Rapid, Single-Step Protein Encapsulation via Flash NanoPrecipitation


Abstract
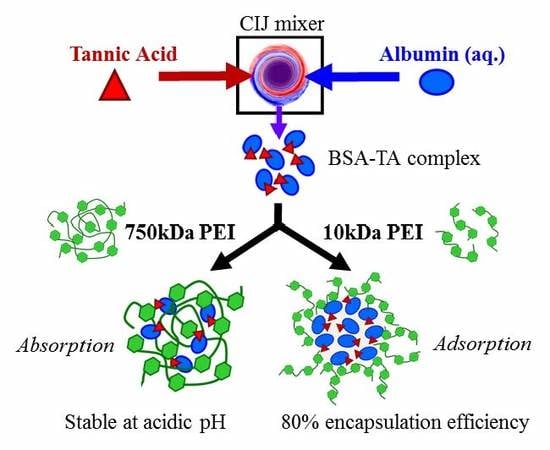
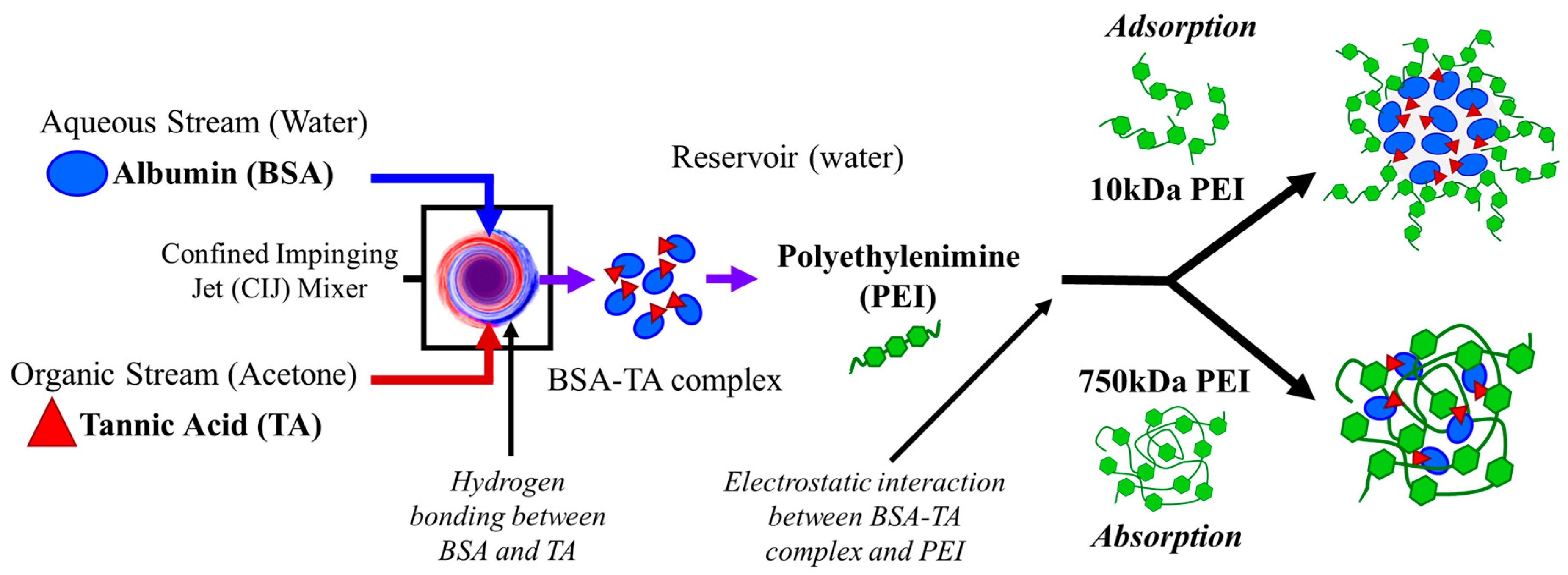
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoparticle Preparation
2.3. Nanoparticle Characterization
2.4. Protein Quantification
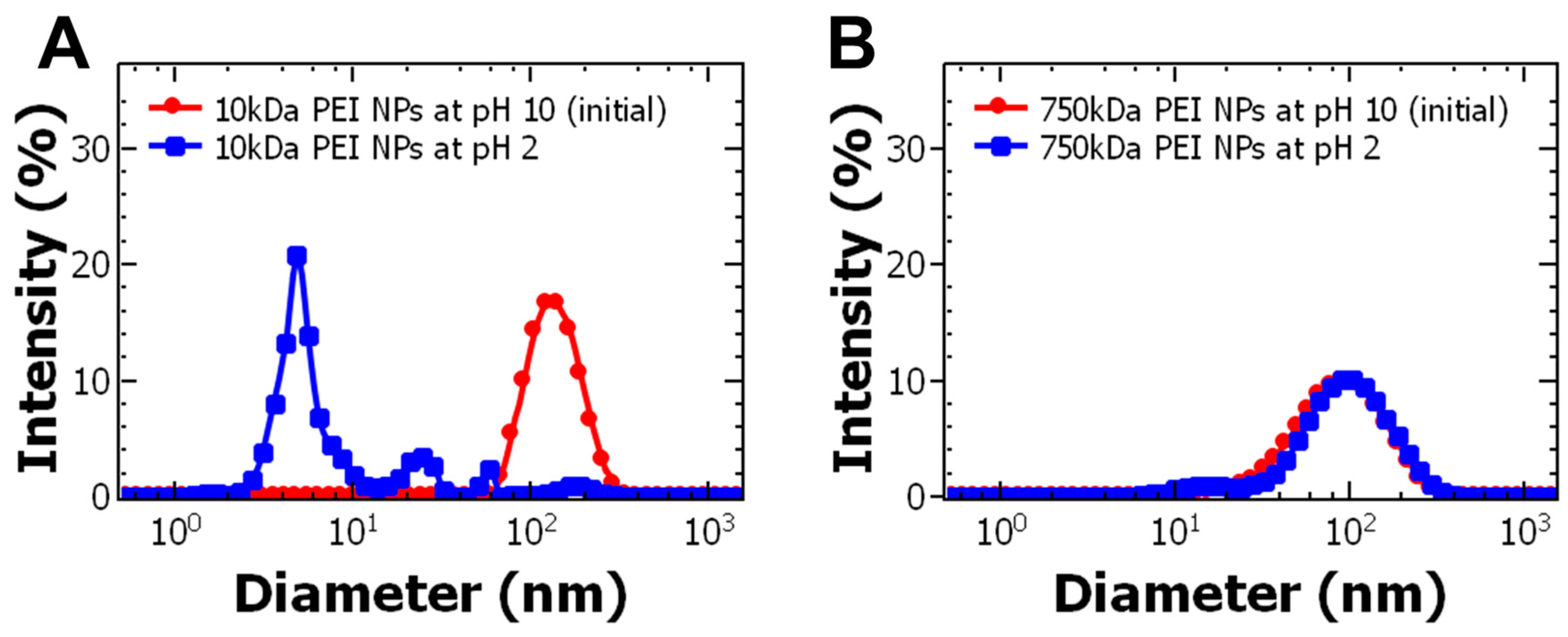
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tang, C.; Prud’homme, R.K. Targeted Theragnostic Nanoparticles Via Flash Nanoprecipitation: Principles of Material Selection. In Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development; Vauthier, C., Ponchel, G., Eds.; Springer: Basel, Switzerland, 2016; pp. 55–85. ISBN 978-3-319-41421-8. [Google Scholar] [CrossRef]
- Zhang, C.; Pansare, V.J.; Prud’homme, R.K.; Zhang, C.; Priestley, R.D. Flash nanoprecipitation of polystyrene nanoparticles. Soft Matter 2012, 8, 86–93. [Google Scholar] [CrossRef]
- Pustulka, K.M.; Wohl, A.R.; Lee, H.S.; Michel, A.R.; Han, J.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C.W. Flash nanoprecipitation: Particle structure and stability. Mol. Pharm. 2013, 10, 4367–4377. [Google Scholar] [CrossRef] [PubMed]
- Calo-Fernández, B.; Martínez-Hurtado, J.L. Biosimilars: Company Strategies to Capture Value from the Biologics Market. Pharmaceuticals 2012, 5, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol 2006, 17, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Challenges in Development of Nanoparticle-Based Therapeutics. AAPS J 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkerton, N.M.; Behar, L.; Hadri, K.; Amouroux, B.; Mingotaud, C.; Talham, D.R.; Chassaing, S.; Marty, J.-D. Ionic Flash NanoPrecipitation (iFNP) for the facile, one-step synthesis of inorganic–organic hybrid nanoparticles in water. Nanoscale 2017, 9, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, N.M.; Hadri, K.; Amouroux, B.; Behar, L.; Mingotaud, C.; Destarac, M.; Kulai, I.; Mazières, S.; Chassaing, S.; Marty, J.-D. Quench ionic flash nano precipitation as a simple and tunable approach to decouple growth and functionalization for the one-step synthesis of functional LnPO 4 -based nanoparticles in water. Chem. Commun. 2018, 54, 9438–9441. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, N.M.; Grandeury, A.; Fisch, A.; Brozio, J.; Riebesehl, B.U.; Prud’homme, R.K. Formation of Stable Nanocarriers by in Situ Ion Pairing during Block-Copolymer-Directed Rapid Precipitation. Mol. Pharm. 2013, 10, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Amin, D.; Messersmith, P.B.; Anthony, J.E.; Prud’homme, R.K. Polymer Directed Self-Assembly of pH-Responsive Antioxidant Nanoparticles. Langmuir 2015, 31, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Pagels, R.F.; Prud’homme, R.K. Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics. J. Control. Release 2015, 219, 519–535. [Google Scholar] [CrossRef]
- Markwalter, C.E.; Prud’homme, R.K. Design of a Small-Scale Multi-Inlet Vortex Mixer for Scalable Nanoparticle Production and Application to the Encapsulation of Biologics by Inverse Flash NanoPrecipitation. J. Pharm. Sci. 2018, 107, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Ren, Y.; Vandermark, J.; Archang, M.M.; Williford, J.-M.; Liu, H.-W.; Lee, J.; Wang, T.-H.; Mao, H.-Q. Continuous Production of Discrete Plasmid DNA-Polycation Nanoparticles Using Flash Nanocomplexation. Small 2016, 12, 6214–6222. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, J.P.; Robinson, W.B. Formation of Complexes between Protein and Tannic Acid. J. Agric. Food Chem. 1969, 17, 772–777. [Google Scholar] [CrossRef]
- Hagerman, A.E.; Butler, L.G. Determination of Protein in Tannin-Protein Precipitates. J. Agric. Food Chem. 1980, 28, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Rockholm, D.C.; Martin, J.S. Effects of surfactants, pH, and certain cations on precipitation of proteins by tannins. J. Chem. Ecol. 1985, 11, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Levit, S.L.; Stwodah, R.M.; Tang, C. Rapid, Room Temperature Nanoparticle Drying and Low-Energy Reconstitution via Electrospinning. J. Pharm. Sci. 2018, 107, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Nanocomposix’s Guide to Dynamic Light Scattering Measurement and Analysis 2015. Available online: http://50.87.149.212/sites/default/files/nanoComposix%20Guidelines%20for%20DLS%20Measurements%20and%20Analysis.pdf (accessed on 24 July 2019).
- Johnson, B.K.; Prud homme, R.K. Flash NanoPrecipitation of Organic Actives and Block Copolymers using a Confined Impinging Jets Mixer. Aust. J. Chem. 2003, 56, 1021–1024. [Google Scholar] [CrossRef]
- Meechai, N.; Jamieson, A.M.; Blackwell, J. Translational Diffusion Coefficients of Bovine Serum Albumin in Aqueous Solution at High Ionic Strength. J. Colloid Interface Sci. 1999, 218, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Prud’homme, R.K. Mechanism for Rapid Self-Assembly of Block Copolymer Nanoparticles. Phys. Rev. Lett. 2003, 91, 118302. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Sosa, C.L.; Pagels, R.F.; Priestley, R.D.; Prud’homme, R.K. Efficient preparation of size tunable PEGylated gold nanoparticles. J. Mater. Chem. B 2016, 4, 4813–4817. [Google Scholar] [CrossRef]
- Saad, W.S.; Prud’homme, R.K. Principles of nanoparticle formation by flash nanoprecipitation. Nano Today 2016, 11, 212–227. [Google Scholar] [CrossRef]
- Zhu, Z. Effects of amphiphilic diblock copolymer on drug nanoparticle formation and stability. Biomaterials 2013, 34, 10238–10248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Wang, L.; Riebe, M.; Tung, H.-H.; Prud’homme, R.K. Formulation and stability of itraconazole and odanacatib nanoparticles: Governing physical parameters. Mol. Pharm. 2009, 6, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Neubrand, A. Effects of Particle Size and Molecular Weight of Polyethylenimine on Properties of Nanoparticulate Silicon Dispersions. J. Am. Ceram. Soc. 2001, 84, 806–812. [Google Scholar] [CrossRef]
- Lindquist, G.M.; Stratton, R.A. The role of polyelectrolyte charge density and molecular weight on the adsorption and flocculation of colloidal silica with polyethylenimine. J. Colloid Interface Sci. 1976, 55, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Zhou, Y.; Sun, Y. Influence of pH and ionic strength on the steric mass-action model parameters around the isoelectric point of protein. Biotechnol. Prog. 2005, 21, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.A.; Miller, D.; Millard, P.; Basu, S.; Horkay, F.; Chandran, P.L. Unusual Salt and pH Induced Changes in Polyethylenimine Solutions. PLoS ONE 2016, 11, 1–20. [Google Scholar] [CrossRef]
- Jo, A.; Zhang, R.; Allen, I.C.; Riffle, J.S.; Davis, R.M. Design and Fabrication of Streptavidin-Functionalized, Fluorescently Labeled Polymeric Nanocarriers. Langmuir 2018, 34, 15783–15794. [Google Scholar] [CrossRef]
- Pagels, R.F.; Edelstein, J.; Tang, C.; Prud’homme, R.K. Controlling and Predicting Nanoparticle Formation by Block Copolymer Directed Rapid Precipitations. Nano Lett. 2018, 18, 1139–1144. [Google Scholar] [CrossRef]
- Zhu, Z.; Margulis-Goshen, K.; Magdassi, S.; Talmon, Y.; Macosko, C.W. Polyelectrolyte stabilized drug nanoparticles via flash nanoprecipitation: A model study with beta-carotene. J. Pharm. Sci. 2010, 99, 4295–4306. [Google Scholar] [CrossRef]
- Kosacheva, E.M.; Kudryavtsev, D.B.; Bakeeva, R.F.; Kuklin, A.I.; Islamov, A.K.; Kudryavtseva, L.A.; Sopin, V.F.; Konovalov, A.I. The aggregation of branched polyethylenimine and cationic surfactants in aqueous systems. Colloid J. 2006, 68, 713–720. [Google Scholar] [CrossRef]
- Zhang, M.; Xue, Y.-N.; Liu, M.; Zhuo, R.-X.; Huang, S.-W. Biocleavable Polycationic Micelles as Highly Efficient Gene Delivery Vectors. Nanoscale Res. Lett. 2010, 5, 1804–1811. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Wu, C. How Does DNA Complex with Polyethylenimine with Different Chain Lengths and Topologies in Their Aqueous Solution Mixtures? Macromolecules 2012, 45, 4346–4353. [Google Scholar] [CrossRef]
- Blocher, W.C.; Perry, S.L. Complex coacervate-based materials for biomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9. [Google Scholar] [CrossRef]
- Salt-Induced Charge Screening and Significant Conductivity Enhancement of Conducting Poly(3,4-ethylenedioxythiophene):Poly(styrenesulfonate)—Macromolecules (ACS Publications). Available online: https://pubs.acs.org/doi/10.1021/ma900327d (accessed on 14 February 2019).
- Donato, L.; Garnier, C.; Doublier, J.-L.; Nicolai, T. Influence of the NaCl or CaCl2 Concentration on the Structure of Heat-Set Bovine Serum Albumin Gels at pH 7. Biomacromolecules 2005, 6, 2157–2163. [Google Scholar] [CrossRef]
- Meng, F.; Zhong, Y.; Cheng, R.; Deng, C.; Zhong, Z. pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: Concept and recent advances. Nanomedicine 2014, 9, 487–499. [Google Scholar] [CrossRef]
- Lynn, D.M.; Amiji, M.M.; Langer, R. pH-Responsive Polymer Microspheres: Rapid Release of Encapsulated Material within the Range of Intracellular pH. Angew. Chem. Int. Ed. 2001, 40, 1707–1710. [Google Scholar] [CrossRef]
- Du, J.-Z.; Du, X.-J.; Mao, C.-Q.; Wang, J. Tailor-Made Dual pH-Sensitive Polymer–Doxorubicin Nanoparticles for Efficient Anticancer Drug Delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef]
- Kumar, V.; Prud’homme, R.K. Thermodynamic limits on drug loading in nanoparticle cores. J. Pharm. Sci. 2008, 97, 4904–4914. [Google Scholar] [CrossRef]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method. Drug Dev. Ind. Pharm. 1999, 25, 471–476. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Zeta Potential (mV) |
|---|---|
| BSA-TA precipitate | −13.1 ± 0.6 |
| BSA-TA with PS-b-PEG | −18.0 ± 3.0 |
| PEI | +34. 3 ± 4.2 |
| BSA-TA with PEI | +18.8 ± 0.9 |
| Sample | Initial | 7 days | ||||
|---|---|---|---|---|---|---|
| Zeta Potential (mV) | Diameter (nm) | PDI | Zeta Potential (mV) | Diameter (nm) | PDI | |
| 10 kDa PEI | +15.7 ± 1.0 | 153 ± 7 | 0.125 ± 0.022 | +14.4 ± 1.9 | 152 ± 1 | 0.055 ± 0.013 |
| 750 kDa PEI | +18.5 ± 1.3 | 107 ± 5 | 0.285 ± 0.004 | +18.5 ± 1.3 | 94 ± 3 | 0.259 ± 0.011 |
| Salt Added | Concentration (mM) | Ionic Strength (M) | Diameter (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|---|
| Initial 10 kDa PEI | 0 | 0 | 146 ± 2 | 0.125 ± 0.020 | 15.7 ± 2.0 |
| NaCl | 10 | 0.01 | 145 ± 2 | 0.065 ± 0.019 | 13.5 ± 2.6 |
| 30 | 0.03 | 139 ± 2 | 0.069 ± 0.007 | 13.4 ± 2.0 | |
| 100 | 0.1 | 194 ± 3 | 0.035 ± 0.023 | 14.3 ± 0.3 | |
| 300 | 0.3 | 290 ± 6 | 0.110 ± 0.024 | 11.8 ± 1.4 | |
| CaCl2 | 10 | 0.03 | 138 ± 6 | 0.351 ± 0.019 | 15.8 ± 1.0 |
| 100 | 0.3 | 188 ± 7 | 0.148 ± 0.022 | 16.1 ± 0.5 |
| Sample | Condition | Encapsulation Efficiency (EE%) | Drug Loading (DL%) |
|---|---|---|---|
| 10 kDa PEI NPs | no TA | 8% ± 3% | 1% ± 0% |
| with TA | 79% ± 7% | 13% ± 1% | |
| 750 kDa PEI NPs | no TA | 74% ± 6% | 12% ± 1% |
| with TA | 50% ± 10% | 8% ± 2% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levit, S.L.; Walker, R.C.; Tang, C. Rapid, Single-Step Protein Encapsulation via Flash NanoPrecipitation. Polymers 2019, 11, 1406. https://doi.org/10.3390/polym11091406
Levit SL, Walker RC, Tang C. Rapid, Single-Step Protein Encapsulation via Flash NanoPrecipitation. Polymers. 2019; 11(9):1406. https://doi.org/10.3390/polym11091406
Chicago/Turabian StyleLevit, Shani L., Rebecca C. Walker, and Christina Tang. 2019. "Rapid, Single-Step Protein Encapsulation via Flash NanoPrecipitation" Polymers 11, no. 9: 1406. https://doi.org/10.3390/polym11091406
APA StyleLevit, S. L., Walker, R. C., & Tang, C. (2019). Rapid, Single-Step Protein Encapsulation via Flash NanoPrecipitation. Polymers, 11(9), 1406. https://doi.org/10.3390/polym11091406