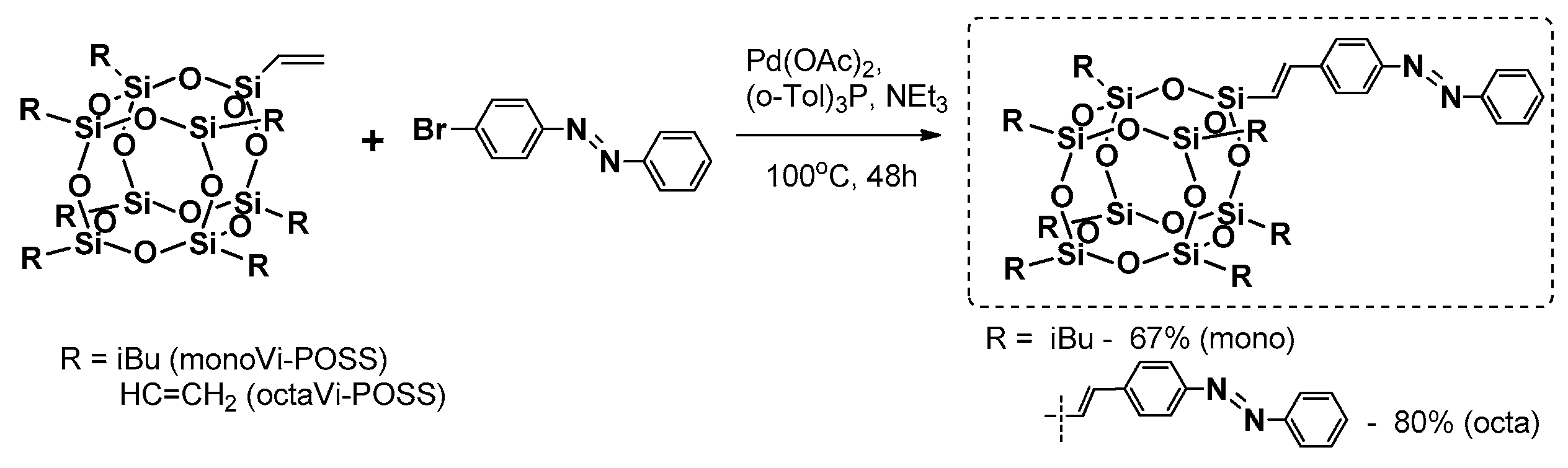{kind=link}
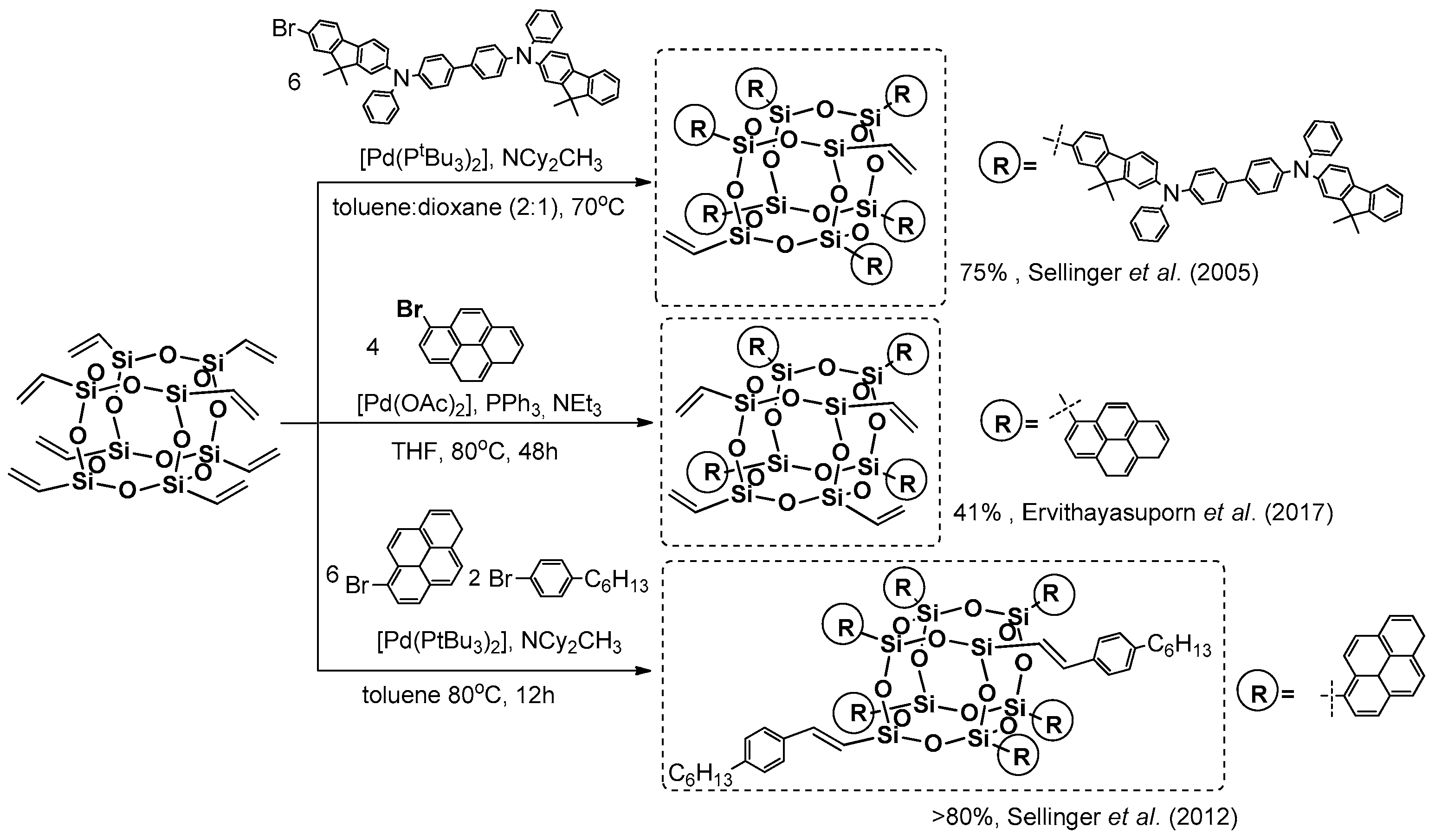{kind=link}
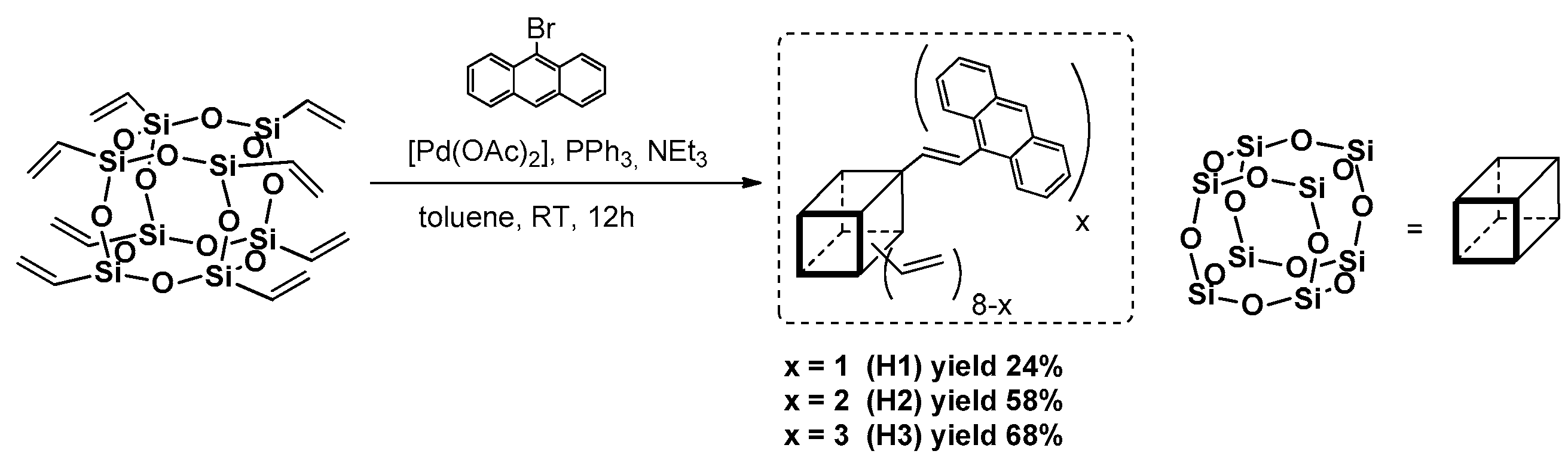{kind=link}
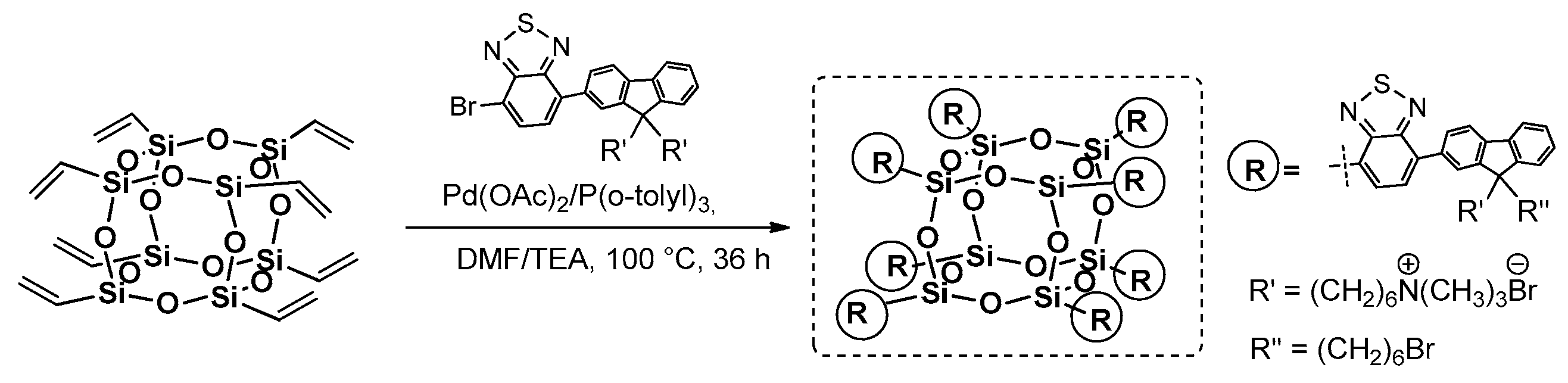{kind=link}
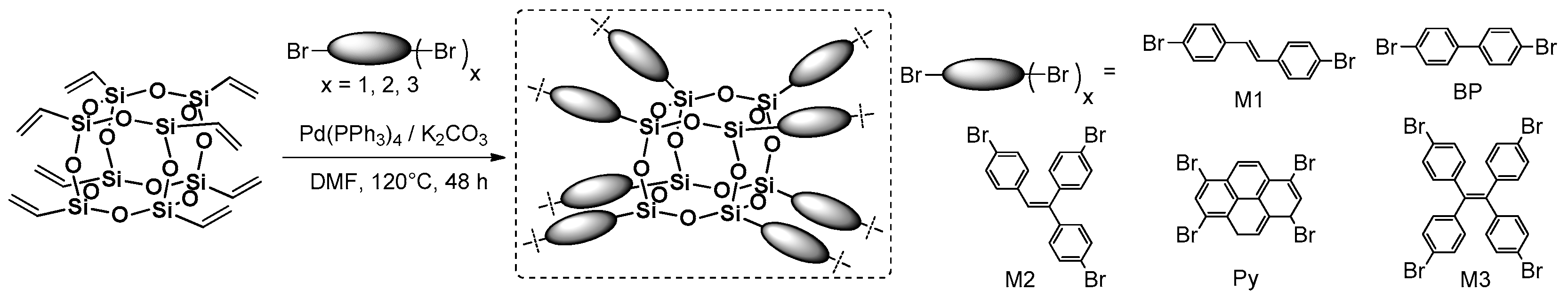{kind=link}
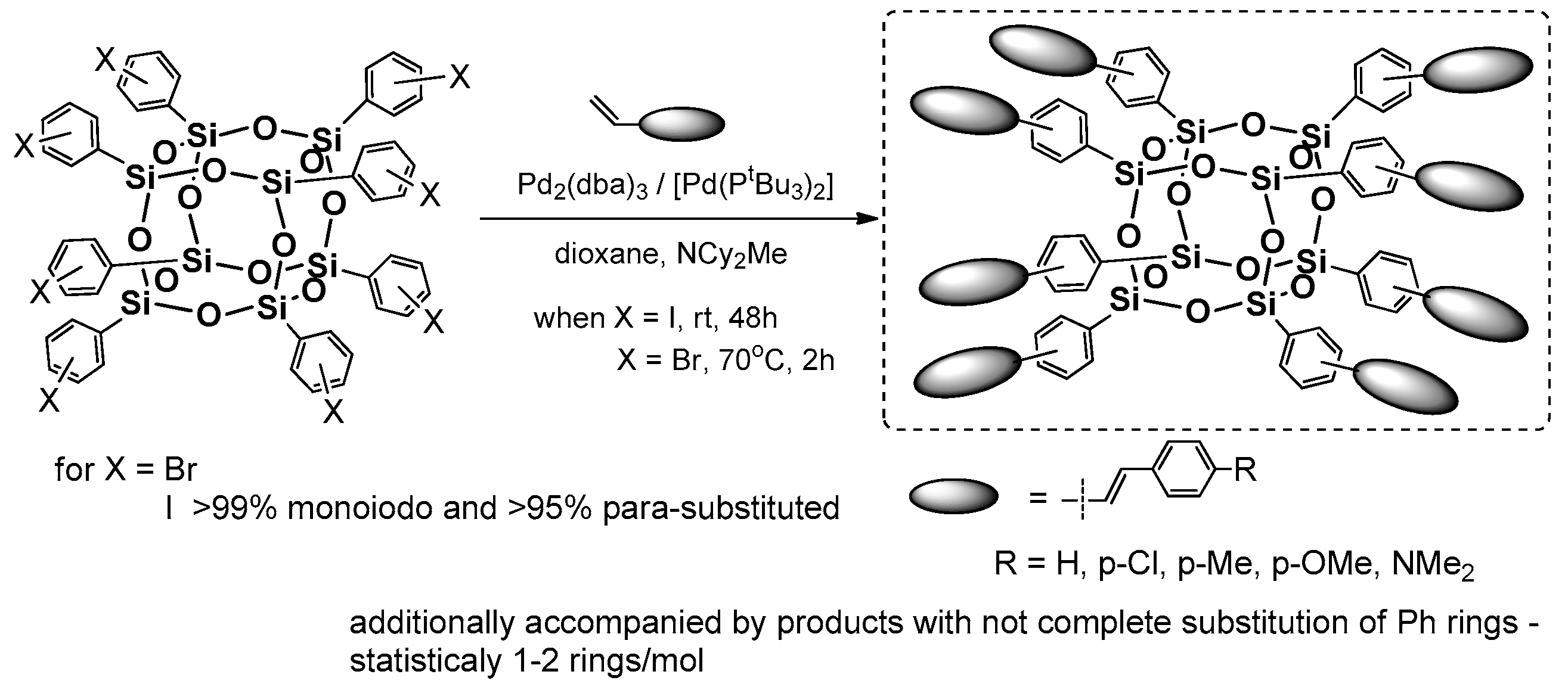{kind=link}
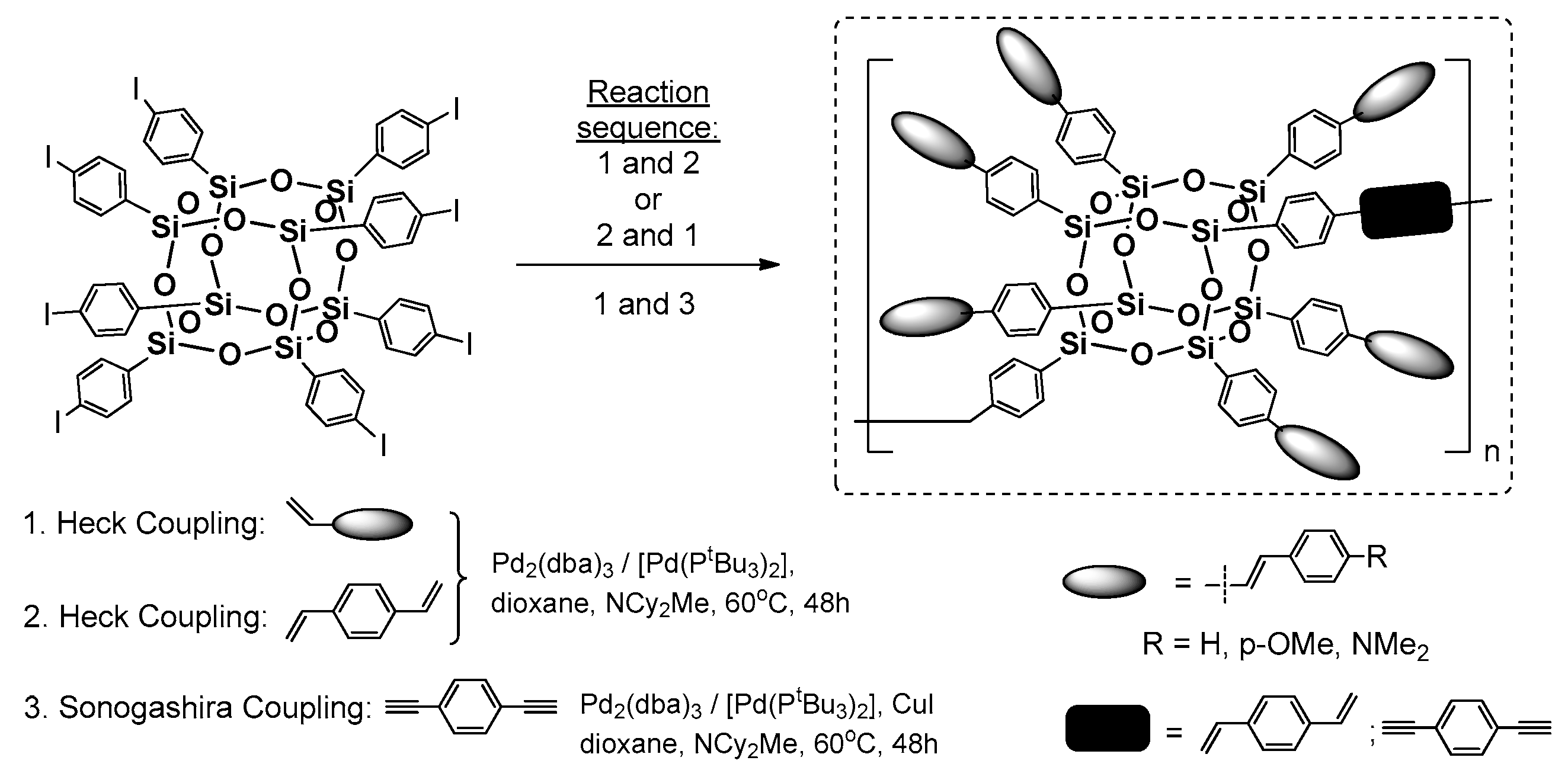{kind=link}
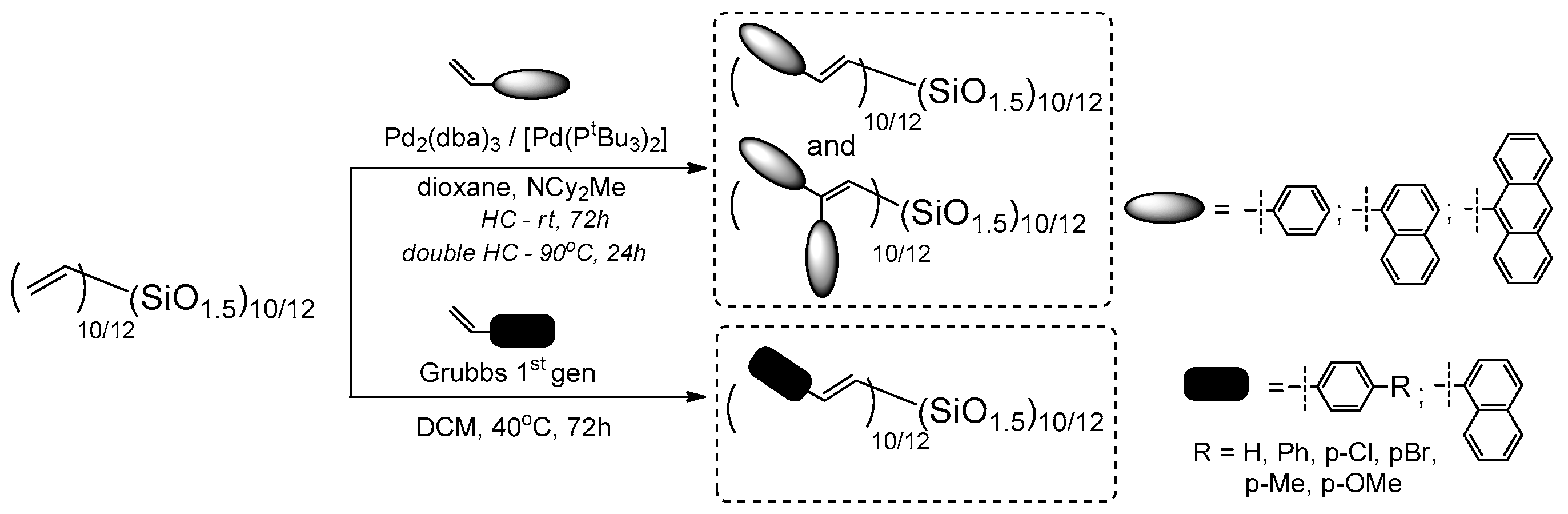{kind=link}
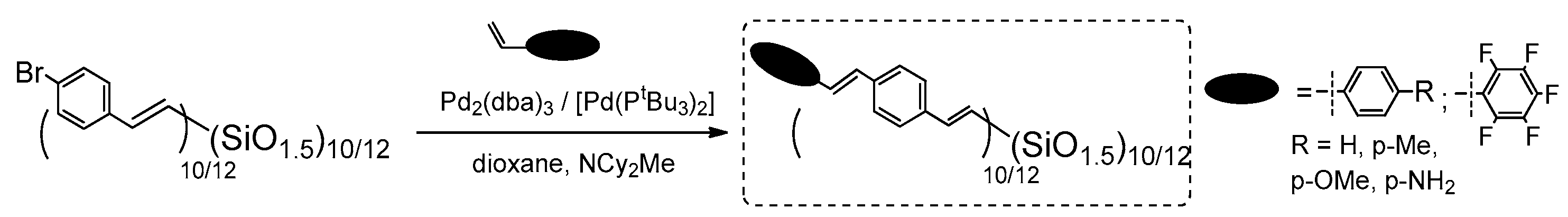{kind=link}
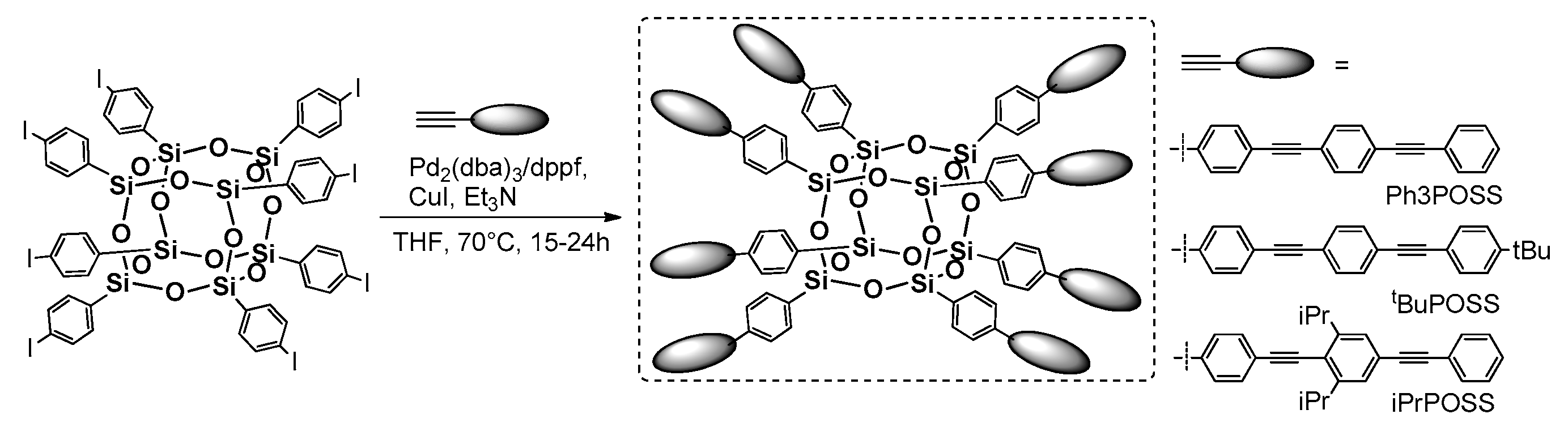{kind=link}
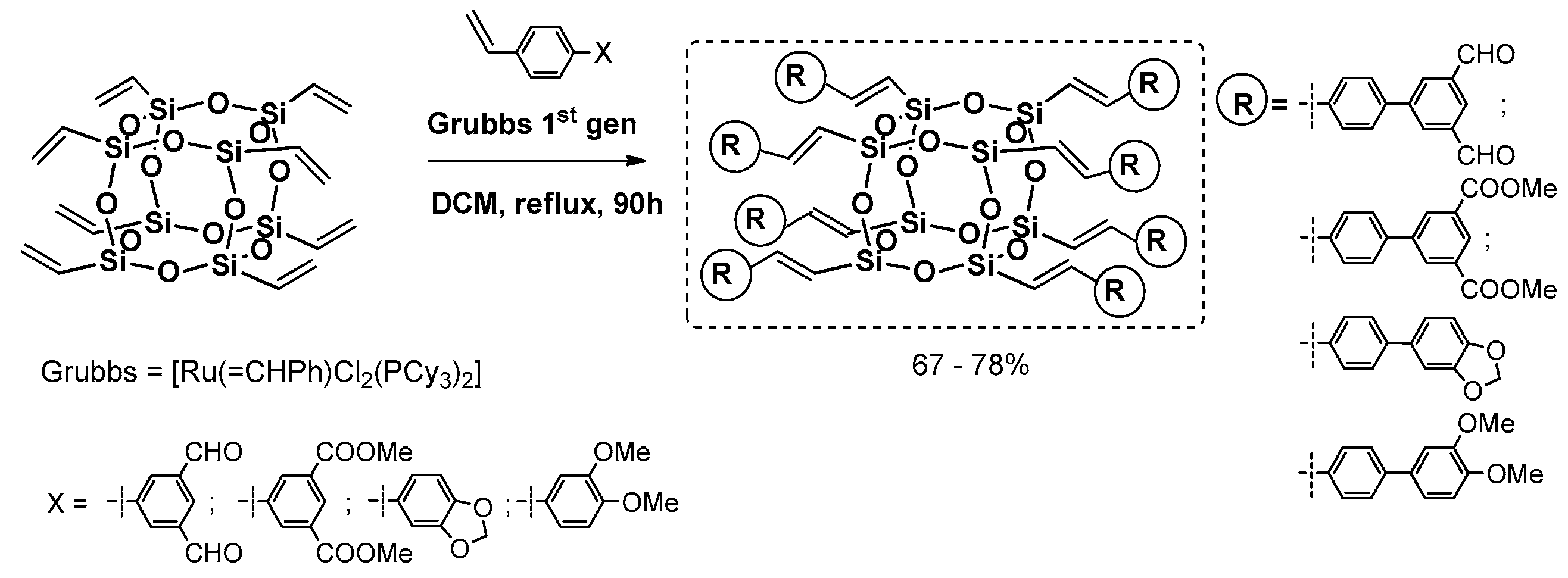{kind=link}
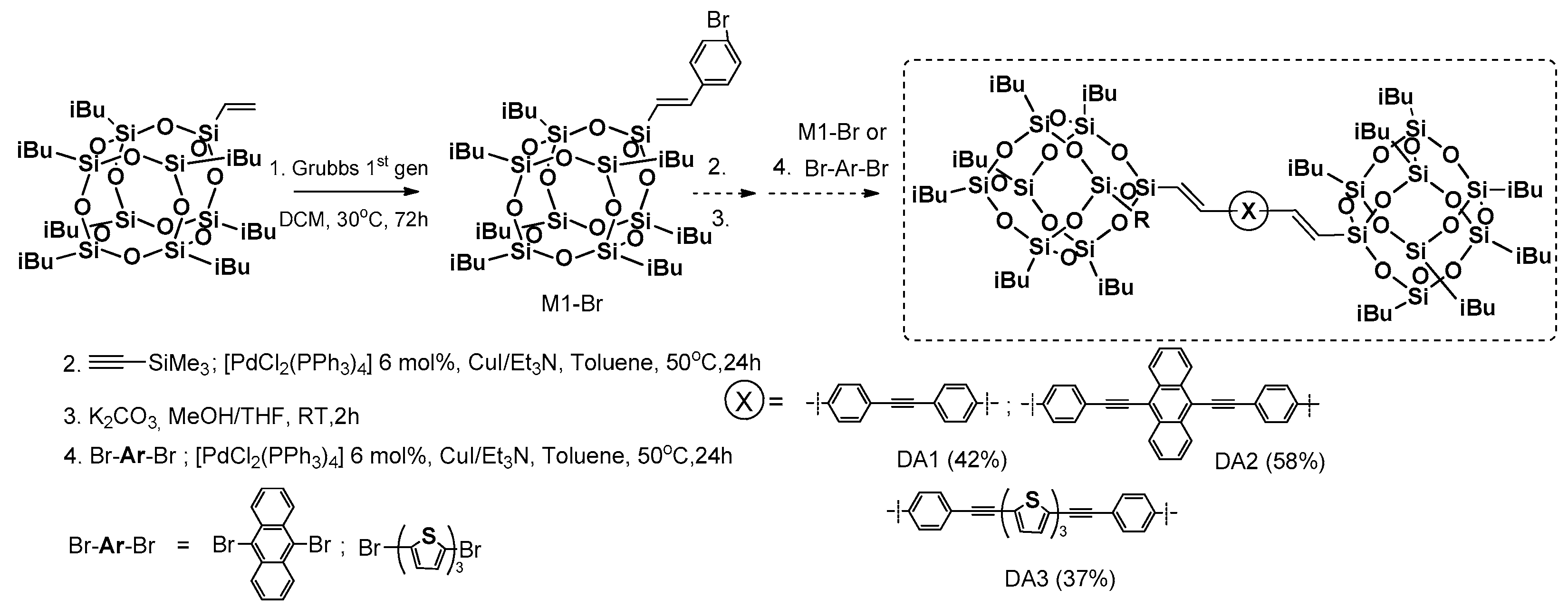{kind=link}
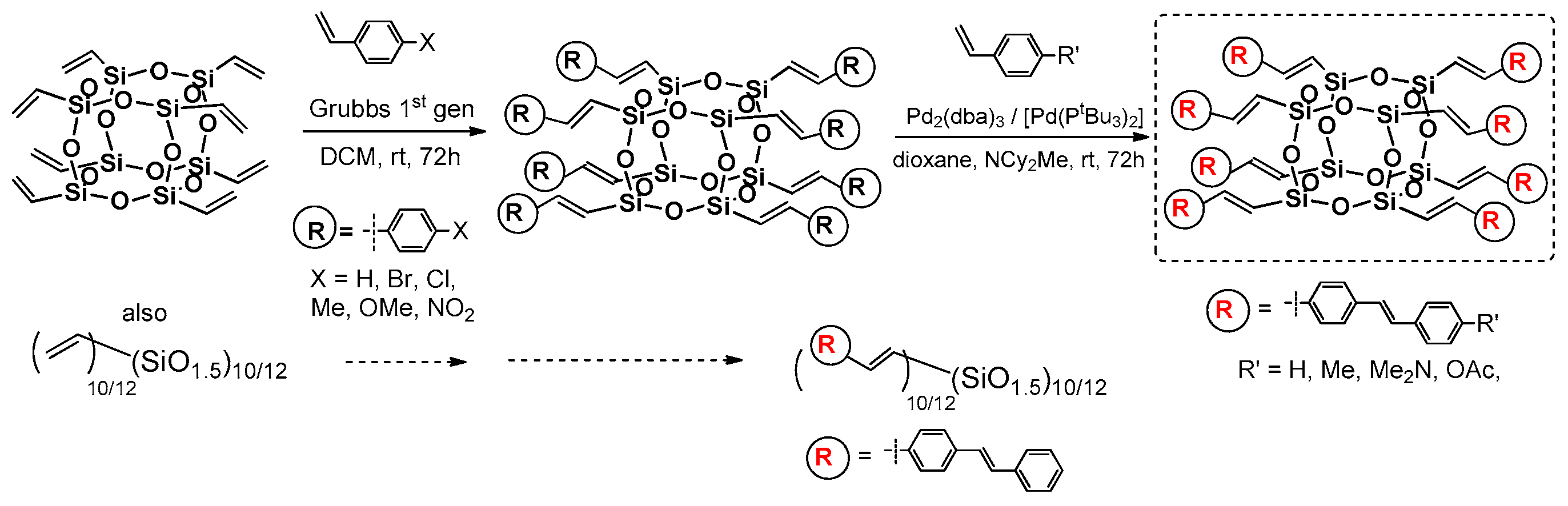{kind=link}
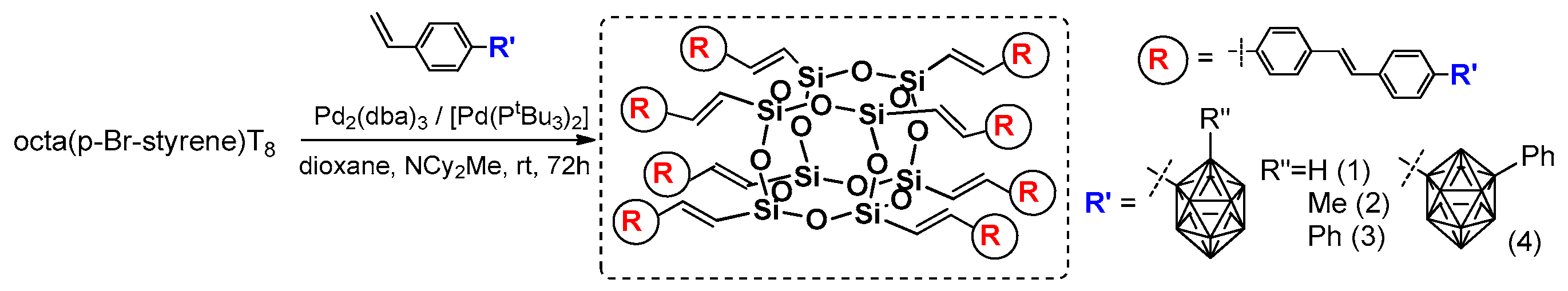{kind=link}
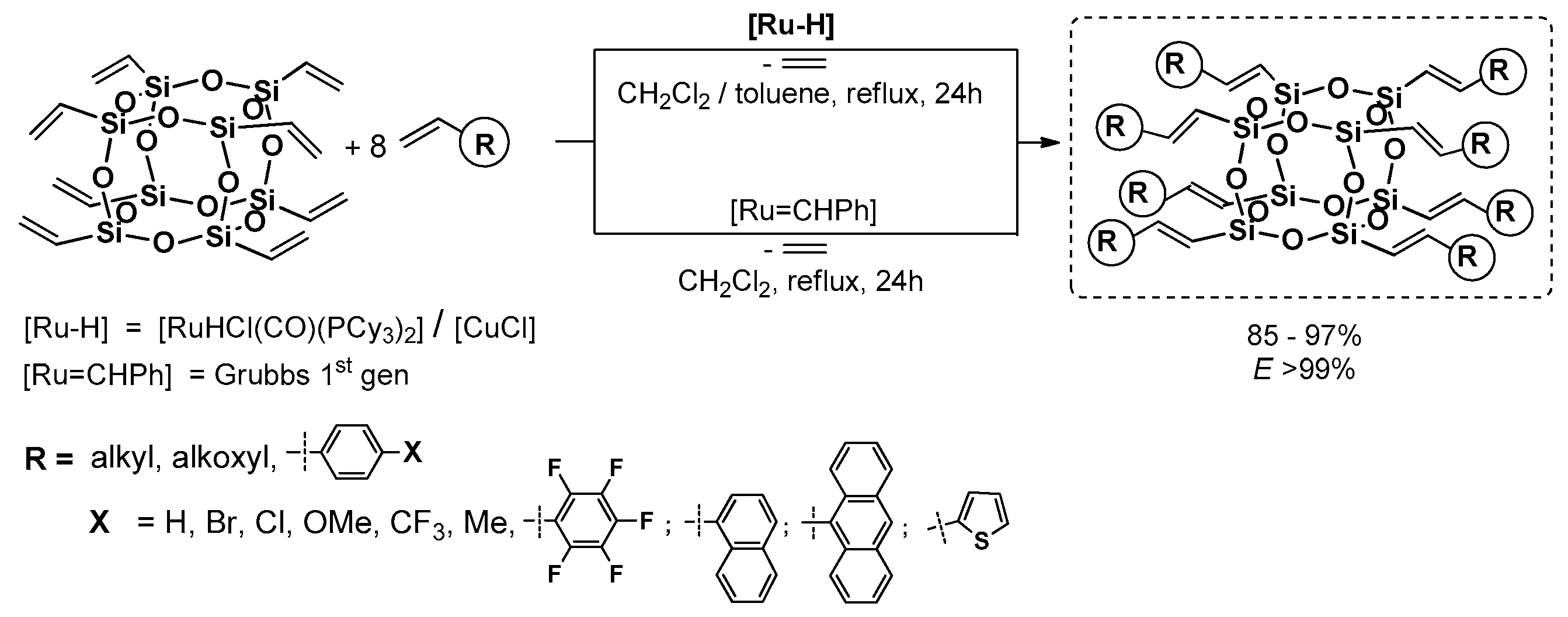{kind=link}
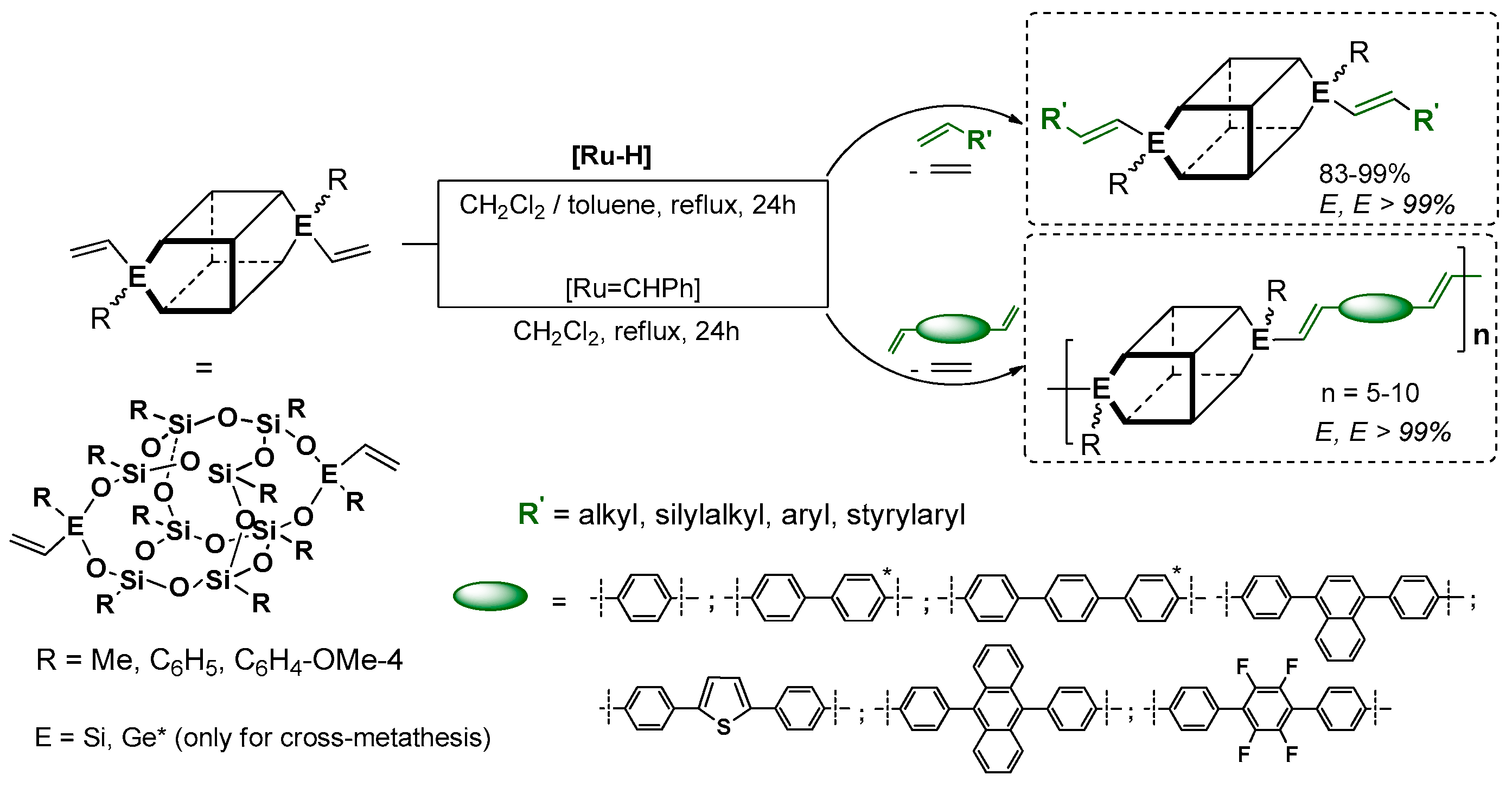{kind=link}
{kind=link}
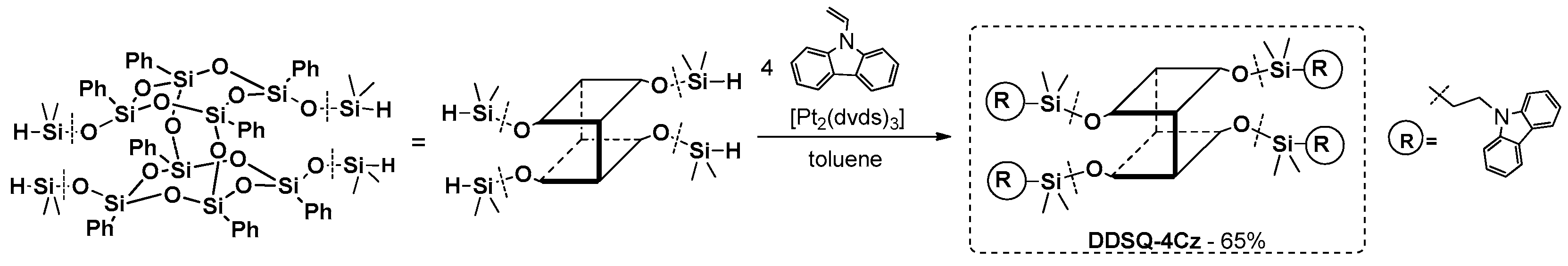{kind=link}
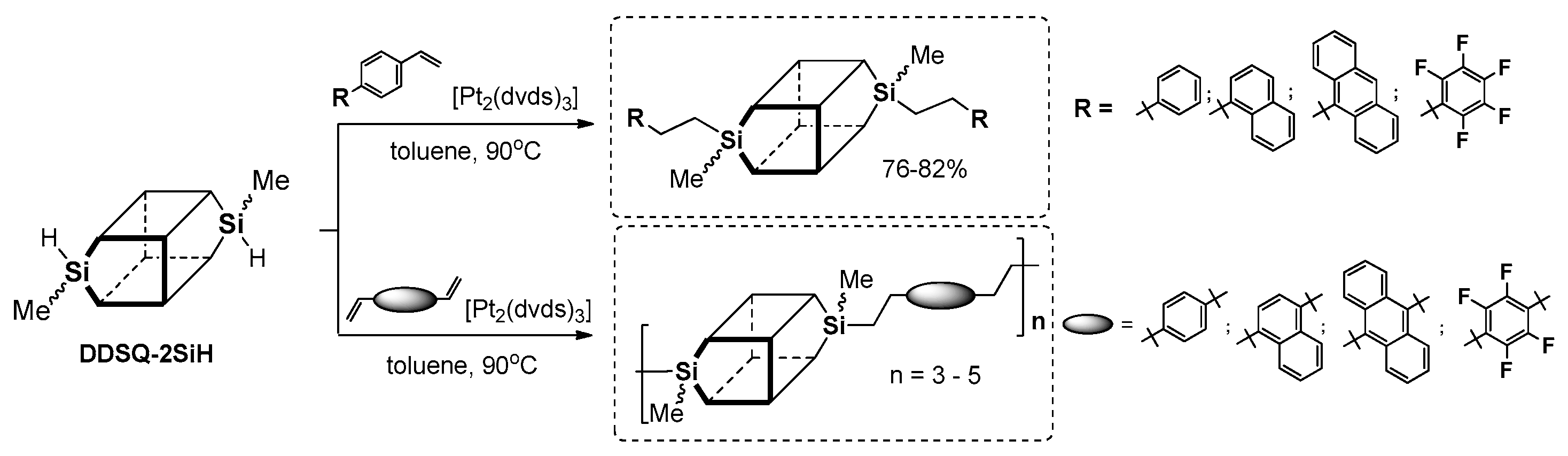{kind=link}
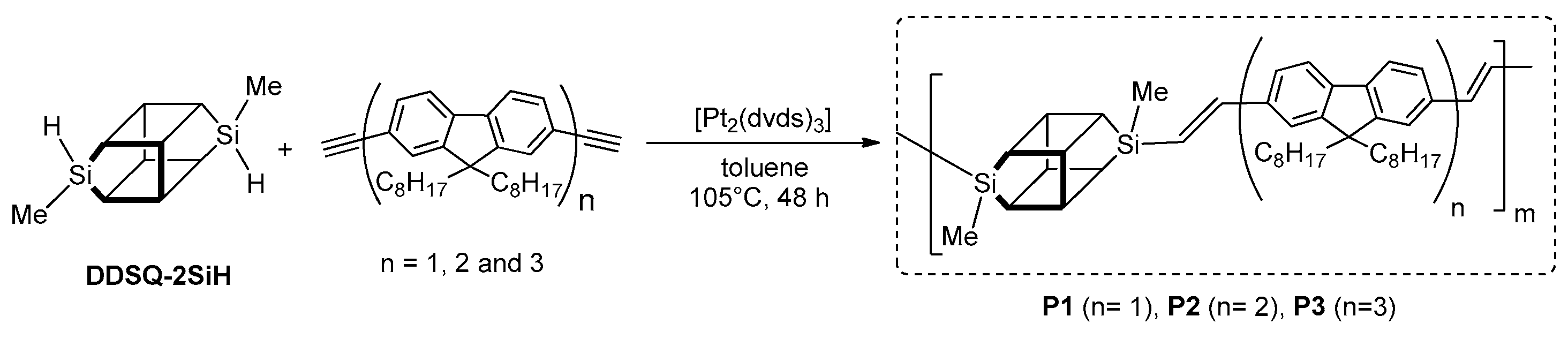{kind=link}
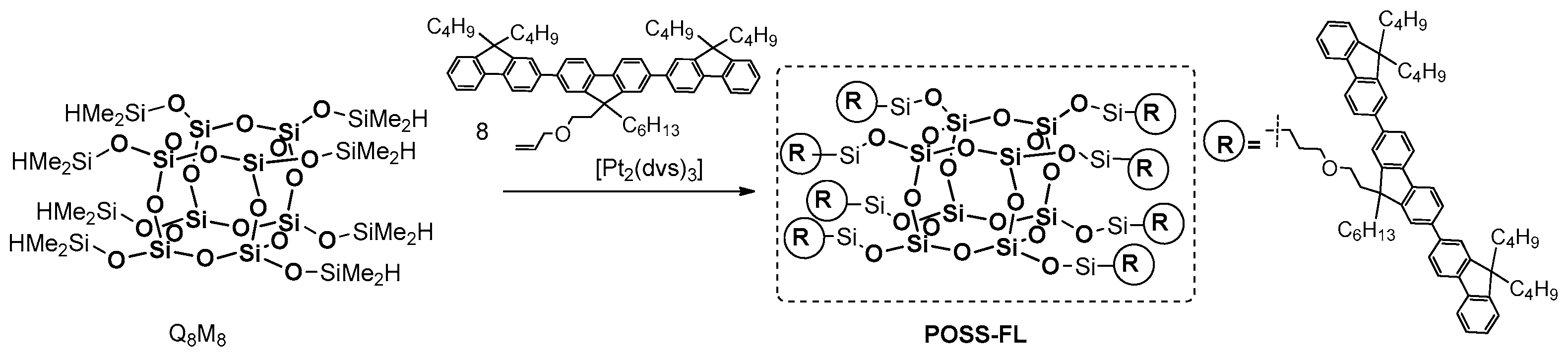{kind=link}
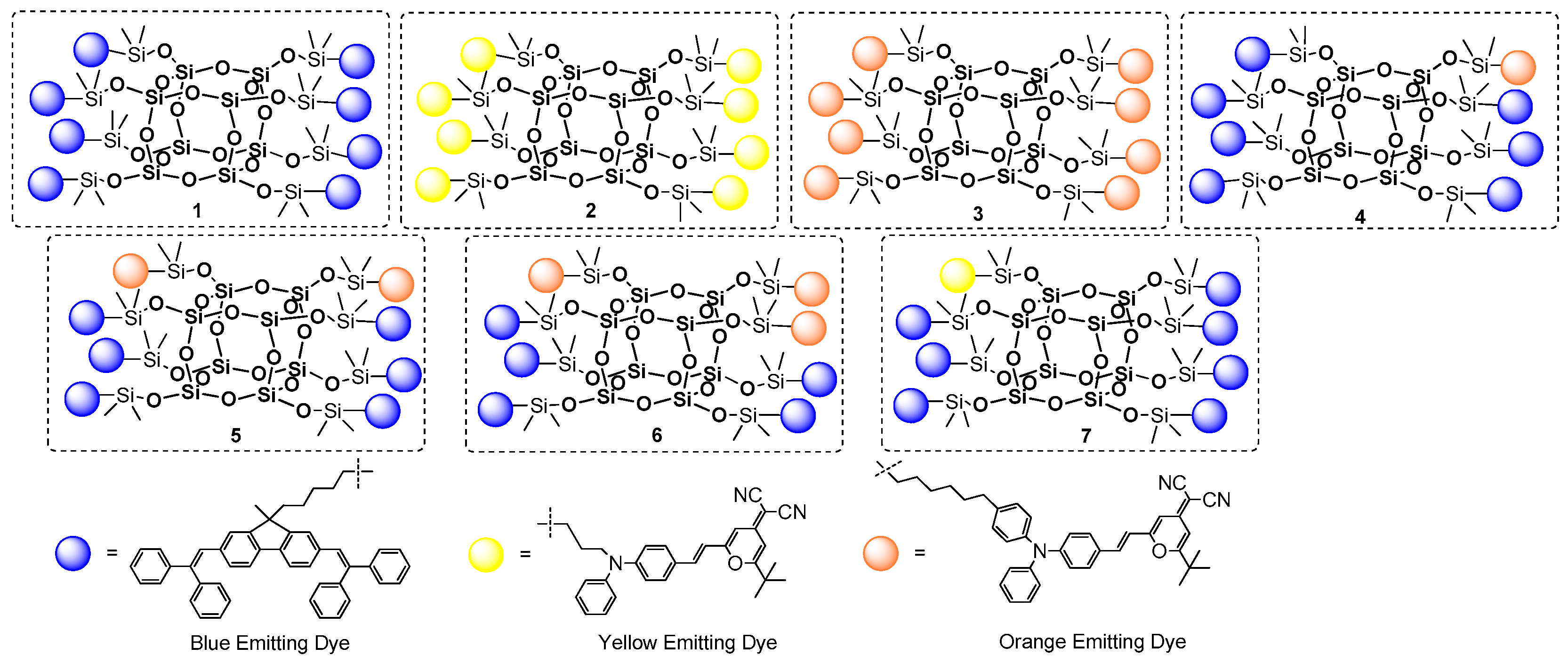{kind=link}
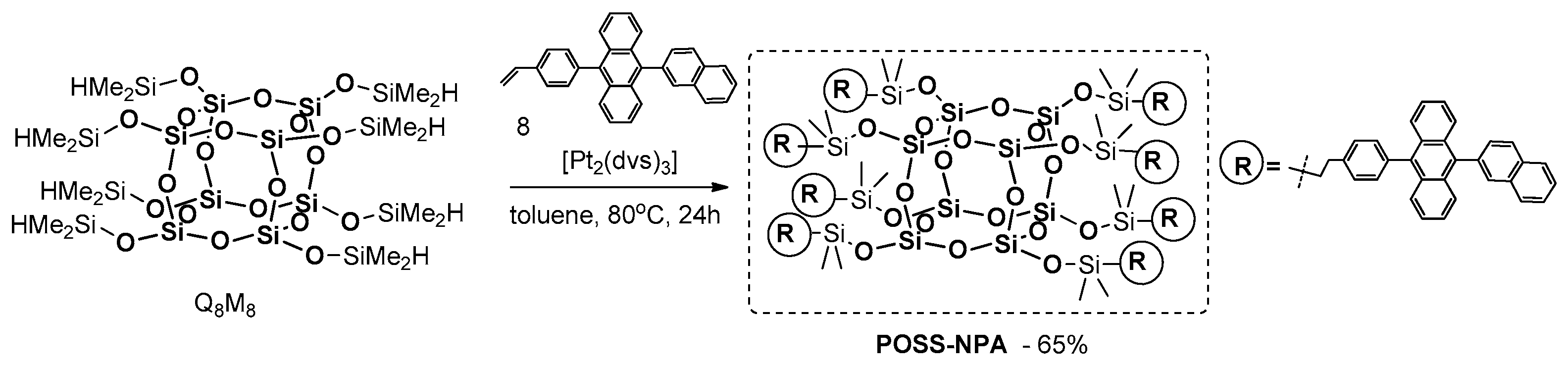{kind=link}
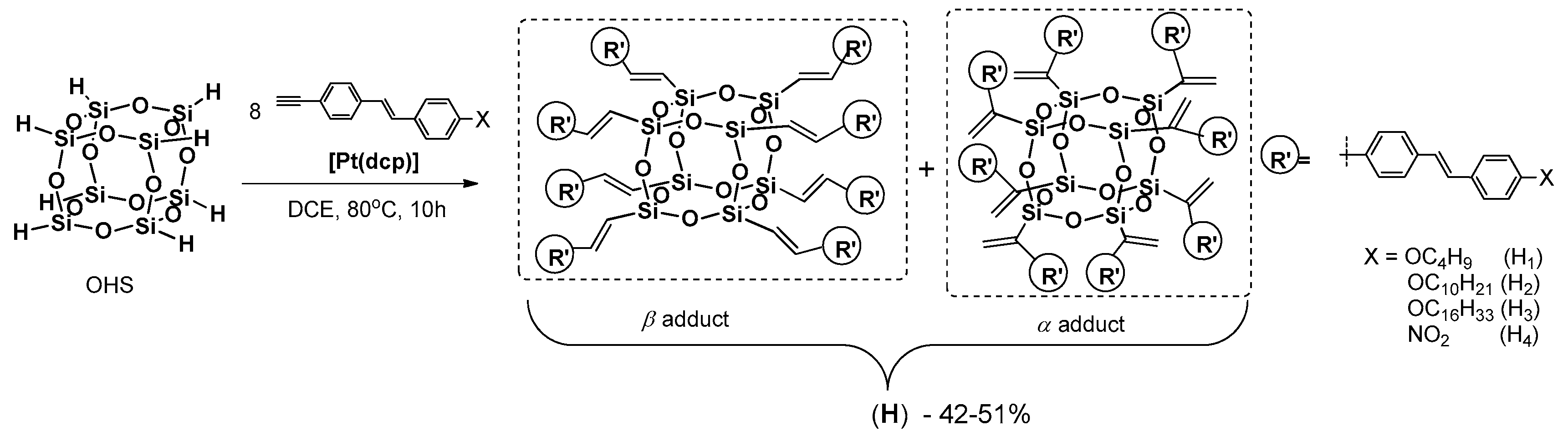{kind=link}
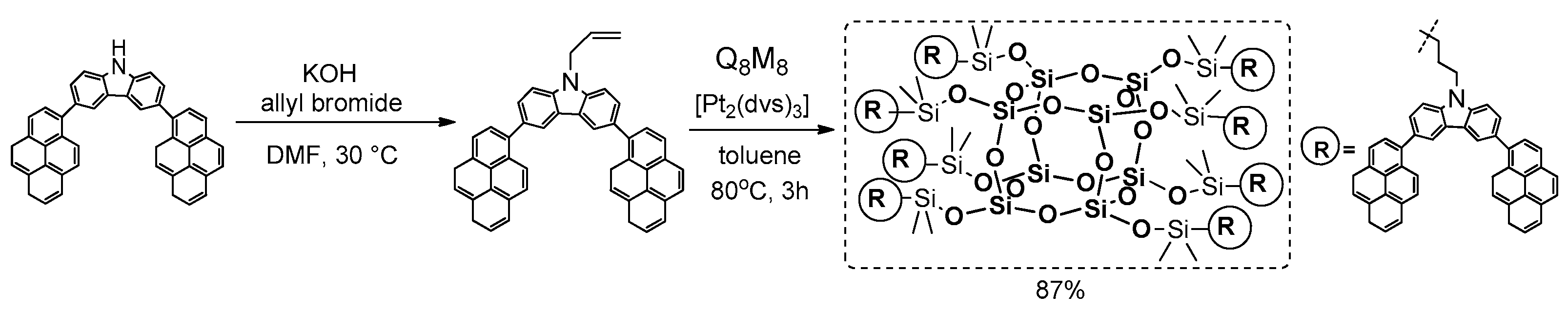{kind=link}
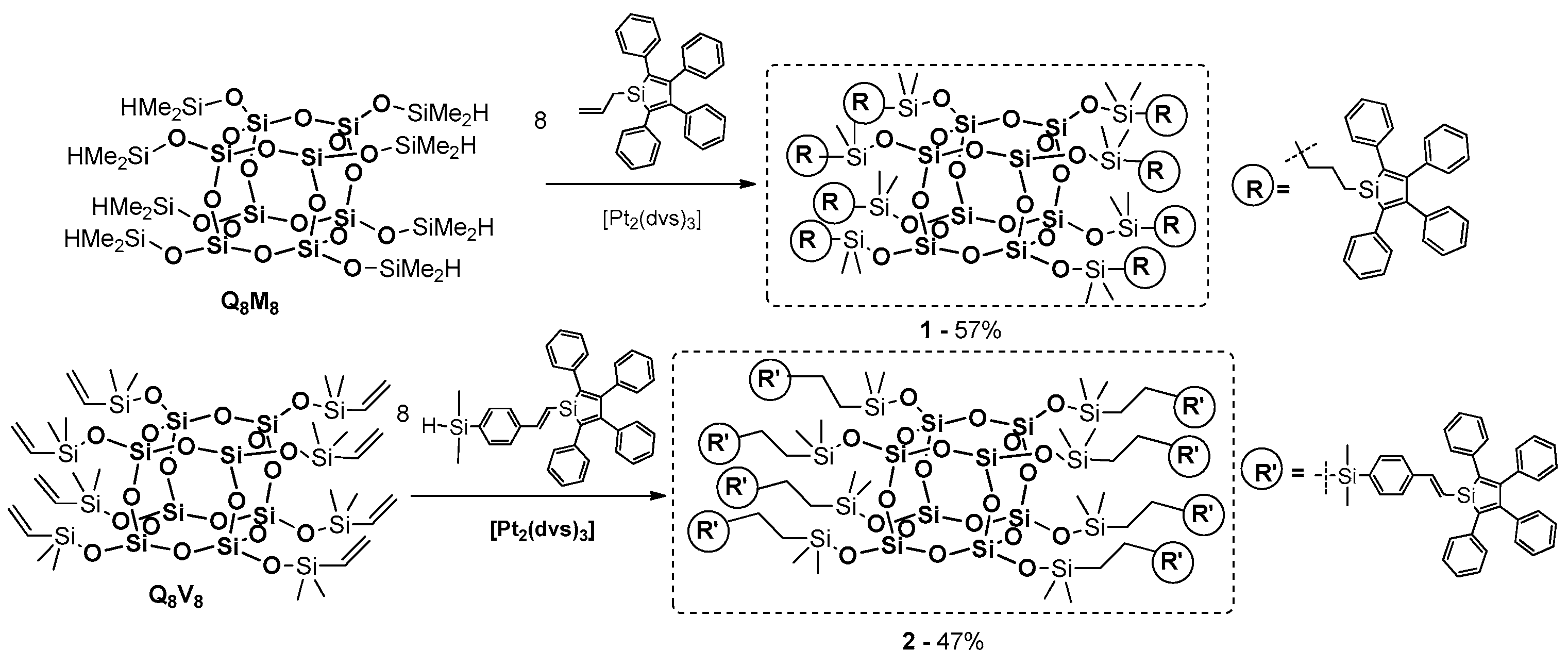{kind=link}
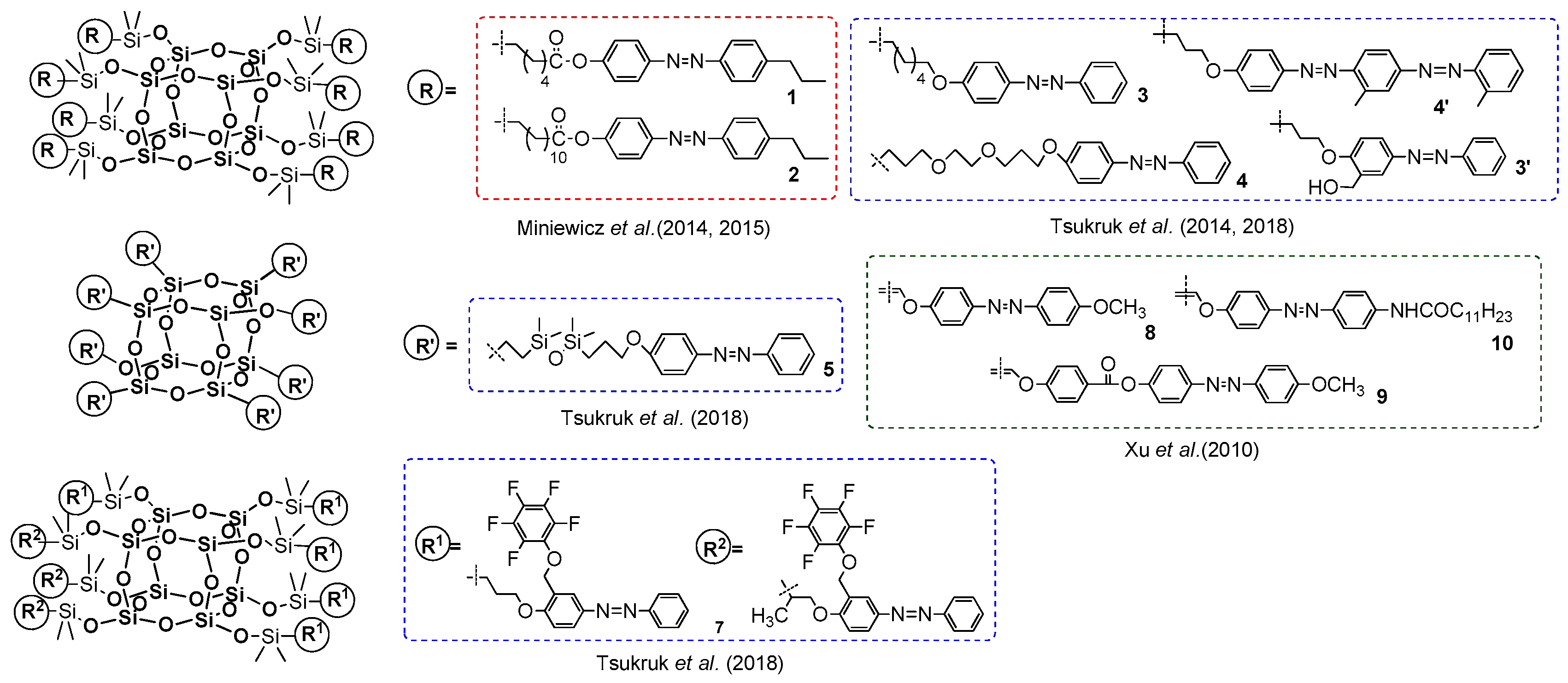{kind=link}
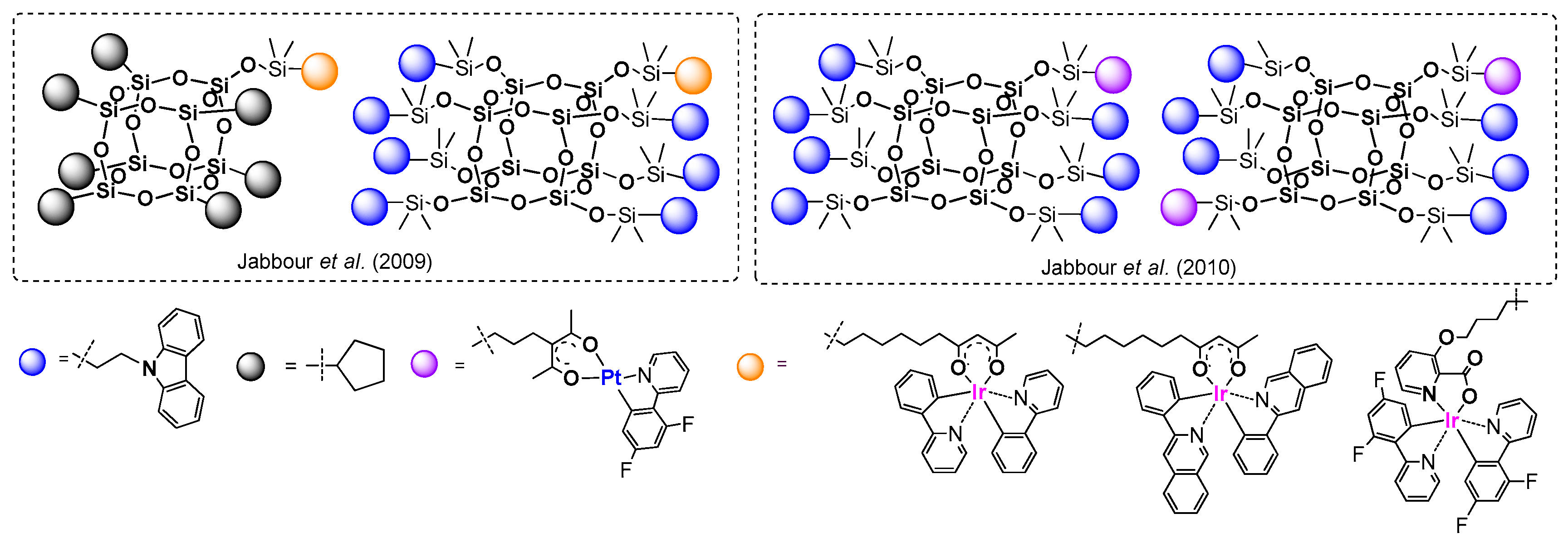{kind=link}
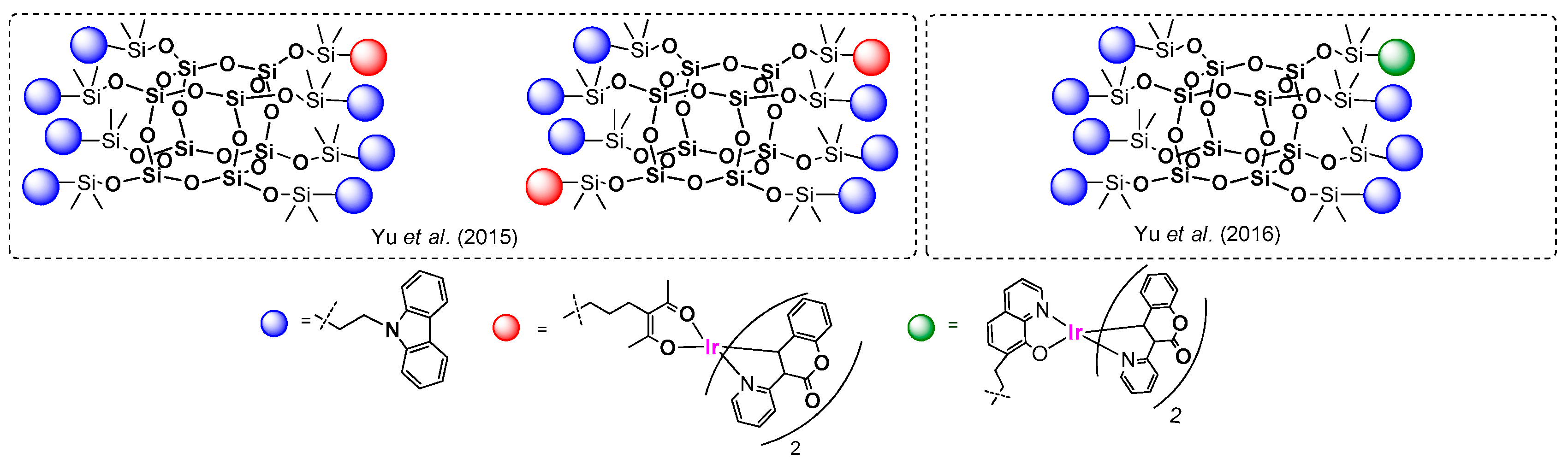{kind=link}
{kind=link}
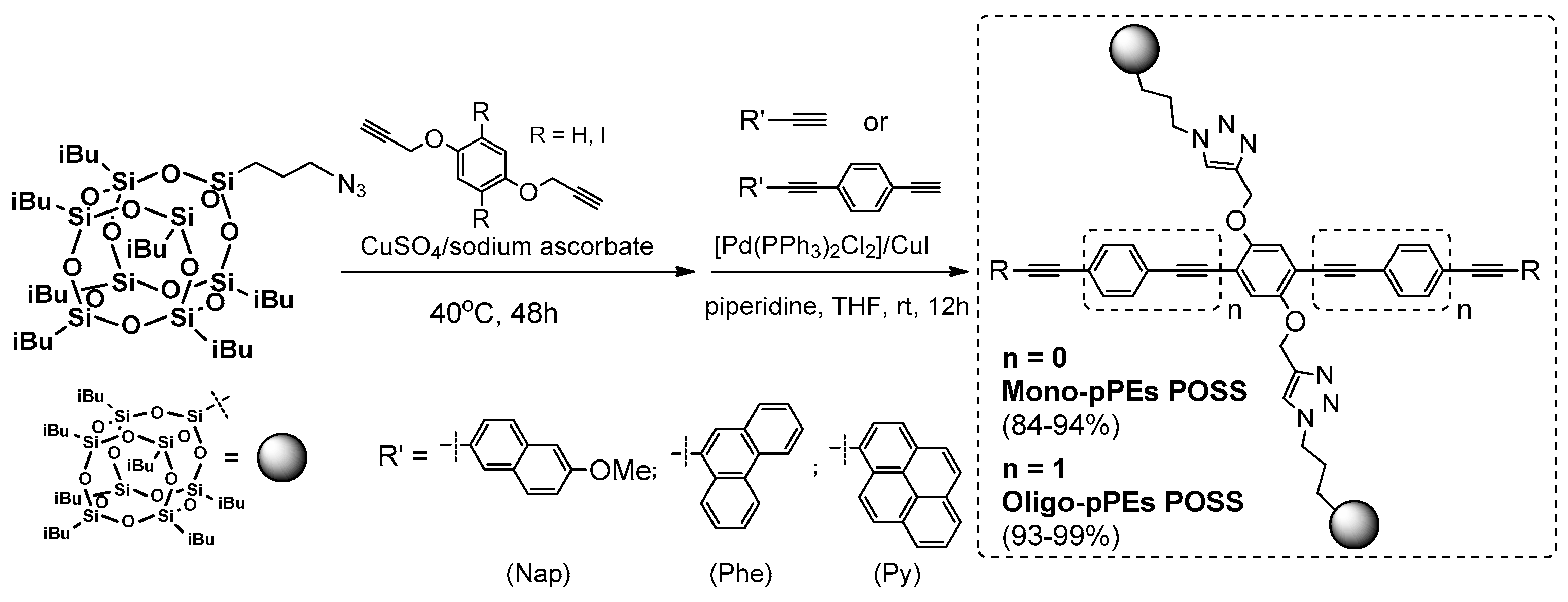{kind=link}
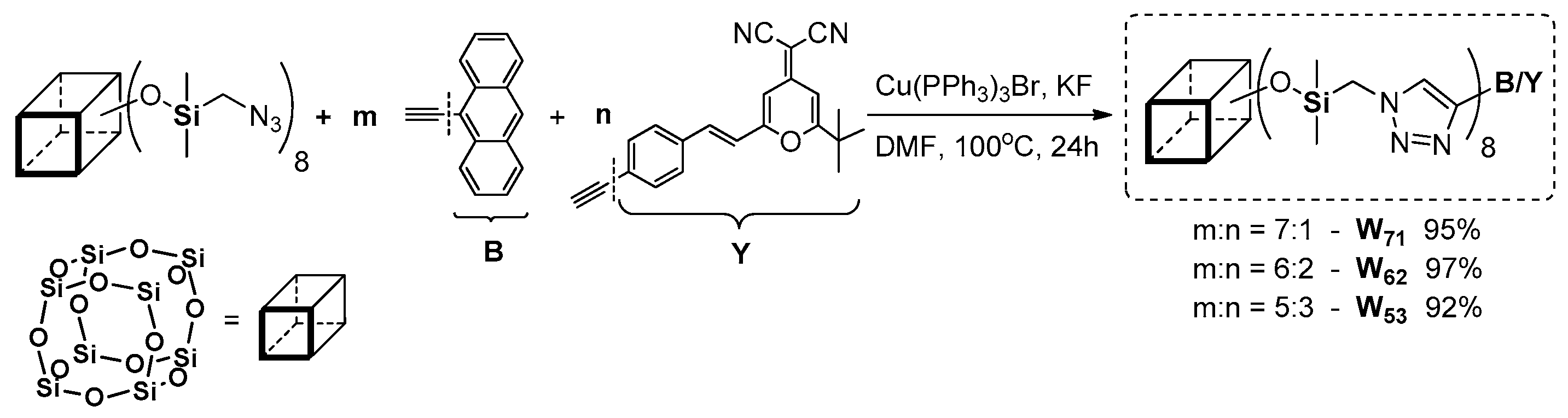{kind=link}
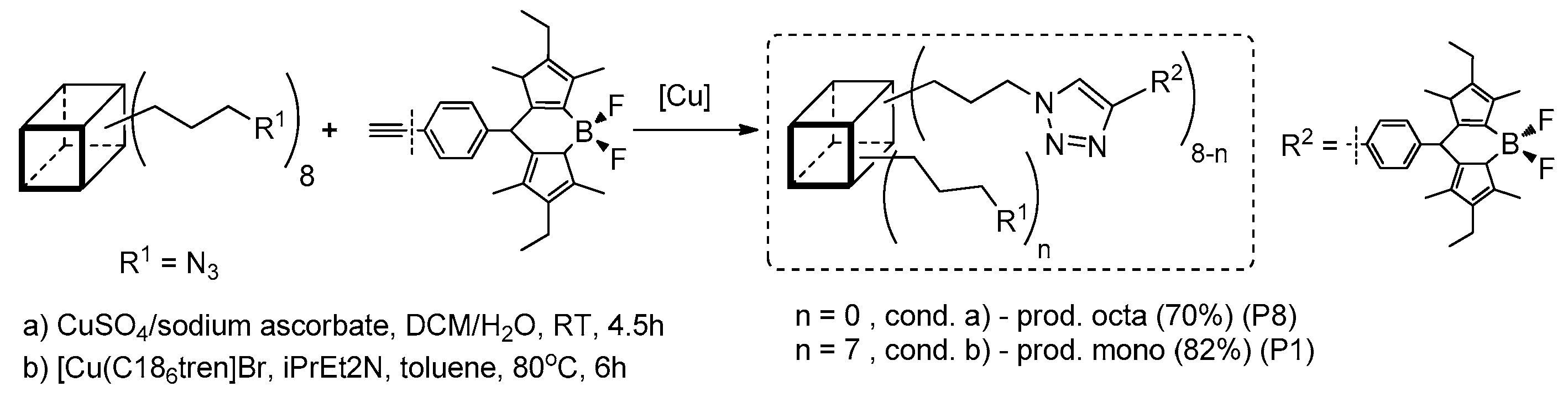{kind=link}
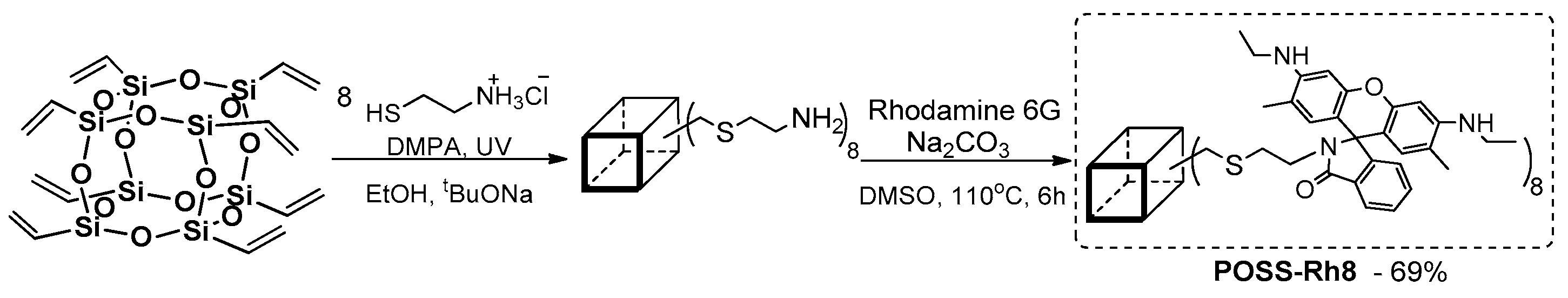{kind=link}
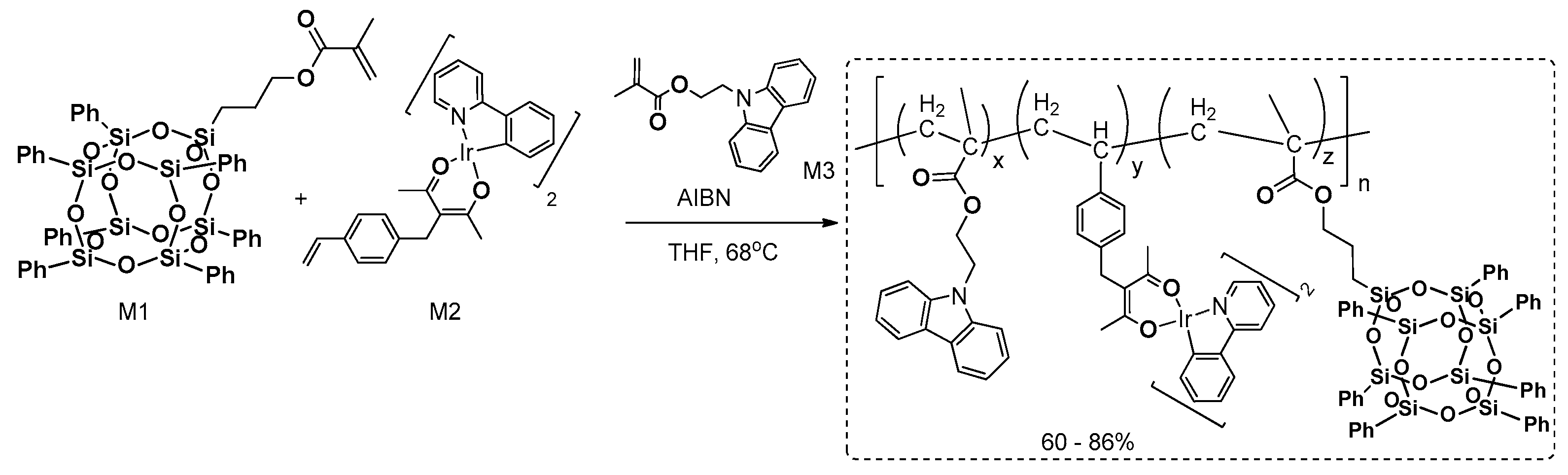{kind=link}







































