Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications



Abstract
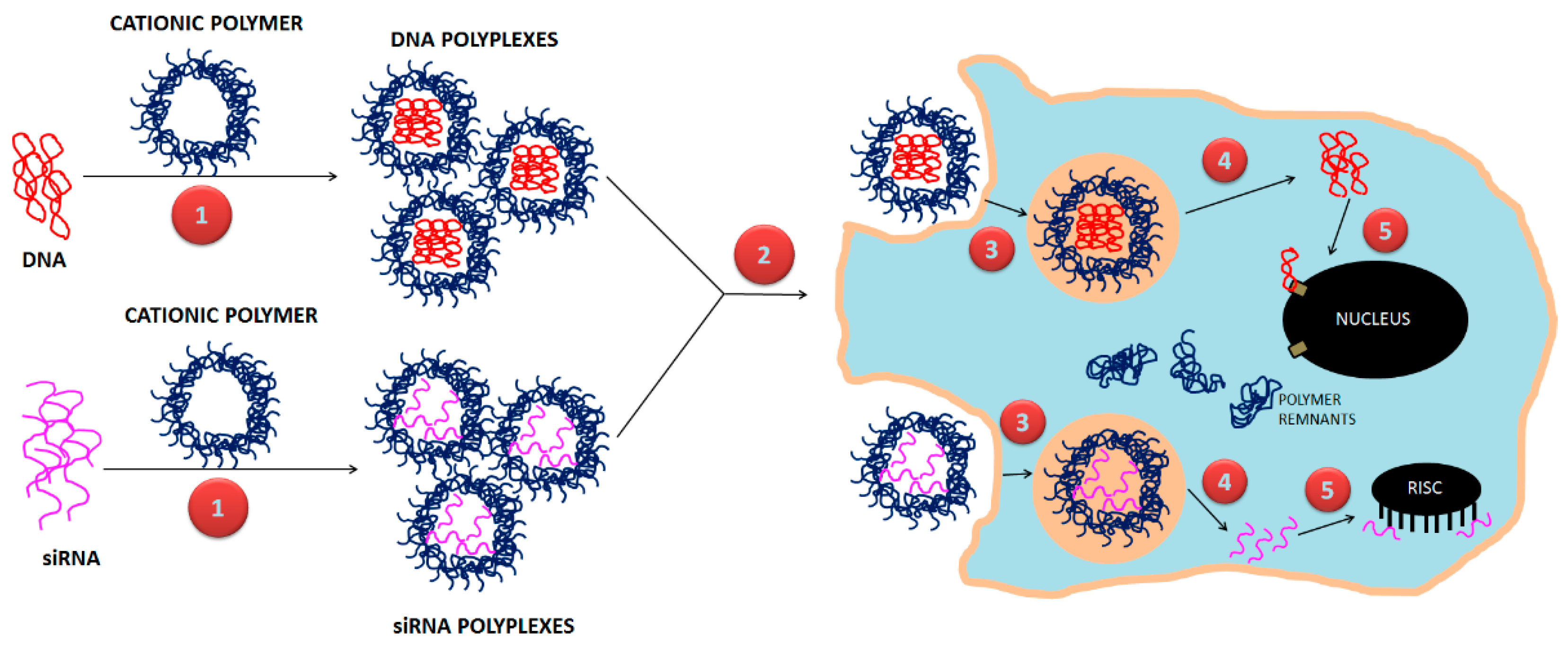
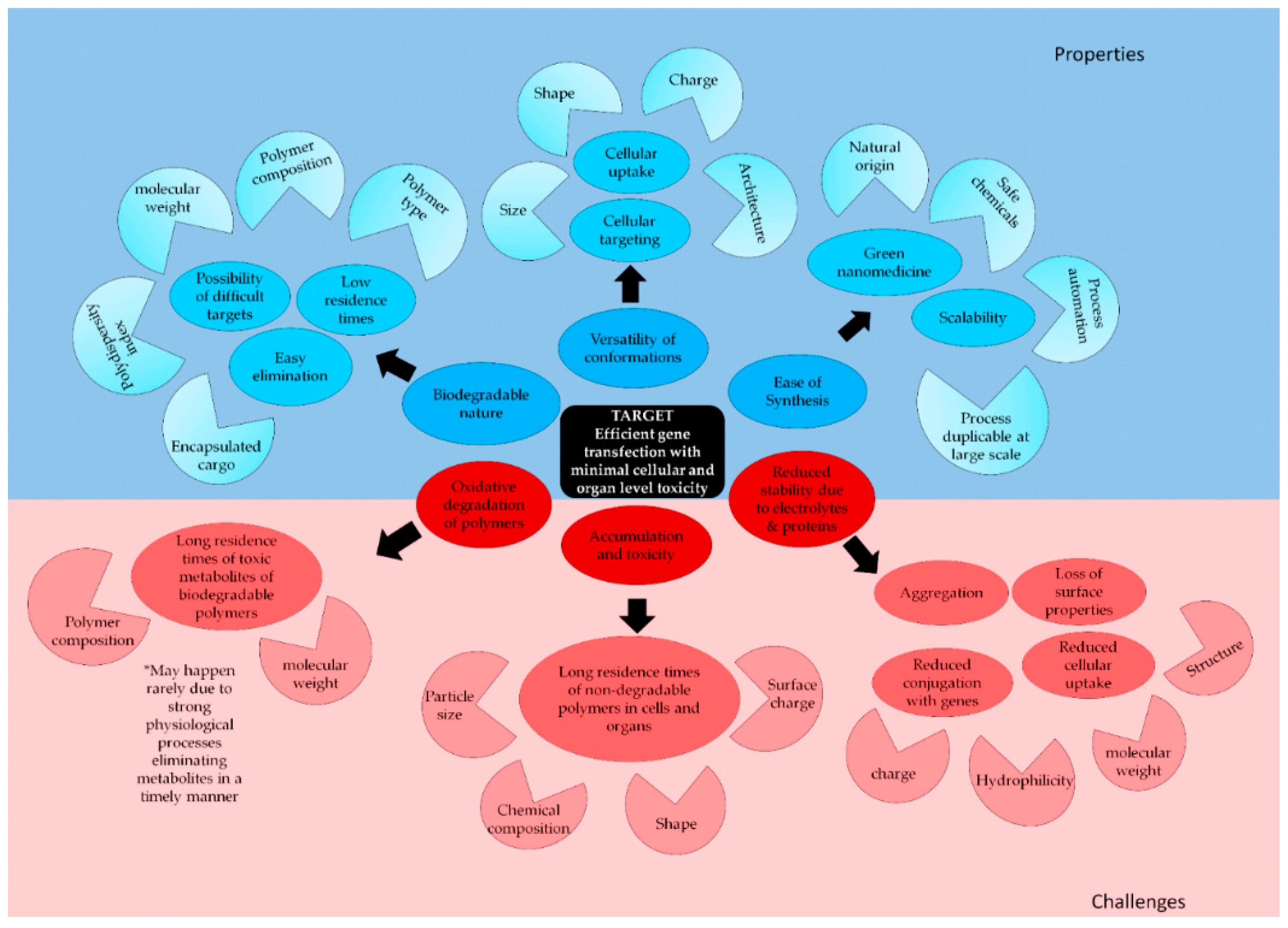
:1. Introduction
2. Properties of Polymeric Nanoparticles Advantageous for Biomedical Use
2.1. Biodegradability
2.2. Facile Chemistry
2.2.1. Versatality of Functionalization
2.2.2. Ease of Synthesis
2.3. Scalable Production
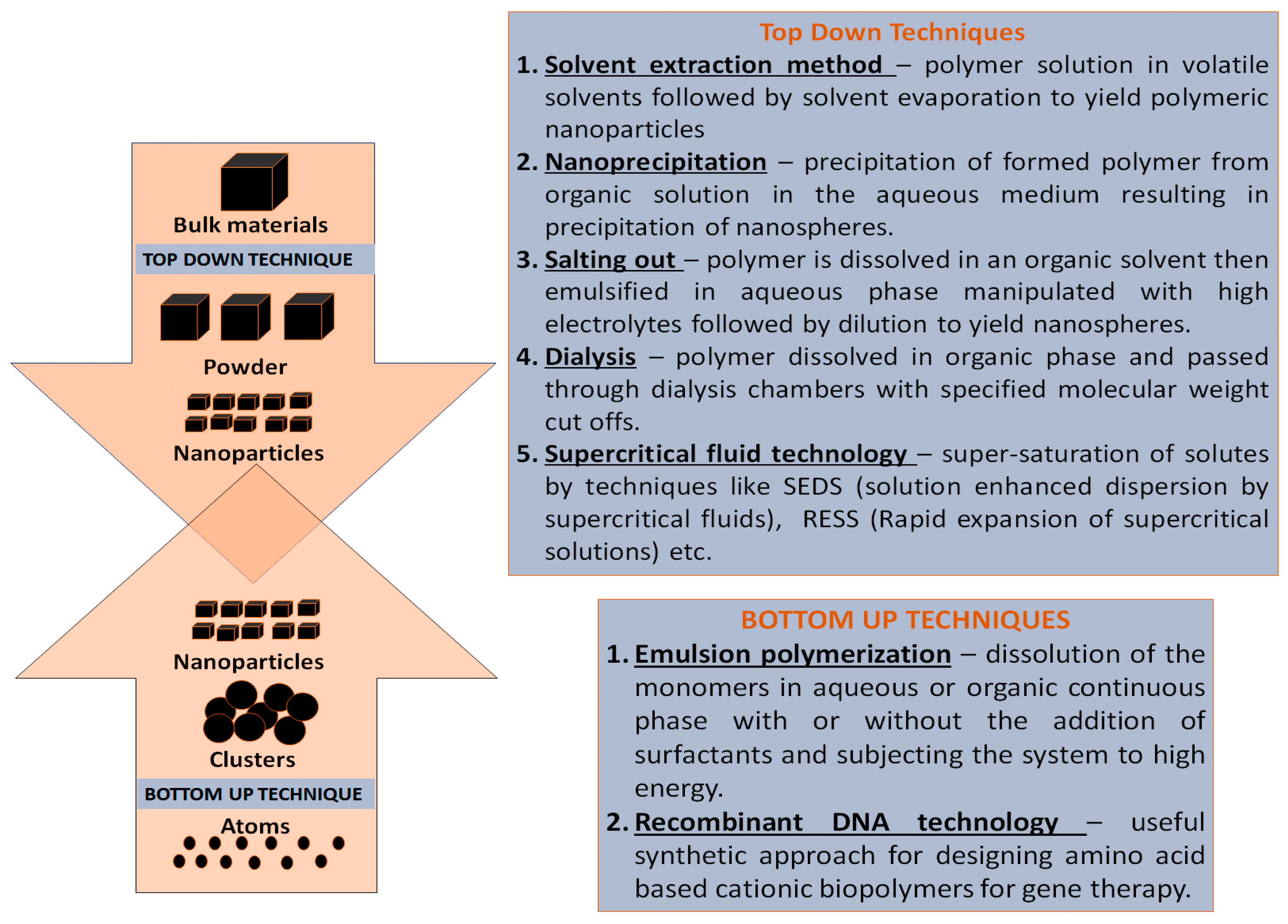
3. Strategies for Designing Polymeric Nanoparticles
3.1. Top-Down Strategy for Polymer Synthesis
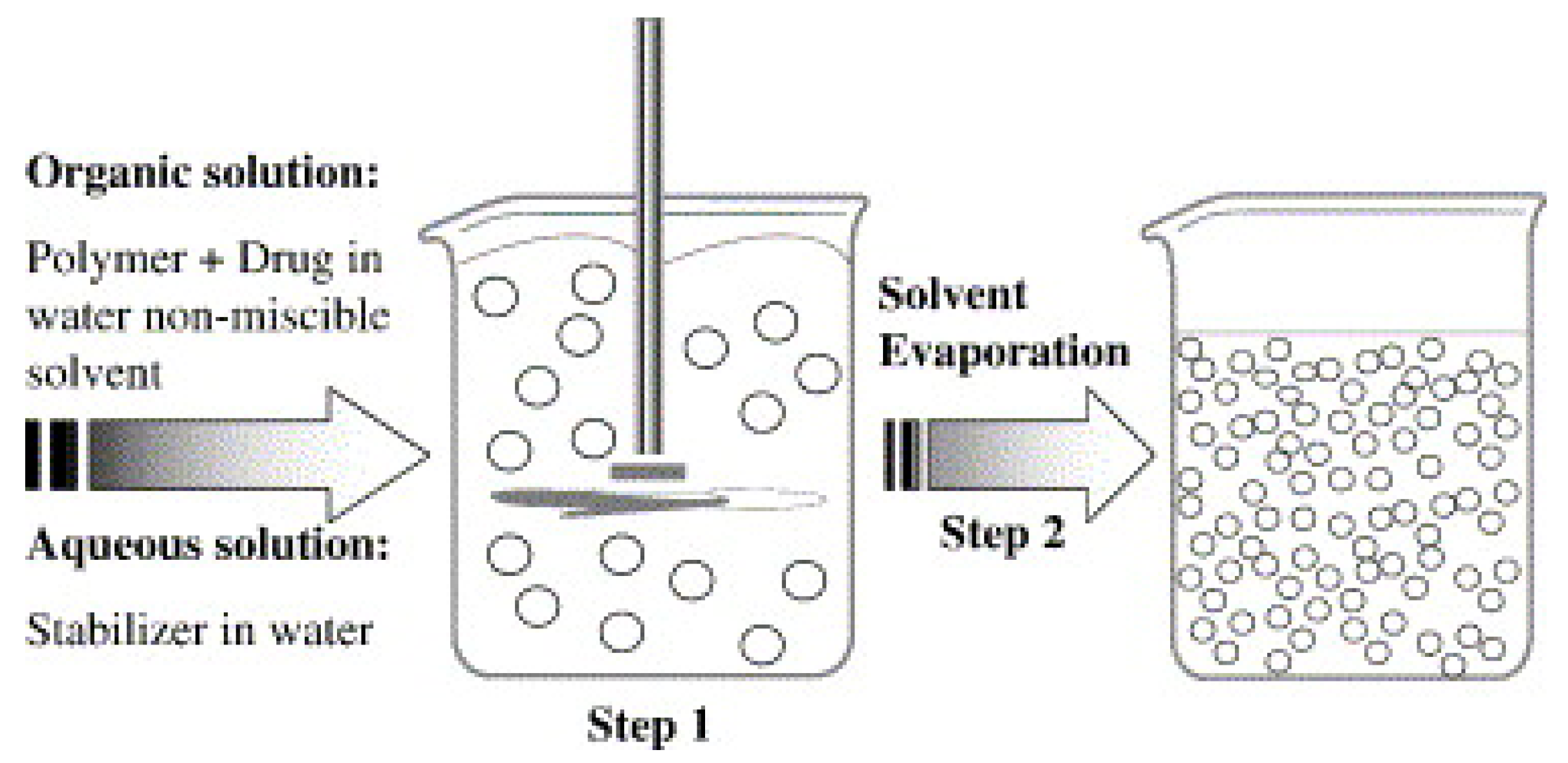
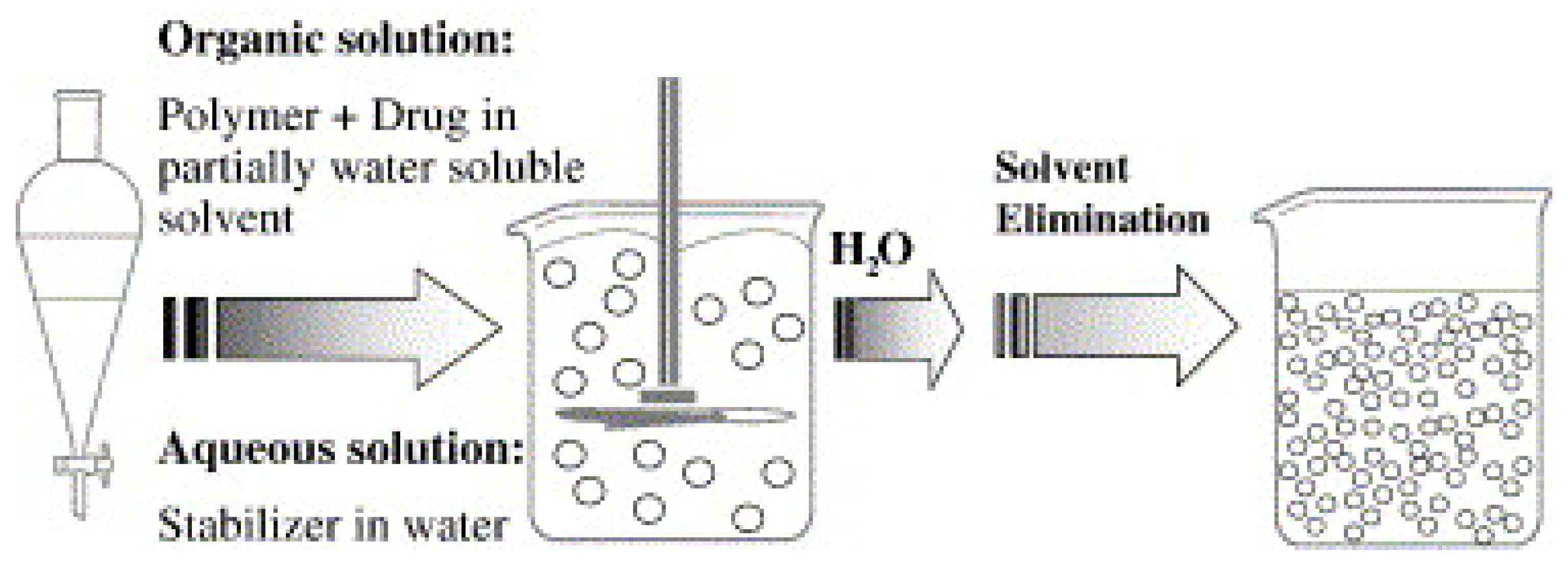
3.1.1. Solvent Evaporation Method
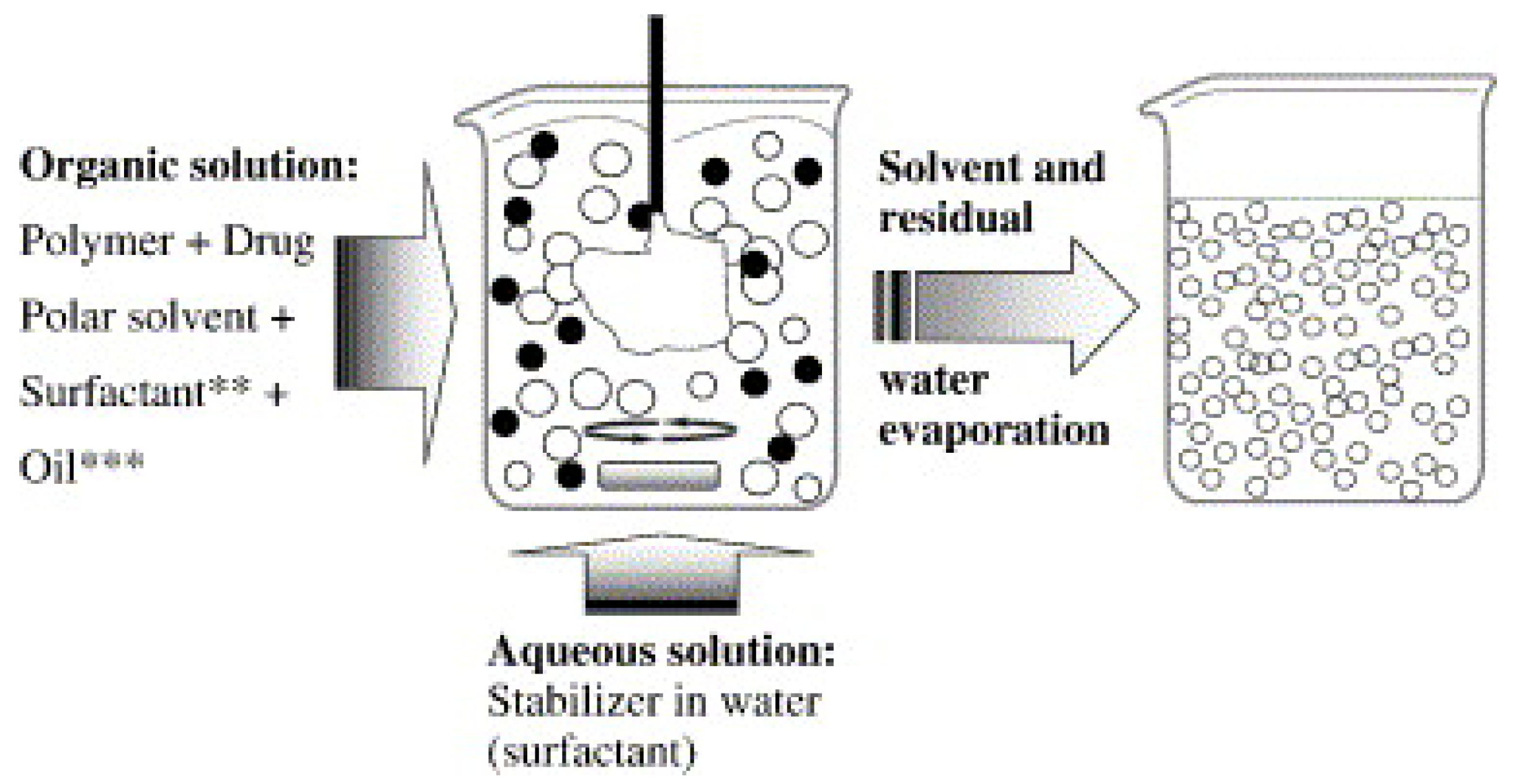
3.1.2. Solvent Displacement Method
3.1.3. Salting Out
3.1.4. Dialysis
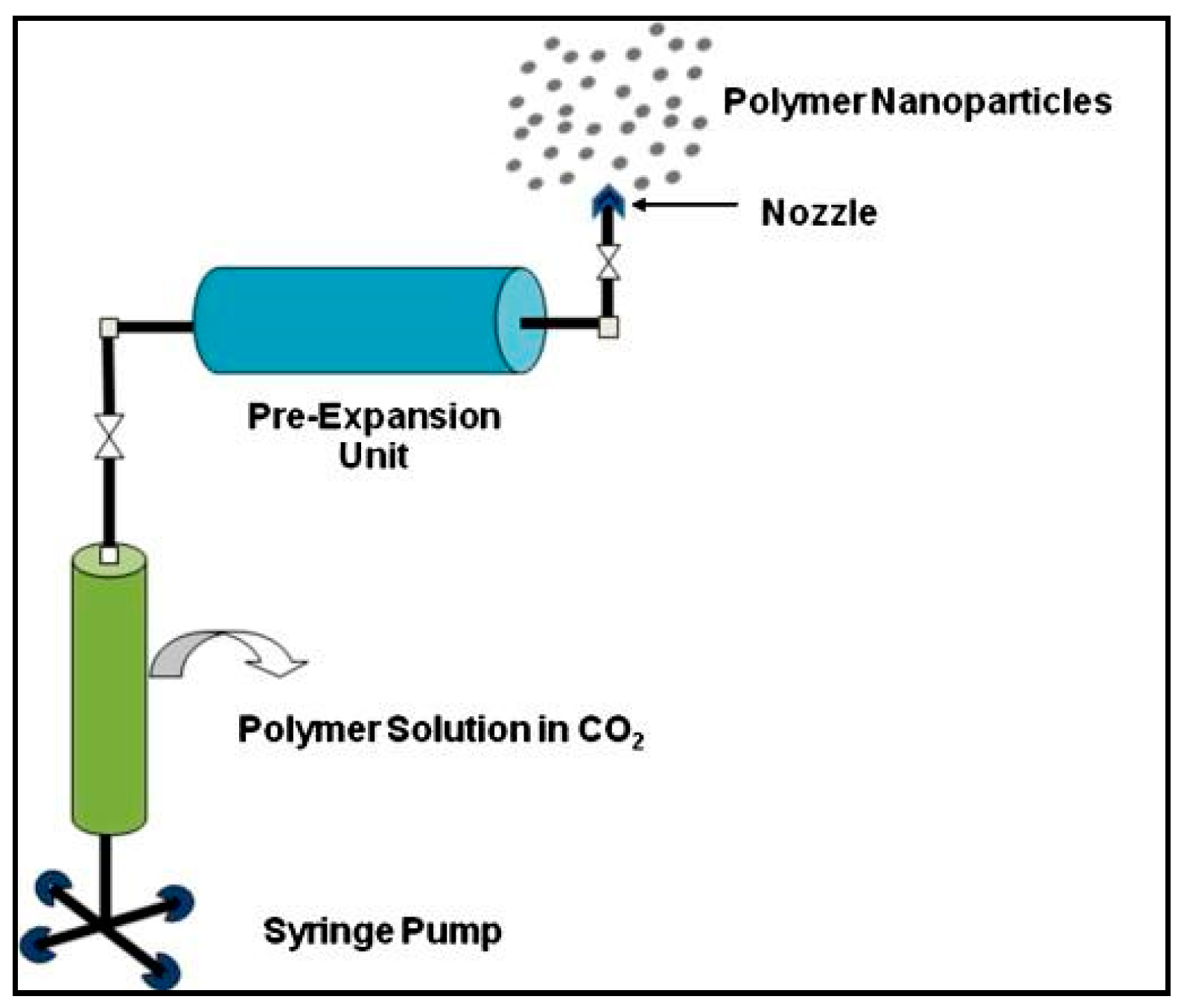
3.1.5. Supercritical Fluid Technology
3.2. Bottom-Up Strategies for the Preparation of Polymer Nanoparticles
3.2.1. Emulsion Polymerization
3.2.2. Recombinant Technology
3.3. Polymerization Chemistries for Common Synthetic Polymers
3.3.1. Poly(Lactic Acid)
- Direct condensation of lactic acid: It is the conventional method of synthesis utilizing solvents and exhibiting high reaction times [86]. It has been done using diphenyl ether as a solvent in the presence of tin (II) chloride as the catalyst. The process is strictly dependent on the polymerization temperature and pressure. An increase in temperature leads to a high molecular weight PLA [87]. Other solvent systems like p-xylene [88] have also been employed. Solid-state direct poly-condensation without the use of a solvent was proposed utilizing this reaction chemistry. A pre-polymer product was formed first, using p-toluene sulfonic acid without the addition of any catalyst. This product was then subjected to solid-state polymerization under high temperature and pressure conditions [89].
- Ring opening polymerization of lactide: This process is completed in two steps. In the first step, lactic acid cyclizes into lactide (a close chain lactone di-ester) under heat and a vacuum. A nitrogen-controlled inert environment is used to speed up the removal of water vapors, enhancing cyclization. The second step involves disruption of the cyclic ring, followed by the union of open chains forming the polymer. This step is catalyzed by stannous octoate to promote formation of ester linkages [86,90]. Process parameters and solvents used in this approach fulfills the requirements of “green chemistry” [90], a novel advancement in polymer nanoscience, which was discussed previously.
3.3.2. Poly-l-Lysine
3.3.3. Poly(Amidoamine)
3.3.4. Poly(Methyl-Methacrylate)
3.3.5. Poly(Ethylene-Imine)
3.3.6. Poly(Lactic-co-Glycolide)
- PLGA containing drugs
- i)
- Vivitrol (naltrexone) intramuscular (IM)
- ii)
- Zoladex (gorserelin acetate) subcutaneous (SC)
- iii)
- Lupron depot, Lupron (leuprolide acetate) IM, and Lupaneta pack (leuprolide acetate and norethindrone) oral and IM
- iv)
- Sandostatin LAR (octreotide) SC
- v)
- Trelstar (triptorelin pamoate) IM
- vi)
- Arestin (minocycline HCL) periodontal
- vii)
- Risperidal Consta (risperidone) IM
- viii)
- Ozurdex (dexamethasone) SC
- ix)
- Bydureon (exanatide) tablets oral
- x)
- Signifor LAR (pasireotide pamoate) IM
- PLA containing drugs
- i)
- Lurpon depot
- ii)
- Atridox (doxycycline) periodontal
4. Challenges Associated with the Use of Polymers in Nanomedicine
4.1. Stability of PNPs in an Electrolyte and Protein-Rich Biological Medium
4.2. Accumulation and Toxicity of Polymeric Nanoparticles
4.3. Oxidative Degradation of Polymers—Generation of Toxic Metabolites
4.4. High Cost Associated with Biological Analyses and Process Development
5. Biodistribution and Cellular Interaction of Polymeric Nanoparticles
5.1. Biodistribution of the PEGylated Polymers
5.2. Translational Concerns Regarding PEGylation of Gene Complexes
- As PEG-based dendrimers and block copolymers are designed, it is imperative to control the aggregation and micelle formation that can compromise the biocompatibility and in vivo stability of the formulation [180]. For instance, injectable products should have a particle size restricted to <0.5 µm with a maximum aggregation limit of 5 µm to avoid serious embolic episodes.
- For PEGylated proteins and peptides, it is considered that PEG may cause partial fragmentation of proteins giving rise to new epitopes, or the methoxyl terminus of the PEG molecule can cause antigenicity in the blood.
- Difficulty in characterizing PEGylated drugs also remains a concern [179].
- Remnants of reaction chemicals used in PEGylation can be a concern for sensitive drugs and biomolecules [185].
- Patients with renal or liver insufficiency may be exposed to an increased risk of toxicity. For example, 240 grams of PEG 400 exposed a patient who was concomitantly taking lorazepam to acute renal tubular necrosis; therefore, continuous drug monitoring is required for such patients who are receiving a cocktail of medications [179].
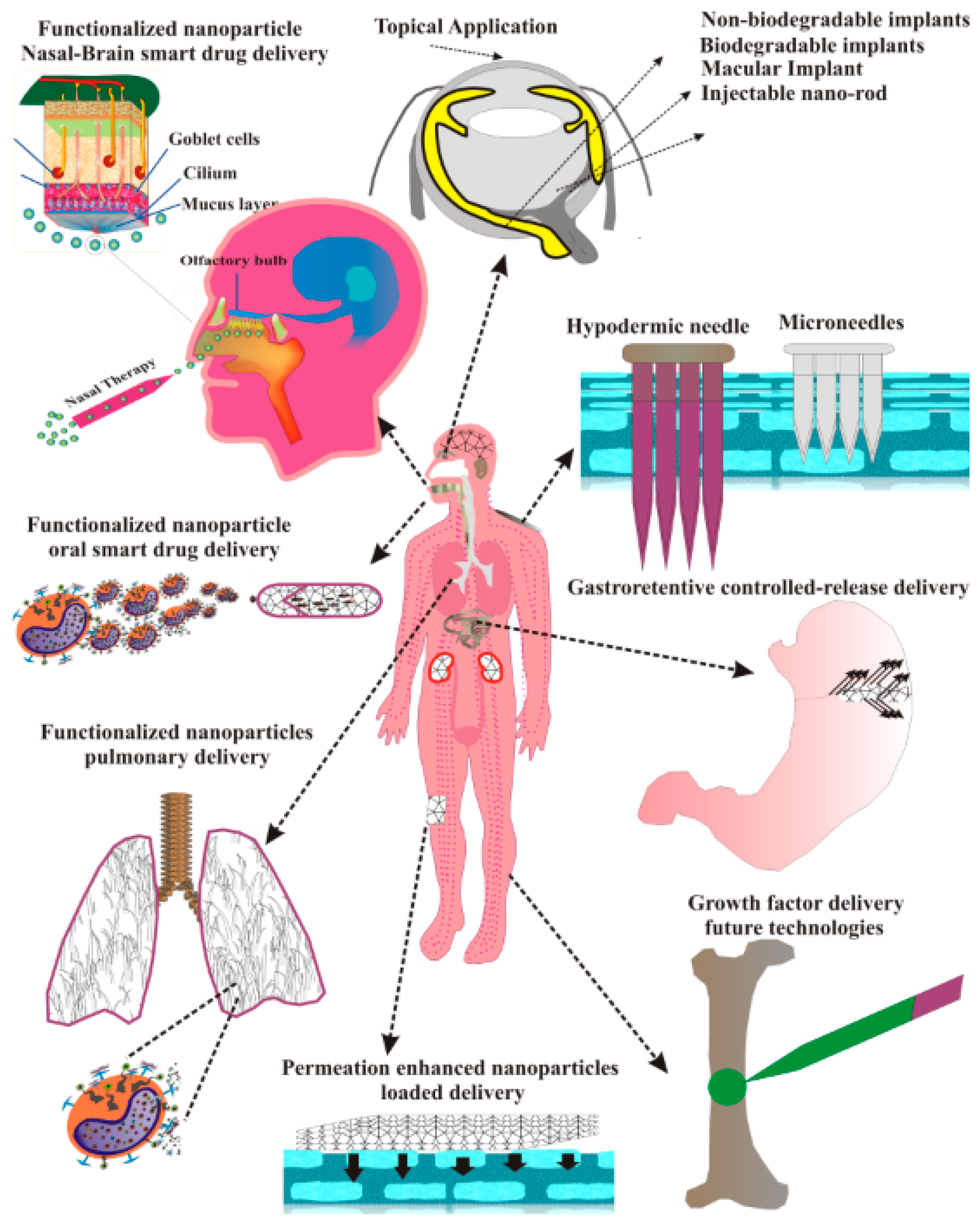
6. Non-Invasive Routes of Administration of PNPs
6.1. PNPs for Oral Administration
6.2. PNPs for Topical Therapeutics
7. Non-Conventional Polymeric Designs for Gene Delivery
- -
- Achieving higher gene transfection as compared to conventional PNPs
- -
- Achieving sustained and controllable release of therapeutic genes
- -
- Achieving cellular targeting to improve localization of polyplexes within in vivo tumor models.
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| CD | cyclodextrin |
| NP | nanoparticle |
| PAMAM | polyamidoamine |
| PCA | poly-cyanoacrylate |
| PCL | poly-ε-caprolactone |
| PEG | polyethylene glycol |
| PLA | poly-lactic-acid |
| PLCL | poly (lactide-co-caprolactone) |
| PLG | poly-d-l-glycolide |
| PLGA | poly-d-l-lactide-co-glycolide |
| PLL | poly-l-lysine |
| PNP | polymeric nanoparticles |
| PVP | polyvinylpyrrolidone |
References
- Yin, H.; Kanasty, R.; Eltoukhy, A.; Vegas, A.R.; Dorkin, J.; Anderson, D. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Abedini, F.; Ou, K.-L.; Domb, A. Polymers in gene therapy technology. Polym. Adv. Technol. 2015, 26, 198–211. [Google Scholar] [CrossRef]
- Coll, J.; Chollet, P.; Brambilla, E.; Desplanques, D.; Behr, J.; Favrot, M. In vivo delivery to tumors of DNA complexed with linear polyethylenimine. Hum. Gene 1999, 10, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Alnylam Pharmaceuticals Inc. Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO™ (patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults. Available online: http://investors.alnylam.com/news-releases/news-release-details/alnylam-announces-first-ever-fda-approval-rnai-therapeutic (accessed on 23 April 2019).
- Felgner, P.; Gadek, T.; Holm, M.; Roman, R.; Chan, H.; Wenz, M.; Northrop, J.; Ringold, G.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Samal, S.; Dash, M.; Van Vlierberghe, S.; Kaplan, D.; Chiellini, E.; van Blitterswijk, C.; Moroni, L.; Dubruel, P. Cationic polymers and their therapeutic potential. Chem. Soc. Rev. 2012, 41, 7147–7194. [Google Scholar] [CrossRef]
- Lechardeur, D.; Verkman, A.; Lukacs, G. Intracellular routing of plasmid DNA during non-viral gene transfer. Adv. Drug. Deliv. Rev. 2005, 57, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Bielinska, A.; Shivdasani, R.; Zhang, L.; Nabel, G. Regulation of gene expression with double-stranded phosphorothioate oligonucleotides. Science 1990, 250, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Zanta, M.; Behr, J. Optimized galenics improve in vitro gene transfer with cationic molecules up to 1000-fold. Gene 1996, 3, 1074–1080. [Google Scholar]
- Erbacher, P.; Zou, S.; Bettinger, T.; Steffan, A.; Remy, J. Chitosan-based vector/DNA complexes for gene delivery: Biophysical characteristics and transfection ability. Pharm. Res. 1998, 15, 1332–1339. [Google Scholar] [CrossRef]
- Ferrari, S.; Moro, E.; Pettenazzo, A.; Behr, J.; Zacchello, F.; Scarpa, M. ExGen 500 is an efficient vector for gene delivery to lung epithelial cells in vitro and in vivo. Gene 1997, 4, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.; Baker, H.; Curiel, D.; Jiang, W.; Conry, R. Humoral and cellular immune responses of dogs immunized with a nucleic acid vaccine encoding human carcinoembryonic antigen. Gene 1998, 5, 865–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Lee, J.; Cho, M.; Sheares, V. Synthesis of amine-functionalized diene-based polymers as novel gene delivery vectors. Macromolecules 2006, 39, 8625–8631. [Google Scholar] [CrossRef]
- Wood, K.; Azarin, S.; Arap, W.; Pasqualini, R.; Langer, R.; Hammond, P. Tumor-targeted gene delivery using molecularly engineered hybrid polymers functionalized with a tumor-homing peptide. Bioconjug. Chem. 2008, 19, 403–405. [Google Scholar] [CrossRef]
- Bolhassani, A.; Javanzad, S.; Saleh, T.; Hashemi, M.; Aghasadeghi, M.; Sadat, S. Polymeric nanoparticles potent vectors for vaccine delivery targeting cancer and infectious diseases. Hum. Vaccin. Immunother. 2014, 10, 321–332. [Google Scholar] [CrossRef]
- Mahapatro, A.; Singh, D. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J. Nanobiotechnol. 2011, 9, 55. [Google Scholar] [CrossRef]
- Makadia, H.; Siegel, S. Poly Lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Houchin, M.; Topp, E. Physical properties of PLGA films during polymer degradation. J. Appl. Polym. Sci. 2009, 114, 2848–2854. [Google Scholar] [CrossRef]
- Siegel, S.; Kahn, J.; Metzger, K.; Winey, K.; Werner, K.; Dan, N. Effect of drug type on the degradation rate of PLGA matrices. Eur. J. Pharm. Biopharm. 2006, 64, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Brem, H.; Gabikian, P. Biodegradable polymer implants to treat brain tumors. J. Control. Release 2001, 74, 63–67. [Google Scholar] [CrossRef]
- Siparsky, G.; Voorhees, K.J.; Miao, F. Hydrolysis of polylactic acid (PLA) and polycaprolactone (PCL) in aqueous acetonitrile solutions: Autocatalysis. J. Environ. Polym. Degrad. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Rancan, F.; Papakostas, D.; Hadam, S.; Hackbarth, S.; Delair, T.; Primard, C.; Verrier, B.; Sterry, W.; Blume-Peytavi, U.; Vogt, A. Investigation of polylactic acid (PLA) nanoparticles as drug delivery systems for local dermatotherapy. Pharm. Res. 2009, 26, 2027–2036. [Google Scholar] [CrossRef]
- Han, M.; Kim, S.; Liu, S. Synthesis and degradation behavior of poly(ethyl cyanoacrylate). Polym. Degrad. Stab. 2008, 93, 1243–1251. [Google Scholar] [CrossRef]
- Uhrich, K.; Cannizzaro, S.; Langer, R.; Shakesheff, K. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Fang, Y.-P.; Wu, P.-C.; Huang, Y.-B.; Tzeng, C.-C.; Chen, Y.-H.; Hung, Y.-H.; Tsai, M.-J.; Tsai, Y.-H. Modification of polyethylene glycol onto solid lipid nanoparticles encapsulating a novel chemotherapeutic agent (PK-L4) to enhance solubility for injection delivery. Int. J. Nanomed. 2012, 7, 4995–5005. [Google Scholar] [CrossRef]
- Zubris, K.; Khullar, O.; Griset, A.; Gibbs-Strauss, S.; Frangioni, J.; Colson, Y.; Grinstaff, M. Ease of synthesis, controllable sizes, and in vivo large animal lymph migration of polymeric nanoparticles. ChemMedChem 2010, 5, 1435–1438. [Google Scholar] [CrossRef]
- Watkins, R.; Wu, L.; Zhang, C.; Davis, R.; Xu, B. Natural product-based nanomedicine: Recent advances and issues. Int. J. Nanomed. 2015, 10, 6055–6074. [Google Scholar]
- Jahangirian, H.; Lemraski, E.; Webster, T.; Rafiee-Moghaddam, R.; Abdollahi, Y. A review of drug delivery systems based on nanotechnology and green chemistry: Green nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef]
- Zhao, M.; Cheng, J.; Yan, J.; Chen, F.; Sheng, J.; Sun, D.; Chen, J.; Miao, J.; Zhang, R.; Zheng, C.; et al. Hyaluronic acid reagent functional chitosan-PEI conjugate with AQP2-siRNA suppressed endometriotic lesion formation. Int. J. Nanomed. 2016, 11, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Luo, Z.; Zhang, J.; Luo, T.; Zhou, J.; Zhao, X.; Cai, K. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials 2016, 83, 51–65. [Google Scholar] [CrossRef]
- Maksimović, M.; Omanović-Mikličanin, E. Towards green nanotechnology: Maximizing benefits and minimizing harm. In CMBEBIH 2017. IFMBE Proceedings; Springer: Singapore, 2017; Volume 62, pp. 164–170. [Google Scholar]
- Prat, D.; Hayler, J.; Wells, A. A survey of solvent selection guides. Green Chem. 2014, 16, 4546–4551. [Google Scholar] [CrossRef]
- Precision Nanosystems. Product Suite and Solutions Overview. Available online: https://www.precisionnanosystems.com/our-technology/product-comparison (accessed on 23 April 2019).
- Meyer, R.; Meyer, R.; Green, J. An automated multidimensional thin film stretching device for the generation of anisotropic polymeric micro- and nanoparticles. J. Biomed. Mater. Res. A 2015, 103, 2747–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Luft, J.; Yi, X.; Tian, S.; Owens, G.; Wang, J.; Johnson, A.; Berglund, P.; Smith, J.; Napier, M.; et al. RNA replicon delivery via lipid-complexed PRINT protein particles. Mol. Pharm. 2013, 10, 3366–3374. [Google Scholar] [CrossRef]
- Jiang, X.; Leong, D.; Ren, Y.; Li, Z.; Torbenson, M.; Mao, H. String-like micellar nanoparticles formed by complexation of PEG-b-PPA and plasmid DNA and their transfection efficiency. Pharm. Res. 2011, 28, 1317–1327. [Google Scholar] [CrossRef]
- Barua, S.; Yoo, J.; Kolhar, P.; Wakankar, A.; Gokarn, Y.; Mitragotri, S. Particle shape enhances specificity of antibody-displaying nanoparticles. Proc. Natl. Acad. Sci. USA 2013, 110, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Florez, L.; Herrmann, C.; Cramer, J.; Hauser, C.; Koynov, K.; Landfester, K.; Crespy, D.; Mailänder, V. How shape influences uptake: Interactions of anisotropic polymer nanoparticles and human mesenchymal stem cells. Small 2012, 8, 2222–2230. [Google Scholar] [CrossRef]
- Rolland, J.; Maynor, B.; Euliss, L.; Exner, A.; Denison, G.; DeSimone, J. Direct fabrication and harvesting of monodisperse, shape-specific nanobiomaterials. J. Am. Chem. Soc. 2005, 127, 10096–10100. [Google Scholar] [CrossRef]
- Behrendt, J.; Guzman, J.; Purdie, L.; Willcock, H.; Morrison, J.; Foster, A.; O’Reilly, R.; McCairn, M.; Turner, M. Scalable synthesis of multicolour conjugated polymer nanoparticles via Suzuki-Miyaura polymerisation in a miniemulsion and application in bioimaging. React. Funct. Polym. 2016, 107, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Boylan, N.; Cai, S.; Miao, B.; Patel, H.; Hanes, J. Scalable method to produce biodegradable nanoparticles that rapidly penetrate human mucus. J. Control. Release 2013, 170, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Geckeler, K.; Stirn, J. Polyreaktionen-Mechanismen, Systematik, Relevanz. Naturwissenschaften 1993, 80, 487–500. [Google Scholar] [CrossRef]
- Zhang, X.; Geng, X.; Jiang, H.; Li, J.; Huang, J. Synthesis and characteristics of chitin and chitosan with the (2-hydroxy-3-trimethylammonium)propyl functionality, and evaluation of their antioxidant activity in vitro. Carbohydr. Polym. 2012, 89, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Sun, G.; Zhang, X.D.; Liu, Y.; He, C.; Leong, K. PEI-g-chitosan, a novel gene delivery system with transfection efficiency comparable to polyethylenimine in vitro and after liver administration in vivo. Bioconjug. Chem. 2006, 17, 152–158. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Azzam, T.; Tabata, Y.; Domb, A. Dextran-spermine polycation: An efficient nonviral vector for in vitro and in vivo gene transfection. Gene 2004, 11, 194–203. [Google Scholar] [CrossRef]
- Kritchenkov, A.; Andranovitš, S.; Skorik, Y. Chitosan and its derivatives: Vectors in gene therapy. Russ. Chem. Rev. 2017, 86, 231. [Google Scholar] [CrossRef]
- Santos-Carballal, B.; Fernández, E.; Goycoolea, F. Chitosan in non-viral gene delivery: Role of structure, characterization methods, and insights in cancer and rare diseases therapies. Polymers 2018, 10, 444. [Google Scholar] [CrossRef]
- Alleman, E.; Gurny, R.; Doelker, E.; Allémann, E.; Deolker, E. Drug-loaded nanoparticles—Preparation methods and drug targeting issues. Eur. J. Pharm. Biopharm. 1993, 39, 173–191. [Google Scholar]
- Anton, N.; Benoit, J.; Saulnier, P. Design and production of nanoparticles formulated from nano-emulsion templates—A review. J. Control. Release 2008, 128, 185–199. [Google Scholar] [CrossRef]
- Nagavarma, B.; Yadav, H.; Ayaz, A.; Vasudha, L.; Shivakumar, H. Different techniques for preparation of polymeric nanoparticles- A review. Asian J. Pharm. Clin. Res. 2012, 5, 16–23. [Google Scholar]
- Zambaux, M.; Bonneaux, F.; Gref, R.; Maincent, P.; Dellacherie, E.; Alonso, M.; Labrude, P.; Vigneron, C. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J. Control. Release 1998, 50, 31–40. [Google Scholar] [CrossRef]
- Lemoine, D.; Préat, V. Polymeric nanoparticles as delivery system for influenza virus glycoproteins. J. Control. Release 1998, 54, 15–27. [Google Scholar] [CrossRef]
- Reis, C.; Neufeld, R.; Ribeiro, A.; Veiga, F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine 2006, 2, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Fessi, H.; Puisieux, F.; Devissaguet, J.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Barichello, J.; Morishita, M.; Takayama, K.; Nagai, T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev. Ind. Pharm. 1999, 25, 471–476. [Google Scholar] [CrossRef]
- Galindo-Rodriguez, S.; Allémann, E.; Fessi, H.; Doelker, E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm. Res. 2004, 21, 1428–1439. [Google Scholar] [CrossRef]
- François, G.; Katz, J. Nanoparticles and nanocapsules created using the Ouzo effect: Spontaneous emulisification as an alternative to ultrasonic and high-shear devices. Chemphyschem 2005, 6, 209–216. [Google Scholar]
- Quintanar-Guerrero, D.; Allémann, E.; Fessi, H.; Doelker, E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev. Ind. Pharm. 1998, 24, 1113–1128. [Google Scholar] [CrossRef]
- Allémann, E.; Leroux, J.; Gurny, R. Polymeric nano- and microparticles for the oral delivery of peptides and peptidomimetics. Adv. Drug Deliv. Rev. 1998, 34, 171–189. [Google Scholar] [CrossRef]
- Zhang, Z.; Grijpma, D.; Feijen, J. Poly(trimethylene carbonate) and monomethoxy poly(ethylene glycol)-block-poly(trimethylene carbonate) nanoparticles for the controlled release of dexamethasone. J. Control. Release 2006, 111, 263–270. [Google Scholar] [CrossRef]
- Song, C.; Labhasetwar, V.; Murphy, H.; Qu, X.; Humphrey, W.; Shebuski, R.; Levy, R. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J. Control. Release 1997, 43, 197–212. [Google Scholar] [CrossRef]
- Couvreur, P.; Dubernet, C.; Puisieux, F. Controlled drug delivery with nanoparticles: Current possibilities and future trends. Eur. J. Pharm. Biopharm. 1995, 41, 2–13. [Google Scholar]
- Reis, C.; Neufeld, R.; Veiga, F. Preparation of Drug-Loaded Polymeric Nanoparticles. In Nanomedicine in Cancer; Pan Stanford Publishing Pte. Ltd.: Singapore, 2017; pp. 171–198. [Google Scholar]
- Jeong, Y.; Cho, C.; Kim, S.; Ko, K.; Kim, S.; Shim, Y.-H.; Nah, J.-W. Preparation of poly(DL-lactide-co-glycolide) nanoparticles without surfactant. J. Appl. Polym. Sci. 2001, 80, 2228–2236. [Google Scholar] [CrossRef]
- Jeon, H.; Jeong, Y.; Jang, M.; Park, Y.; Nah, J. Effect of solvent on the preparation of surfactant-free poly(DL-lactide-co-glycolide) nanoparticles and norfloxacin release characteristics. Int. J. Pharm. 2000, 207, 99–108. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, Z.; Wang, X.; Xu, J.; Yang, K.; Cui, Q.; Chen, X.; Cao, M.; Weng, J.; Zhang, Q. Formation of poly(l,d-lactide) spheres with controlled size by direct dialysis. Polymers 2007, 48, 5767–5779. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, Y.; Kim, Y.; Kim, S. Self-assembled polymeric nanoparticles of poly(ethylene glycol) grafted pullulan acetate as a novel drug carrier. Arch. Pharm. Res. 2004, 27, 562–569. [Google Scholar] [CrossRef]
- Hornig, S.; Heinze, T. Nanoscale structures of dextran esters. Carbohydr. Polym. 2007, 68, 280–286. [Google Scholar] [CrossRef]
- Heinze, T.; Michealis, N.; Schubert, S. Reactive polymeric nanoparticles based on unconventional dextran derivatives. Eur. Polym. J. 2007, 43, 697–703. [Google Scholar] [CrossRef]
- Park, K.; Song, H.; Na, K.; Bom, H.; Lee, K.; Kim, S.; Kang, D.; Lee, D. Ionic strength-sensitive pullulan acetate nanoparticles (PAN) for intratumoral administration of radioisotope: Ionic strength-dependent aggregation behavior and (99m)Technetium retention property. Colloids Surf. B Biointerfaces 2007, 59, 16–23. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J. Design of surface-modified poly(D,L-lactide-co-glycolide) nanoparticles for targeted drug delivery to bone. J. Control. Release 2007, 122, 24–30. [Google Scholar] [CrossRef]
- Lee, J.; Cho, E.; Cho, K. Incorporation and release behavior of hydrophobic drug in functionalized poly(D,L-lactide)-block-poly(ethylene oxide) micelles. J. Control. Release 2004, 94, 323–335. [Google Scholar] [CrossRef]
- Rao, J.; Geckeler, K. Polymer nanoparticles: Preparation techniques and size-control parameters. Prog. Polym. Sci. 2011, 36, 887–913. [Google Scholar] [CrossRef]
- York, P. Strategies for particle design using supercritical fluid technologies. Pharm. Sci. Technol. Today 1999, 2, 430–440. [Google Scholar] [CrossRef]
- Kawashima, Y. Nanoparticulate systems for improved drug delivery. Adv. Drug Deliv. Rev. 2001, 47, 1–2. [Google Scholar] [CrossRef]
- Keshavarz, A.; Karimi-Sabet, J.; Fattahi, A.; Golzary, A.; Rafiee-Tehrani, M.; Dorkoosh, F.A. Preparation and characterization of raloxifene nanoparticles using rapid expansion of supercritical solution (RESS). J. Supercrit. Fluids 2012, 63, 169–179. [Google Scholar] [CrossRef]
- Meziani, M.; Pathak, P.; Hurezeanu, R.; Thies, M.; Enick, R.; Sun, Y. Supercritical-fluid processing technique for nanoscale polymer particles. Angew. Chem. Int. Ed. 2004, 43, 704–707. [Google Scholar] [CrossRef]
- Puig, J. Microemulsion polymerization. J. Polym. Mat. Encycl. 1996, 6, 4333–4341. [Google Scholar]
- Canine, B.; Hatefi, A. Development of recombinant cationic polymers for gene therapy research. Adv. Drug Deliv. Rev. 2010, 62, 1524–1529. [Google Scholar] [CrossRef]
- Arís, A.; Feliu, J.; Knight, A.; Coutelle, C.; Villaverde, A. Exploiting viral cell-targeting abilities in a single polypeptide, non-infectious, recombinant vehicle for integrin-mediated DNA delivery and gene expression. Biotechnol. Bioeng. 2000, 68, 689–696. [Google Scholar] [CrossRef]
- Villaverde, A.; Feliu, J.; Harbottle, R.; Benito, A.; Coutelle, C. A recombinant, arginine-glycine-aspartic acid (RGD) motif from foot-and-mouth disease virus binds mammalian cells through vitronectin and, to a lower extent, fibronectin receptors. Gene 1996, 180, 101–106. [Google Scholar] [CrossRef]
- Chen, T.; Bae, Y.; Furgeson, D. Intelligent biosynthetic nanobiomaterials (IBNs) for hyperthermic gene delivery. Pharm. Res. 2008, 25, 683–691. [Google Scholar] [CrossRef]
- Meyer, D.; Chilkoti, A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: Examples from the elastin-like polypeptide system. Biomacromolecules 2002, 3, 357–367. [Google Scholar] [CrossRef]
- Hatefi, A.; Megeed, Z.; Ghandehari, H. Recombinant polymer-protein fusion: A promising approach towards efficient and targeted gene delivery. J. Gene Med. 2006, 8, 468–476. [Google Scholar] [CrossRef]
- Behr, J. The proton sponge: A trick to enter cells the viruses did not exploit. Chim. Int. J. Chem. 1997, 51, 34–36. [Google Scholar]
- Lasprilla, A.; Martinez, J.; Lunelli, B.; Figueroa, J.; Jardini, A.; Filho, R. Synthesis and characterization of poly (lactic acid) for use in biomedical field. Chem. Eng. Trans. 2011, 24, 985. [Google Scholar]
- Laonuad, P.; Chaiyut, N.; Ksapabutr, B. Poly(lactic acid) preparation by polycondensation method. Optoelectron. Adv. Mater. Rapid Commun. 2010, 4, 1200–1202. [Google Scholar]
- Zhang, J.; Krishnamachari, P.; Lou, J.; Shahbazi, A. Synthesis of Poly(L(+) Lactic Acid) by Polycondensation Method in Solution. In Proceedings of the 2007 National Conference on Environmental Science and Technology; Springer-Verlag: New York, NY, USA, 2009; pp. 3–8. [Google Scholar]
- Pivsa-Art, S.; Tong-ngok, T.; Junngam, S.; Wongpajan, R.; Pivsa-Art, W. Synthesis of poly(D-lactic acid) using a 2-steps direct polycondensation process. Energy Procedia 2013, 34, 604–609. [Google Scholar] [CrossRef]
- Robert, J.; Aubrecht, K. Ring-opening polymerization of lactide to form a biodegradable polymer. J. Chem. Educ. 2008, 85, 258. [Google Scholar] [CrossRef]
- Kadlecova, Z.; Baldi, L.; Hacker, D.; Wurm, F.; Klok, H. Comparative study on the in vitro cytotoxicity of linear, dendritic, and hyperbranched polylysine analogues. Biomacromolecules 2012, 13, 3127–3137. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Kawano, T.; Shiba, K.; Mori, T.; Katayama, Y.; Niidome, T. Structural advantage of dendritic poly(L-lysine) for gene delivery into cells. Bioorg. Med. Chem. 2007, 15, 526–532. [Google Scholar] [CrossRef]
- Ohsaki, M.; Okuda, T.; Wada, A.; Hirayama, T.; Niidome, T.; Aoyagi, H. In vitro gene transfection using dendritic poly(L-lysine). Bioconjug. Chem. 2002, 13, 510–517. [Google Scholar] [CrossRef]
- Kadlecova, Z.; Rajendra, Y.; Matasci, M.; Baldi, L.; Hacker, D.; Wurm, F.; Klok, H. DNA delivery with hyperbranched polylysine: A comparative study with linear and dendritic polylysine. J. Control. Release 2013, 169, 276–288. [Google Scholar] [CrossRef]
- Zu, G.; Liu, M.; Zhang, K.; Hong, S.; Dong, J.; Cao, Y.; Jiang, B.; Luo, L.; Pei, R. Functional hyperbranched polylysine as potential contrast agent probes for magnetic resonance imaging. Biomacromolecules 2016, 17, 2302–2308. [Google Scholar] [CrossRef]
- Wang, H.; Shi, H.; Yin, S. Polyamidoamine dendrimers as gene delivery carriers in the inner ear: How to improve transfection efficiency. Exp. Med. 2011, 2, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.; Rhim, H.; Lee, S.; Ko, K.; Han, J.; Choi, J. Apoptin gene delivery by the functionalized polyamidoamine dendrimer derivatives induces cell death of U87-MG glioblastoma cells. J. Pharm. Sci. 2017, 106, 1618–1633. [Google Scholar] [CrossRef] [PubMed]
- Shadrack, D.M.; Mubofu, E.B.; Nyandoro, S. Synthesis of polyamidoamine dendrimer for encapsulating tetramethylscutellarein for potential bioactivity enhancement. Int. J. Mol. Sci. 2015, 16, 26363–26377. [Google Scholar] [CrossRef]
- Gupta, P.; Elkins, C.; Long, T.; Wilkes, G. Electrospinning of linear homopolymers of poly(methyl methacrylate): Exploring relationships between fiber formation, viscosity, molecular weight and concentration in a good solvent. Polymer 2005, 46, 4799–4810. [Google Scholar] [CrossRef]
- Du, F.; Fischer, J.; Winey, K. Coagulation method for preparing single-walled carbon nanotube/poly(methyl methacrylate) composites and their modulus, electrical conductivity, and thermal stability. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 3333–3338. [Google Scholar] [CrossRef]
- Glusker, D.; Stiles, E.; Yoncoskie, B. The mechanism of the anionic polymerization of methyl methacrylate. I. Quantitative determination of active chains using radioactive terminators. J. Polym. Sci. 1961, 49, 297–313. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef] [Green Version]
- Uhrig, D.; Mays, J. Experimental techniques in high-vacuum anionic polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6179–6222. [Google Scholar] [CrossRef] [Green Version]
- Kiani, K.; Hill, D.; Rasoul, F.; Whittaker, M.; Rintoul, L. Raft mediated surface grafting of t-butyl acrylate onto an ethylene–propylene copolymer initiated by gamma radiation. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1074–1083. [Google Scholar] [CrossRef]
- Nicolay, R.; Kwak, Y.; Matyjaszewski, K. A Green Route to well-defined high-molecular-weight (Co)polymers using ARGET ATRP with alkyl pseudohalides and copper catalysis. J. Ger. Chem. Soc. 2010, 49, 541–544. [Google Scholar] [CrossRef]
- Jäger, M.; Schubert, S.; Ochrimenko, S.; Fischer, D.; Schubert, U. Branched and linear poly(ethylene imine)-based conjugates: Synthetic modification, characterization, and application. Chem. Soc. Rev. 2012, 41, 4755–4767. [Google Scholar]
- Brissault, B.; Kichler, A.; Guis, C.; Leborgne, C.; Danos, O.; Cheradame, H. Synthesis of linear polyethylenimine derivatives for DNA transfection. Bioconjug. Chem. 2003, 14, 581–587. [Google Scholar] [CrossRef]
- Von Harpe, A.; Petersen, H.; Li, Y.; Kissel, T. Characterization of commercially available and synthesized polyethylenimines for gene delivery. J. Control. Release 2000, 69, 309–322. [Google Scholar] [CrossRef]
- Polyplus Transfection®, P. jetPEI® DNA transfection, HTS application. Available online: https://www.polyplus-transfection.com/products/jetpei/ (accessed on 23 April 2019).
- Kitchell, J.; Wise, D. Poly(lactic/glycolic acid) Biodegradable Drug—Polymer Matrix Systems. In Methods in Enzymology; Elsevier Inc.: Amsterdam, The Netherlands, 1985; Volume 112, pp. 436–448. [Google Scholar]
- Wang, Y.; Qu, W.; Choi, S. FDA’s regulatory science program for generic PLA/ PLGA-based drug products. Am. Pharm. Rev. 2016, 19, 5–9. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/188841-FDA-s-Regulatory-Science-Program-for-Generic-PLA-PLGA-Based-Drug-Products/ (accessed on 23 April 2019).
- Oh, J. Polylactide (PLA)-based amphiphilic block copolymers: Synthesis, self-assembly, and biomedical applications. Soft Matter 2011, 7, 5096–5108. [Google Scholar] [CrossRef]
- Squire, M.; Ludwig, B.; Thompson, J.; Jagodzinski, J.; Hall, D.; Andes, D. Premixed antibiotic bone cement: An in vitro comparison of antimicrobial efficacy. J. Arthroplast. 2008, 23, 110–114. [Google Scholar] [CrossRef]
- Lazzari, S.; Moscatelli, D.; Codari, F.; Salmona, M.; Morbidelli, M.; Diomede, L. Colloidal stability of polymeric nanoparticles in biological fluids. J. Nanopart. Res. 2012, 14, 920. [Google Scholar] [CrossRef] [Green Version]
- Cedervall, T.; Lynch, I.; Foy, M.; Berggård, T.; Donnelly, S.; Cagney, G.; Linse, S.; Dawson, K. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew. Chem. Int. Ed. Engl. 2007, 46, 5754–5756. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Hall, J.; McLeland, C.; Dobrovolskaia, M.; McNeil, S. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, H.; Nagasaki, Y.; Kataoka, K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 2003, 55, 403–419. [Google Scholar] [CrossRef]
- Suk, J.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.E.; Peppas, N. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Bisht, S.; Maitra, A. Dextran-doxorubicin/chitosan nanoparticles for solid tumor therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, K.; Tomiyama, N.; Nakashima, M.; Ohya, Y.; Ichikawa, M.; Ouchi, T.; Kanematsu, T. Antitumor activity of dextran derivatives immobilizing platinum complex (II). Anticancer Drugs 2000, 11, 33–38. [Google Scholar] [CrossRef]
- Dhaneshwar, S.; Kandpal, M.; Gairola, N.; Kadam, S. Dextran: A promising macromolecular drug carrier. Indian J. Pharm. Sci. 2006, 68, 705–714. [Google Scholar] [CrossRef]
- Lam, W.; Leung, C.; Chan, H.; Fong, W. Toxicity and DNA binding of dextran-doxorubicin conjugates in multidrug-resistant KB-V1 cells: Optimization of dextran size. Anticancer Drugs 2000, 11, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Madhunapantula, S.; Robertson, G. Toxicological considerations when creating nanoparticle-based drugs and drug delivery systems. Expert Opin. Drug Metab. Toxicol. 2012, 8, 47–69. [Google Scholar] [CrossRef]
- Brooks, D.; Okeefe, P.; Buncke, H. Dextran-induced acute renal failure after microvascular muscle transplantation. Plast. Reconstr. Surg. 2001, 108, 2057–2060. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Torchilin, V.; Kennel, S.; Huang, L. Use of N-terminal modified poly(L-lysine)-antibody conjugate as a carrier for targeted gene delivery in mouse lung endothelial cells. Bioconjug. Chem. 1992, 3, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Liu, F.; Kim, J.; Choi, Y.; Park, J.; Kim, S. Polyethylene glycol-grafted poly-L-lysine as polymeric gene carrier. J. Control. Release 1998, 54, 39–48. [Google Scholar] [CrossRef]
- Guo, L.; Chen, B.; Liu, R.; Xia, G.; Wang, Y.; Li, X.; Wei, C.; Wang, X.; Jiang, H. Biocompatibility assessment of polyethylene glycol-poly L-lysine-poly lactic-co-glycolic acid nanoparticles in vitro and in vivo. J. Nanosci. Nanotechnol. 2015, 15, 3710–3719. [Google Scholar] [CrossRef] [PubMed]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Godbey, W.; Wu, K.; Mikos, A. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Kunath, K.; von Harpe, A.; Fischer, D.; Petersen, H.; Bickel, U.; Voigt, K.; Kissel, T. Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: Comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. J. Control. Release 2003, 89, 113–125. [Google Scholar] [CrossRef]
- Kafil, V.; Omidi, Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in a431 cells. Bioimpacts 2011, 1, 23–30. [Google Scholar] [PubMed]
- Fischer, D.; Bieber, T.; Li, Y.; Elsässer, H.; Kissel, T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: Effect of molecular weight on transfection efficiency and cytotoxicity. Pharm. Res. 1999, 16, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Godbey, W.; Wu, K.; Mikos, A. Poly(ethylenimine)-mediated gene delivery affects endothelial cell function and viability. Biomaterials 2001, 22, 471–480. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Hong, J.; Park, J.; Huh, K.; Chung, H.; Kwon, I.; Jeong, S. PEGylated polyethylenimine for in vivo local gene delivery based on lipiodolized emulsion system. J. Control. Release 2004, 99, 167–176. [Google Scholar] [CrossRef]
- Song, H.; Wang, G.; He, B.; Li, L.; Li, C.; Lai, Y.; Xu, X.; Gu, Z. Cationic lipid-coated PEI/DNA polyplexes with improved efficiency and reduced cytotoxicity for gene delivery into mesenchymal stem cells. Int. J. Nanomed. 2012, 7, 4637–4648. [Google Scholar] [Green Version]
- Mintzer, M.; Grinstaff, M. Biomedical applications of dendrimers: A tutorial. Chem. Soc. Rev. 2011, 40, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.; El-Sayed, M. Dendrimers as carriers for delivery of chemotherapeutic agents. Chem. Rev. 2009, 109, 3141–3157. [Google Scholar] [CrossRef] [PubMed]
- Svenson, S.; Tomalia, D. Dendrimers in biomedical applications--reflections on the field. Adv. Drug Deliv. Rev. 2005, 57, 2106–2129. [Google Scholar] [CrossRef] [PubMed]
- Masotti, A.; Miller, M.; Celluzzi, A.; Rose, L.; Micciulla, F.; Hadoke, P.; Bellucci, S.; Caporali, A. Regulation of angiogenesis through the efficient delivery of microRNAs into endothelial cells using polyamine-coated carbon nanotubes. Nanomedicine 2016, 12, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Jevprasesphant, R.; Penny, J.; Jalal, R.; Attwood, D.; McKeown, N.; D’Emanuele, A. The influence of surface modification on the cytotoxicity of PAMAM dendrimers. Int. J. Pharm. 2003, 252, 263–266. [Google Scholar] [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- El-Sayed, M.; Ginski, M.; Rhodes, C.; Ghandehari, H. Transepithelial transport of poly(amidoamine) dendrimers across Caco-2 cell monolayers. J. Control. Release 2002, 81, 355–365. [Google Scholar] [CrossRef]
- Caster, J.; Yu, S.; Patel, A.; Newman, N.; Lee, Z.; Warner, S.; Wagner, K.; Roche, K.; Tian, X.; Min, Y.; et al. Effect of particle size on the biodistribution, toxicity, and efficacy of drug-loaded polymeric nanoparticles in chemoradiotherapy. Nanomedicine 2017, 13, 1673–1683. [Google Scholar] [CrossRef]
- Singh, R.; Ramarao, P. Accumulated polymer degradation products as effector molecules in cytotoxicity of polymeric nanoparticles. Toxicol. Sci. 2013, 136, 131–143. [Google Scholar] [CrossRef]
- Prencipe, G.; Tabakman, S.; Welsher, K.; Liu, Z.; Goodwin, A.; Zhang, L.; Henry, J.; Dai, H. PEG branched polymer for functionalization of nanomaterials with ultralong blood circulation. J. Am. Chem. Soc. 2009, 131, 4783–4787. [Google Scholar] [CrossRef]
- Forrest, M.; Kwon, G. Clinical developments in drug delivery nanotechnology. Adv. Drug Deliv. Rev. 2008, 60, 861–862. [Google Scholar] [CrossRef]
- Jain, A.; Swarnakar, N.; Godugu, C.; Singh, R.; Jain, S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials 2011, 32, 503–515. [Google Scholar] [CrossRef]
- Jain, R. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Shive, M.; Anderson, J. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar]
- Semete, B.; Booysen, L.; Lemmer, Y.; Kalombo, L.; Katata, L.; Verschoor, J.; Swai, H. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine 2010, 6, 662–671. [Google Scholar] [CrossRef]
- Savic, R.; Luo, L.; Eisenberg, A.; Maysinger, D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science 2003, 300, 615–618. [Google Scholar] [CrossRef]
- Bertram, J.; Jay, S.; Hynes, S.; Robinson, R.; Criscione, J.; Lavik, E. Functionalized poly(lactic-co-glycolic acid) enhances drug delivery and provides chemical moieties for surface engineering while preserving biocompatibility. Acta Biomater 2009, 5, 2860–2871. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.; Farokhzad, O. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Ilium, L.; Hunneyball, I.; Davis, S. The effect of hydrophilic coatings on the uptake of colloidal particles by the liver and by peritoneal macrophages. Int. J. Pharm. 1986, 29, 53–65. [Google Scholar] [CrossRef]
- Peracchia, M.; Fattal, E.; Desmaële, D.; Besnard, M.; Noël, J.; Gomis, J.; Appel, M.; d’Angelo, J.; Couvreur, P. Stealth PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J. Control. Release 1999, 60, 121–128. [Google Scholar] [CrossRef]
- Plard, J.-P.; Bazile, D. Comparison of the safety profiles of PLA50 and Me.PEG-PLA50 nanoparticles after single dose intravenous administration to rat. Colloids Surf. B Biointerfaces 1999, 16, 173–183. [Google Scholar] [CrossRef]
- Carrstensen, H.; Müller, R.; Müller, B. Particle size, surface hydrophobicity and interaction with serum of parenteral fat emulsions and model drug carriers as parameters related to RES uptake. Clin. Nutr. 1992, 11, 289–297. [Google Scholar] [CrossRef]
- Müllera, R.; Wallis, K.; Tröster, S.; Kreuter, J. In vitro characterization of poly(methyl-methaerylate) nanoparticles and correlation to their in vivo fate. J. Control. Release 1992, 20, 237–246. [Google Scholar]
- Norman, M.; Williams, P.; Illum, L. Human serum albumin as a probe for surface conditioning (opsonization) of block copolymer-coated microspheres. Biomaterials 1992, 13, 841–849. [Google Scholar] [CrossRef]
- Roser, M.; Fischer, D.; Kissel, T. Surface-modified biodegradable albumin nano- and microspheres. II: Effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur. J. Pharm. Biopharm. 1998, 46, 255–263. [Google Scholar] [CrossRef]
- Moghimi, S.; Hedeman, H.; Christy, N.; Illum, L.; Davis, S. Enhanced hepatic clearance of intravenously administered sterically stabilized microspheres in zymosan-stimulated rats. J. Leukoc. Biol. 1993, 54, 513–517. [Google Scholar] [CrossRef]
- Moghimi, S.; Hedeman, H.; Muir, I.; Illum, L.; Davis, S. An investigation of the filtration capacity and the fate of large filtered sterically-stabilized microspheres in rat spleen. Biochim. Biophys. Acta 1993, 1157, 233–240. [Google Scholar] [CrossRef]
- Kwon, G. Polymeric micelles for delivery of poorly water-soluble compounds. Crit. Rev. Drug Carr. Syst. 2003, 20, 357–403. [Google Scholar] [CrossRef]
- Jokerst, J.; Lobovkina, T.; Zare, R.; Gambhir, S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Faure, A.; Dufort, S.; Josserand, V.; Perriat, P.; Coll, J.; Roux, S.; Tillement, O. Control of the in vivo biodistribution of hybrid nanoparticles with different poly(ethylene glycol) coatings. Small 2009, 5, 2565–2575. [Google Scholar] [CrossRef]
- Jones, S.; Douglas, K.; Shields, A.; Merkel, O. Correlating quantitative tumor accumulation and gene knockdown using SPECT/CT and bioluminescence imaging within an orthotopic ovarian cancer model. Biomaterials 2018, 178, 183–192. [Google Scholar] [CrossRef]
- Wu, M.; Fan, J.; Gunning, W.; Ratnam, M. Clustering of GPI-anchored folate receptor independent of both cross-linking and association with caveolin. J. Memb. Biol. 1997, 159, 137–147. [Google Scholar] [CrossRef]
- Ross, J.; Chaudhuri, P.; Ratnam, M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer 1994, 73, 2432–2443. [Google Scholar] [CrossRef] [Green Version]
- Elnakat, H.; Ratnam, M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Gref, R.; Domb, A.; Quellec, P.; Blunk, T.; Müller, R.; Verbavatz, J.; Langer, R. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv. Drug Deliv. Rev. 1995, 16, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Veronese, F.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Banerjee, S.; Aher, N.; Patil, R.; Khandare, J. Poly(ethylene glycol)-prodrug conjugates: Concept, design, and applications. J. Drug Deliv. 2012. [Google Scholar] [CrossRef]
- Riber, C.; Zelikin, A. Recent advances in macromolecular prodrugs. Curr. Opin. Colloid Interface Sci. 2017, 31, 1–9. [Google Scholar] [CrossRef]
- Riley, T. The benefits and challenges of PEGylating small molecules. Pharm. Technol. 2008, 32. [Google Scholar]
- Zhang, X.; Wang, H.; Ma, Z.; Wu, B. Effects of pharmaceutical PEGylation on drug metabolism and its clinical concerns. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Scaramuzza, S.; Schiavon, O.; Mendichi, R.; Veronese, F. PEG-epirubicin conjugates with high drug loading. J. Bioact. Comtatible Polym. 2005, 20, 213–230. [Google Scholar] [CrossRef]
- Garay, R.; El-Gewely, R.; Armstrong, J.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.; Saifer, M.; Perez-Ruiz, F. PEG-uricase in the management of treatment-resistant gout and hyperuricemia. Adv. Drug Deliv. Rev. 2008, 60, 59–68. [Google Scholar] [CrossRef]
- Ganson, N.; Kelly, S.; Scarlett, E.; Sundy, J.; Hershfield, M. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. 2006, 8, R12. [Google Scholar] [CrossRef]
- Cheng, T.; Chen, B.; Chern, J.; Wu, M.; Roffler, S. Efficient clearance of poly(ethylene glycol)-modified immunoenzyme with anti-PEG monoclonal antibody for prodrug cancer therapy. Bioconjug. Chem. 2000, 11, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wilson, J.; Zhang, J.; Luo, Y. Characterization of potential degradation products in a PEGylating reagent 20 kDa monomethoxy polyethylene glycol propionaldehyde by RP-HPLC, APCI-MS and NMR. J. Pharm. Biomed. Anal. 2014, 89, 221–226. [Google Scholar] [CrossRef]
- Baker, D.; Levien, T. Irinotecan liposome injection. Hosp. Pharm. 2017, 52, 144–150. [Google Scholar] [CrossRef]
- Batra, J.; Robinson, J.; Mehner, C.; Hockla, A.; Miller, E.; Radisky, D.; Radisky, E. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS ONE 2012, 7, e50028. [Google Scholar] [CrossRef]
- Bailon, P.; Palleroni, A.; Schaffer, C.; Spence, C.; Fung, W.; Porter, J.; Ehrlich, G.; Pan, W.; Xu, Z.; Modi, M.; et al. Rational design of a potent, long-lasting form of interferon: A 40 kDa branched polyethylene glycol-conjugated interferon alpha-2a for the treatment of hepatitis C. Bioconjug. Chem. 2001, 12, 195–202. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, J.; Park, T. PEG grafted polylysine with fusogenic peptide for gene delivery: High transfection efficiency with low cytotoxicity. J. Control. Release 2002, 79, 283–291. [Google Scholar] [CrossRef]
- Foldvari, M.; Chen, D.; Nafissi, N.; Calderon, D.; Narsineni, L.; Rafiee, A. Non-viral gene therapy: Gains and challenges of non-invasive administration methods. J. Control. Release 2016, 240, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Bennet, D.; Kim, S. Polymer Nanoparticles for Smart Drug Delivery. In Application of Nanotechnology in Drug Delivery; IntechOpen: London, UK, 2014; pp. 257–310. [Google Scholar] [CrossRef]
- Page, D.; Cudmore, S. Innovations in oral gene delivery: Challenges and potentials. Drug Discov. Today 2001, 6, 92–101. [Google Scholar] [CrossRef]
- Chen, S.; Jones, D.; Fynan, E.; Farrar, G.; Clegg, J.; Greenberg, H.; Herrmann, J. Protective immunity induced by oral immunization with a rotavirus DNA vaccine encapsulated in microparticles. J. Virol. 1998, 72, 5757–5761. [Google Scholar] [PubMed]
- Liu, C.; Kou, Y.; Zhang, X.; Cheng, H.; Chen, X.; Mao, S. Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin. Drug Deliv. 2018, 15, 223–233. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yin, L.; Song, Y.; Tang, C.; Yin, C. Optimization of multifunctional chitosan-siRNA nanoparticles for oral delivery applications, targeting TNF-α silencing in rats. Acta Biomater. 2015, 17, 98–106. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Lee, S.; Revuri, V.; Hwang, Y.; Kang, S.; Lee, M.; Cho, S.; Cho, K.; Byun, Y.; Bae, Y.; et al. Oral delivery of a therapeutic gene encoding glucagon-like peptide 1 to treat high fat diet-induced diabetes. J. Control. Release 2017, 268, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, M.; Amiji, M. Oral IL-10 gene delivery in a microsphere-based formulation for local transfection and therapeutic efficacy in inflammatory bowel disease. Gene 2008, 15, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Zhang, Z.; Viennois, E.; Kang, Y.; Zhang, M.; Han, M.; Chen, J.; Merlin, D. combination therapy for ulcerative colitis: Orally targeted nanoparticles prevent mucosal damage and relieve inflammation. Theranostics 2016, 6, 2250–2266. [Google Scholar] [CrossRef]
- Kang, S.; Revuri, V.; Lee, S.; Cho, S.; Park, I.; Cho, K.; Bae, W.; Lee, Y. Oral siRNA delivery to treat colorectal liver metastases. ACS Nano 2017, 11, 10417–10429. [Google Scholar] [CrossRef]
- Scheuplein, R.; Blank, I. Permeability of the skin. Physiol. Rev. 1971, 51, 702–747. [Google Scholar] [CrossRef]
- Zhang, Z.; Tsai, P.; Ramezanli, T.; Michniak-Kohn, B. Polymeric nanoparticles-based topical delivery systems for the treatment of dermatological diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Samad, A.; Ullah, Z.; Alam, M.; Wais, M.; Shams, M. Transdermal drug delivery system: Patent reviews. Recent Pat. Drug Deliv. 2009, 3, 143–152. [Google Scholar] [CrossRef]
- Özbaş-Turan, S.; Akbuğa, J. Plasmid DNA-loaded chitosan/TPP nanoparticles for topical gene delivery. Drug Deliv. 2011, 18, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Youngblood, R.; Truong, N.; Segura, T.; Shea, L. It’s all in the delivery: designing hydrogels for cell and non-viral gene therapies. Mol. Ther. 2018, 26, 2087–2106. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, H.; Jia, Y.; Guo, Q.; Qu, Y.; Su, J.; Lu, X.; Zhao, Y.; Qian, Z. A novel gene delivery composite system based on biodegradable folate-poly (ester amine) polymer and thermosensitive hydrogel for sustained gene release. Sci. Rep. 2016, 6, 21402. [Google Scholar] [CrossRef] [Green Version]
- Keeney, M.; Onyiah, S.; Zhang, Z.; Tong, X.; Han, L.; Yang, F. Modulating polymer chemistry to enhance non-viral gene delivery inside hydrogels with tunable matrix stiffness. Biomaterials 2013, 34, 9657–9665. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, A.; Riazi-Rad, F.; Alimohammadian, M.; Gazori, T. Hydrogel nanoparticle encapsulated plasmid as a suitable gene delivery system. Cytol. Genet. 2015, 49, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cai, K.; Luo, Z.; Hu, R. Construction of Polyethyleneimine-β-cyclodextrin/pDNA multilayer structure for improved in situ gene transfection. Adv. Eng. Mater. 2010, 12, 18–25. [Google Scholar] [CrossRef]
- Xu, C.; Wu, Y.-L.; Li, Z.; Loh, X. Cyclodextrin-based sustained gene release systems: A supramolecular solution towards clinical applications. Mater. Chem. Front. 2019, 3, 181–192. [Google Scholar] [CrossRef]
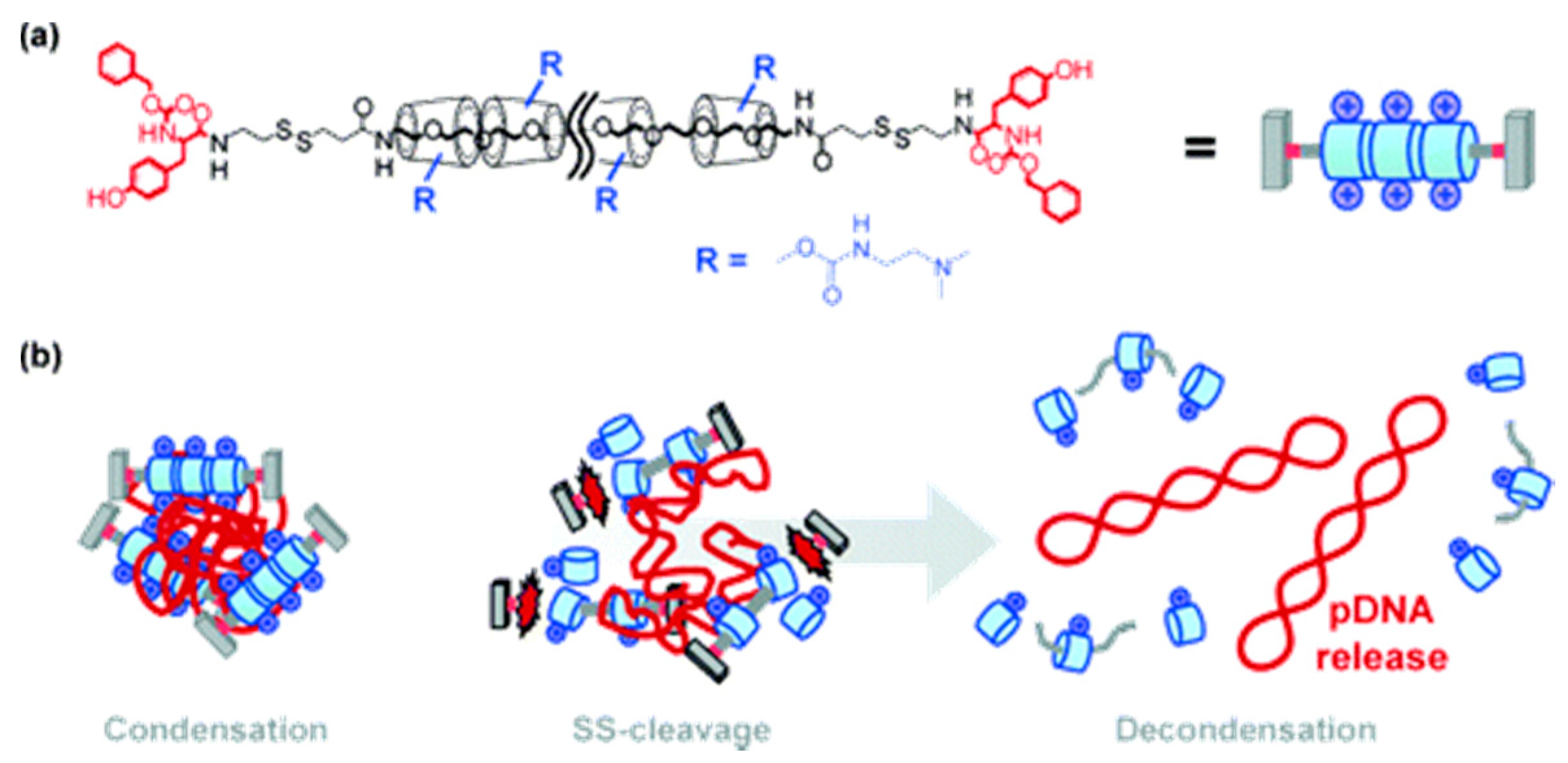
- Ooya, T.; Choi, H.; Yamashita, A.; Yui, N.; Sugaya, Y.; Kano, A.; Maruyama, A.; Akita, H.; Ito, R.; Kogure, K.; et al. Biocleavable polyrotaxane-plasmid DNA polyplex for enhanced gene delivery. J. Am. Chem. Soc. 2006, 128, 3852–3853. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Chua, M.; Li, Z.; Loh, X.; Wu, Y. Injectable Supramolecular hydrogels as delivery agents of Bcl-2 conversion gene for the effective shrinkage of therapeutic resistance tumors. Adv. Healthc. Mater. 2017, 6, 1700159. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, H.; Wang, C.; Li, Y.; Lu, W.; Chen, S.; Luo, J.; Jiang, Y.; Chen, J. Receptor-mediated, tumor-targeted gene delivery using folate-terminated polyrotaxanes. Mol. Pharm. 2012, 9, 1067–1076. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Green Nanotechnology Aspects | Practical Implication of the Aspect |
|---|---|
|
|
| |
| |
|
|
| |
| |
|
|
|
|
| |
|
|
|
| Method of Polymer Synthesis | Example | Average Particle Size (nm) | PDI | Ref. |
|---|---|---|---|---|
| Solvent evaporation method | PLA NPs | 200 | <0.1 | [48] |
| Solvent displacement method | PLGA NPs | 160–170 | <0.2 | [56] |
| Salting out | PTMC NPs | 184 ± 3 | 0.21 | [60] |
| Single emulsion | PTMC NPs | 334 ± 4 | 0.17 | [60] |
| Dialysis | PLA sphere NPs | 198.6 | 0.062 | [66] |
| Supercritical fluid technology (RESS) | Raloxifene NPs | 18.93 ± 3.73 | <0.1 | [19] |
| Supercritical fluid technology (RESOLV) | PHDFDA NPs | <50 | <0.25 | [77] |
| Recombinant technology | K8-ELP(1-60) NPs | <115 | <0.2 | [82] |
| Advantages | Challenges |
|---|---|
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, R.; Alwani, S.; Badea, I. Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications. Polymers 2019, 11, 745. https://doi.org/10.3390/polym11040745
Rai R, Alwani S, Badea I. Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications. Polymers. 2019; 11(4):745. https://doi.org/10.3390/polym11040745
Chicago/Turabian StyleRai, Raj, Saniya Alwani, and Ildiko Badea. 2019. "Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications" Polymers 11, no. 4: 745. https://doi.org/10.3390/polym11040745
APA StyleRai, R., Alwani, S., & Badea, I. (2019). Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications. Polymers, 11(4), 745. https://doi.org/10.3390/polym11040745