Acetoacetate Based Thermosets Prepared by Dual-Michael Addition Reactions



Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
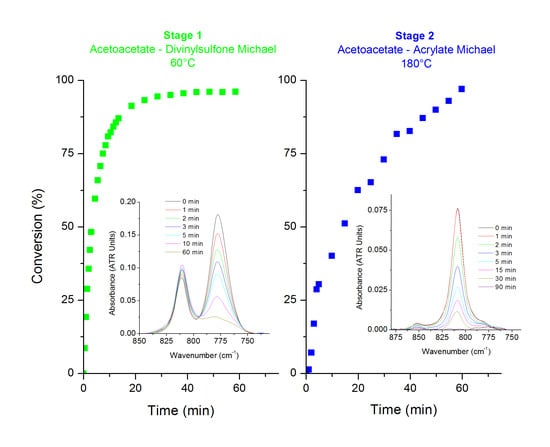
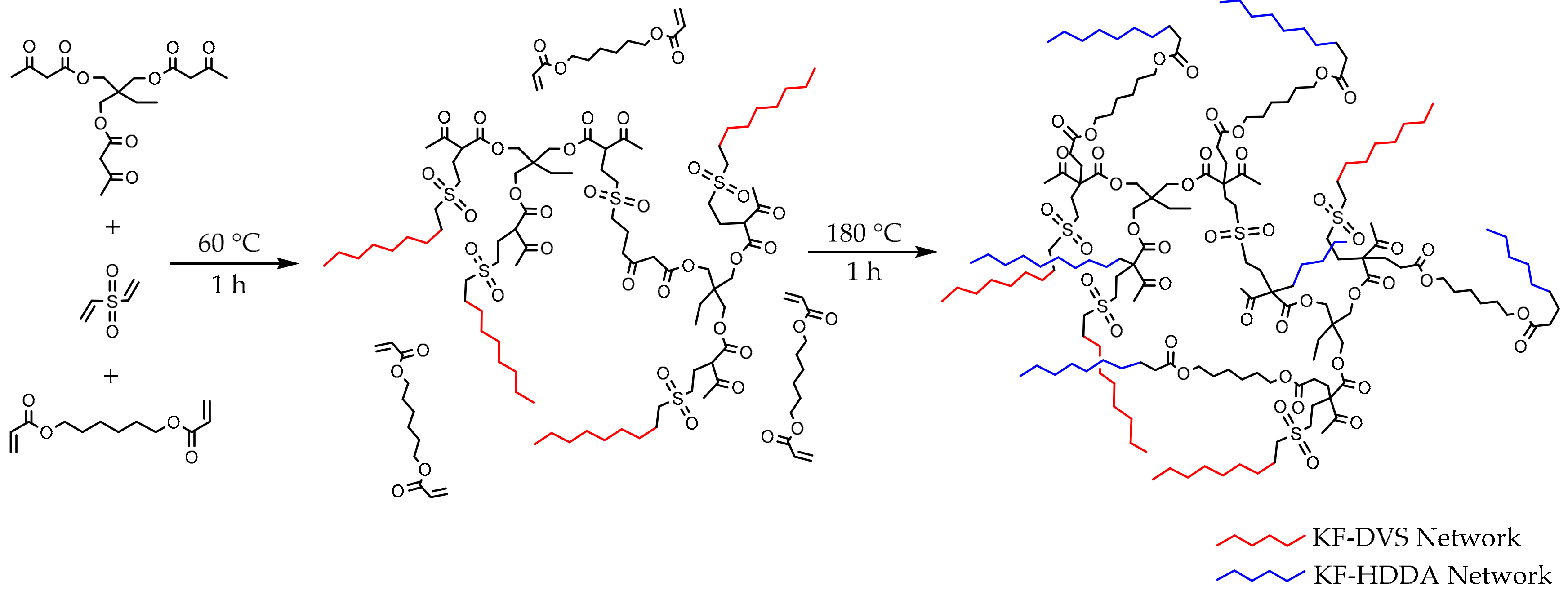
3.1. KF-DVS Pure System (Stage 1)
3.2. Dual System KF-DVS-HDDA (2:1:1 Molar Ratio)
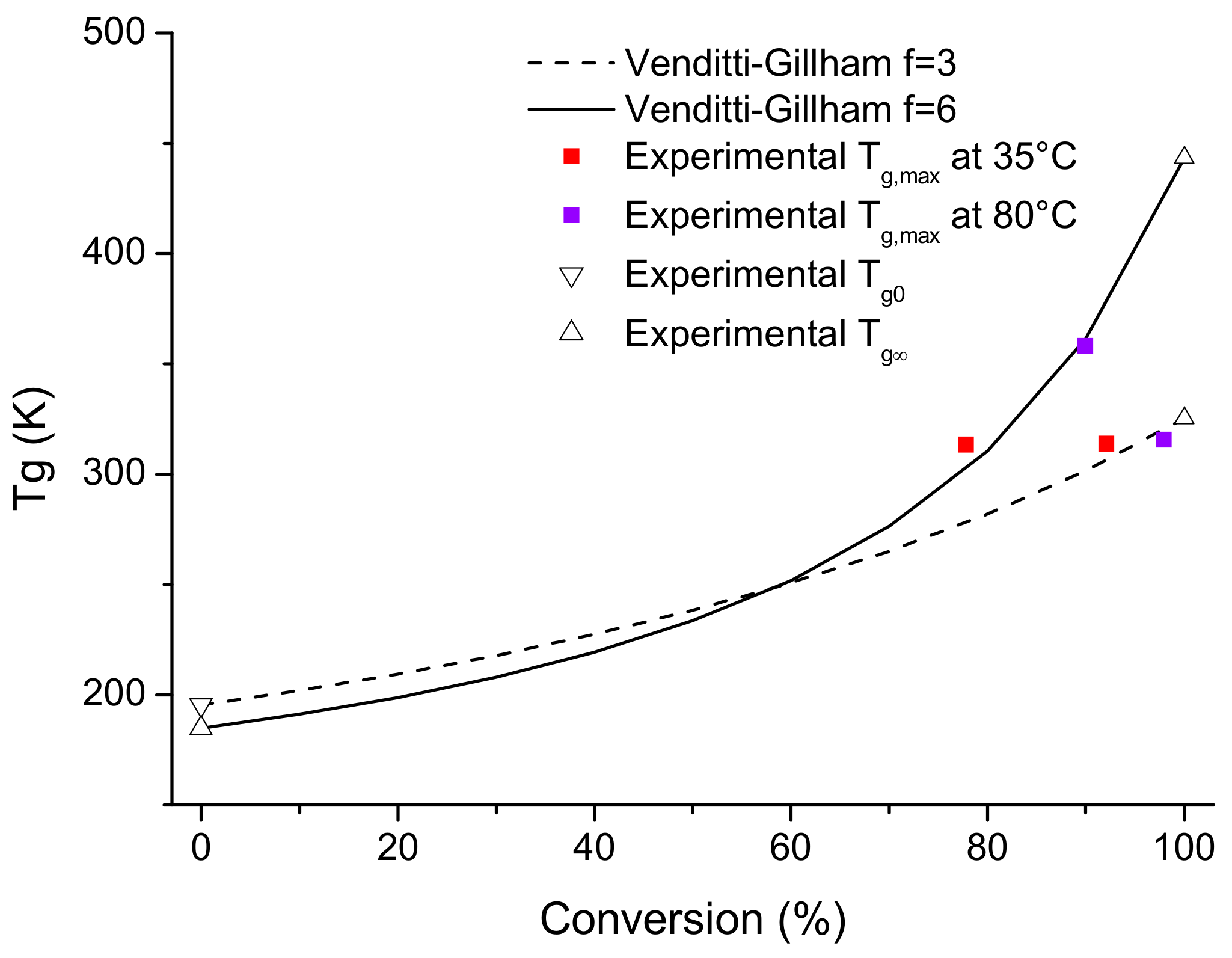
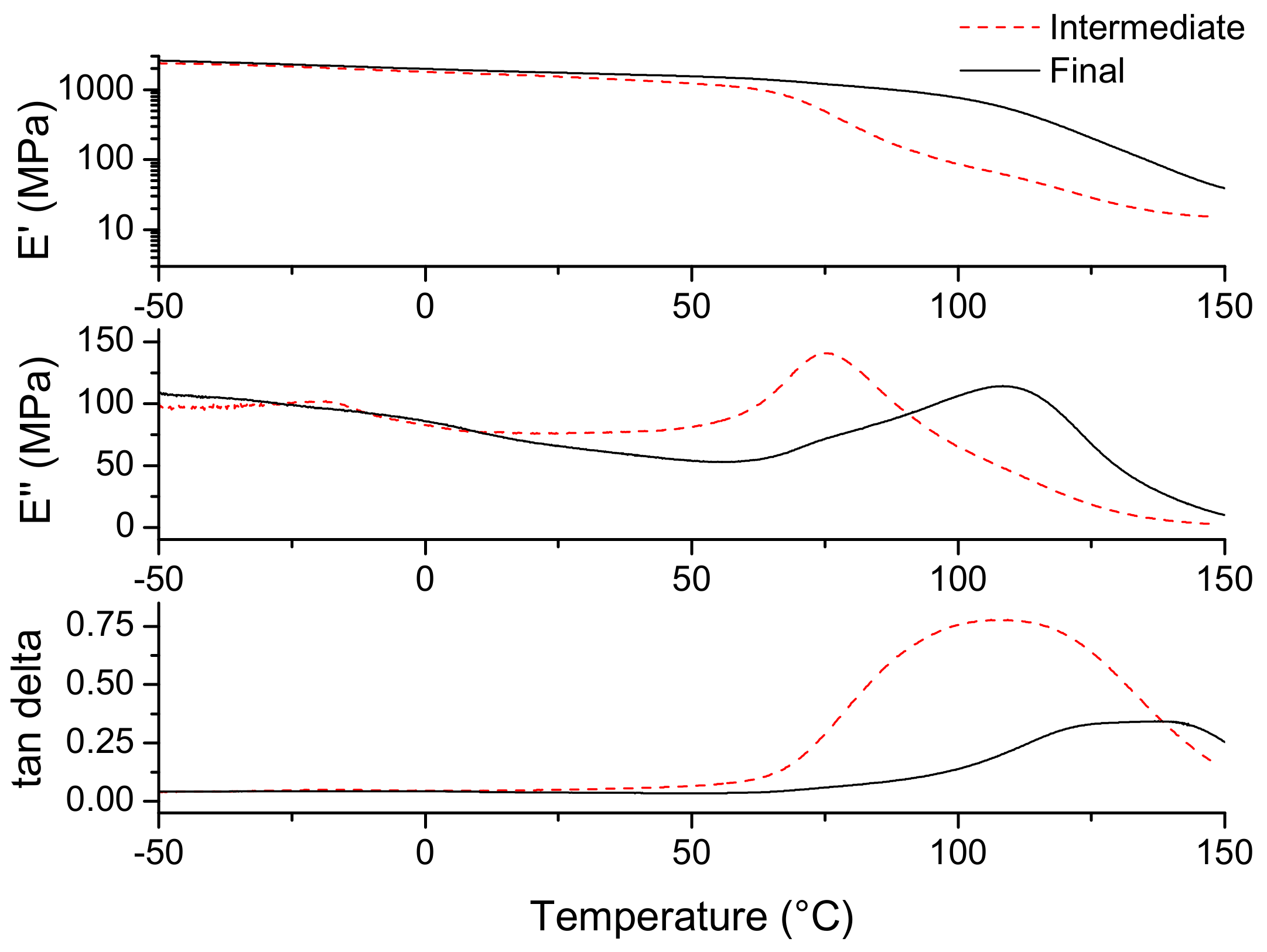
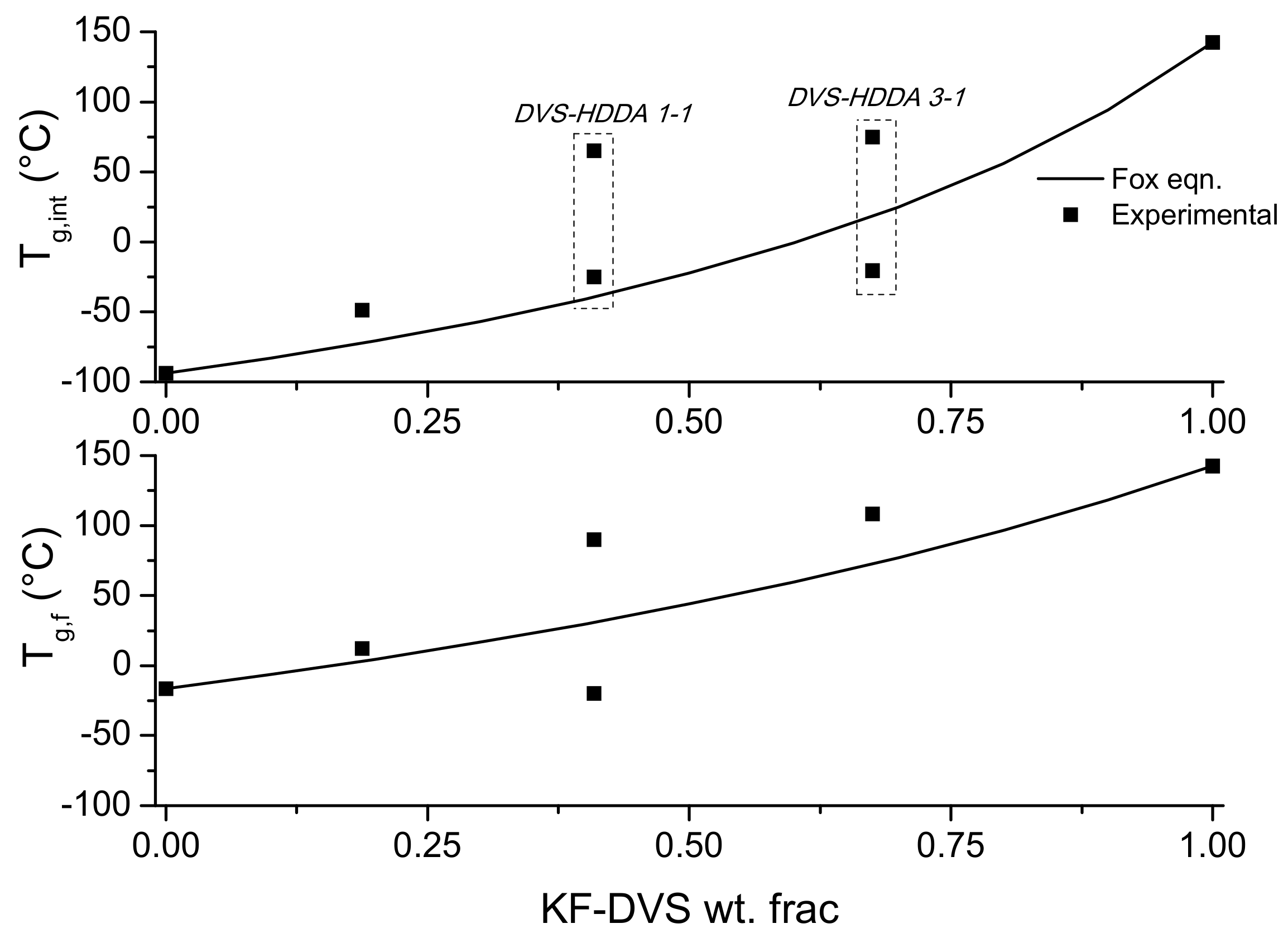
3.3. Material Properties
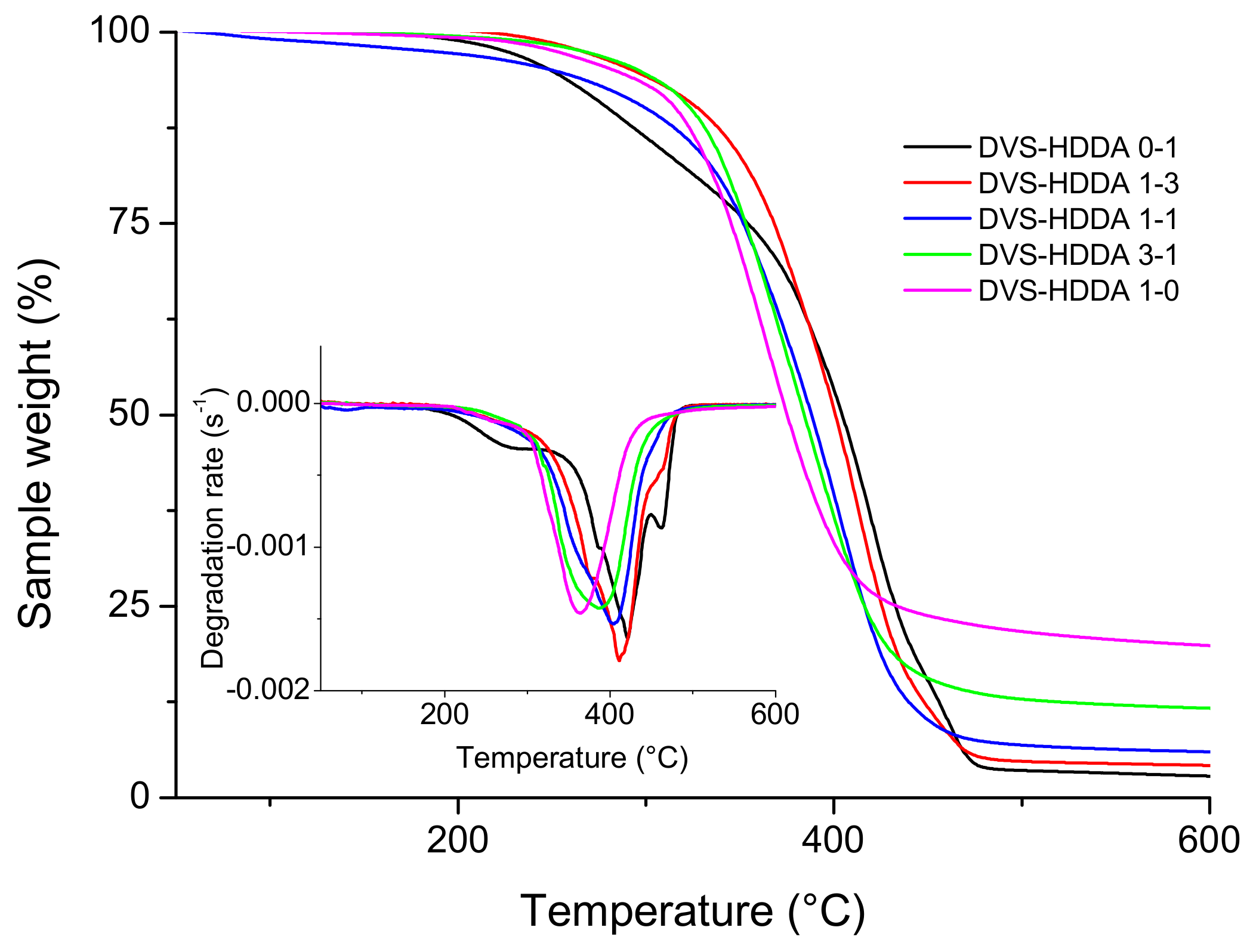
3.4. Thermal Degradation of Materials
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ramis, X.; Fernández-Francos, X.; De La Flor, S.; Ferrando, F.; Serra, À. Click-based dual-curing thermosets and their applications. In Thermosets 2nd edition: Structure, Properties and Application; Guo, Q., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780081010280. [Google Scholar]
- Konuray, O.; Fernández-Francos, X.; Ramis, X.; Serra, À. State of the Art in Dual-Curing Acrylate Systems. Polymers (Basel). 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.P.; Cramer, N.B.; Gaipa, J.C.; McBride, M.K.; Matherly, E.M.; McLeod, R.R.; Shandas, R.; Bowman, C.N. Two-Stage Reactive Polymer Network Forming Systems. Adv. Funct. Mater. 2012, 22, 1502–1510. [Google Scholar] [CrossRef]
- Retailleau, M.; Ibrahim, A.; Croutxé-Barghorn, C.; Allonas, X.; Ley, C.; Le Nouen, D. One-Pot Three-Step Polymerization System Using Double Click Michael Addition and Radical Photopolymerization. ACS Macro Lett. 2015, 4, 1327–1331. [Google Scholar] [CrossRef]
- Peng, H.; Nair, D.P.; Kowalski, B.A.; Xi, W.; Gong, T.; Wang, C.; Cole, M.; Cramer, N.B.; Xie, X.; McLeod, R.R.; et al. High performance graded rainbow holograms via two-stage sequential orthogonal thiol-click chemistry. Macromolecules 2014, 47, 2306–2315. [Google Scholar] [CrossRef]
- Konuray, O.; Areny, N.; Morancho, J.M.; Fernández-Francos, X.; Serra, À.; Ramis, X. Preparation and characterization of dual-curable off-stoichiometric amine-epoxy thermosets with latent reactivity. Polymer (Guildf). 2018, 146, 42–52. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chemie - Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. “Click” chemistry in polymer and material science: An Update. Macromol. Rapid Commun. 2008, 29, 952–981. [Google Scholar] [CrossRef]
- Tunca, U. Orthogonal multiple click reactions in synthetic polymer chemistry. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 3147–3165. [Google Scholar] [CrossRef]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Ramis, X. Curing kinetics and characterization of dual-curable thiol-acrylate-epoxy thermosets with latent reactivity. React. Funct. Polym. 2018, 122, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Chatani, S.; Wang, C.; Podgórski, M.; Bowman, C.N. Triple Shape Memory Materials Incorporating Two Distinct Polymer Networks Formed by Selective Thiol–Michael Addition Reactions. Macromolecules 2014, 47, 4949–4954. [Google Scholar] [CrossRef]
- Peng, H.; Wang, C.; Xi, W.; Kowalski, B.A.; Gong, T.; Xie, X.; Wang, W.; Nair, D.P.; McLeod, R.R.; Bowman, C.N. Facile image patterning via sequential thiol-Michael/thiol-yne click reactions. Chem. Mater. 2014, 26, 6819–6826. [Google Scholar] [CrossRef]
- Jin, K.; Wilmot, N.; Heath, W.H.; Torkelson, J.M. Phase-Separated Thiol-Epoxy-Acrylate Hybrid Polymer Networks with Controlled Cross-Link Density Synthesized by Simultaneous Thiol-Acrylate and Thiol-Epoxy Click Reactions. Macromolecules 2016, 49, 4115–4123. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Serra, À.; Ramis, X. Sequential curing of amine-acrylate-methacrylate mixtures based on selective aza-Michael addition followed by radical photopolymerization. Eur. Polym. J. 2016, 84, 256–267. [Google Scholar] [CrossRef]
- González, G.; Fernández-Francos, X.; Serra, À.; Sangermano, M.; Ramis, X. Environmentally-friendly processing of thermosets by two-stage sequential aza-Michael addition and free-radical polymerization of amine–acrylate mixtures. Polym. Chem. 2015, 6, 6987–6997. [Google Scholar] [CrossRef]
- Retailleau, M.; Pierrel, J.; Ibrahim, A.; Croutxé-Barghorn, C.; Allonas, X. Sequenced click chemistry and photopolymerization: a new approach toward semi-interpenetrating polymer networks. Polym. Adv. Technol. 2017, 28, 491–495. [Google Scholar] [CrossRef]
- Konuray, A.O.; Liendo, F.; Fernández-Francos, X.; Serra, À.; Sangermano, M.; Ramis, X. Sequential curing of thiol-acetoacetate-acrylate thermosets by latent Michael addition reactions. Polymer (Guildf). 2017, 113, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Konuray, A.O.; Ruiz, A.; Morancho, J.M.; Salla, J.M.; Fernández-Francos, X.; Serra, À.; Ramis, X. Sequential dual curing by selective Michael addition and free radical polymerization of acetoacetate-acrylate-methacrylate mixtures. Eur. Polym. J. 2018, 98, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Moszner, N.; Rheinberger, V. Reaction behaviour of monomeric β-ketoesters, 4. Polymer network formation by Michael reaction of multifunctional acetoacetates with multifunctional acrylates. Macromol. Rapid Commun. 1995, 16, 135–138. [Google Scholar]
- Kim, Y.B.; Kim, H.K.; Nishida, H.; Endo, T. Synthesis and characterization of hyperbranched poly(β-ketoester) by the Michael addition. Macromol. Mater. Eng. 2004, 289, 923–926. [Google Scholar] [CrossRef]
- Williams, S.R.; Miller, K.M.; Long, T.E. Michael addition reaction kinetics of acetoacetates and acrylates for the formation of polymeric networks. Prog. React. Kinet. Mech. 2007, 32, 165–194. [Google Scholar] [CrossRef]
- Podgórski, M.; Chatani, S.; Bowman, C.N. Development of glassy step-growth thiol-vinyl sulfone polymer networks. Macromol. Rapid Commun. 2014, 35, 1497–1502. [Google Scholar] [CrossRef]
- Chatani, S.; Nair, D.P.; Bowman, C.N. Relative reactivity and selectivity of vinyl sulfones and acrylates towards the thiol–Michael addition reaction and polymerization. Polym. Chem. 2013, 4, 1048–1055. [Google Scholar] [CrossRef]
- Chatani, S.; Sheridan, R.J.; Podgórski, M.; Nair, D.P.; Bowman, C.N. Temporal control of thiol-click chemistry. Chem. Mater. 2013, 25, 3897–3901. [Google Scholar] [CrossRef]
- Venditti, R.A.; Gillham, J.K. A relationship between the glass transition temperature (Tg) and fractional conversion for thermosetting systems. J. Appl. Polym. Sci. 1997, 64, 3–14. [Google Scholar] [CrossRef]
- Jian, Y.; He, Y.; Sun, Y.; Yang, H.; Yang, W.; Nie, J. Thiol-epoxy/thiol-acrylate hybrid materials synthesized by photopolymerization. J. Mater. Chem. C 2013, 1, 4481–4489. [Google Scholar] [CrossRef]
- Kaya, N.U.; Onen, A.; Guvenilir, Y. Photopolymerization of acrylates by enzymatically synthesized PCL based macrophotoinitiator. Express Polym. Lett. 2017, 11, 493–503. [Google Scholar] [CrossRef]
- Lantean, S.; Roppolo, I.; Sangermano, M.; Fabrizio Pirri, C.; Chiappone, A. Development of New Hybrid Acrylic/Epoxy DLP-3D Printable Materials. Innovations 2018, 3, 29. [Google Scholar] [CrossRef]
- Sangermano, M.; Carbonaro, W.; Malucelli, G.; Priola, A. UV-cured interpenetrating acrylic-epoxy polymer networks: Preparation and characterization. Macromol. Mater. Eng. 2008, 293, 515–520. [Google Scholar] [CrossRef]
- Fox, G.T. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phs. Soc. 1952, 1, 123. [Google Scholar]
- Flores, M.; Fernández-Francos, X.; Ferrando, F.; Ramis, X.; Serra, À. Efficient impact resistance improvement of epoxy/anhydride thermosets by adding hyperbranched polyesters partially modified with undecenoyl chains. Polymer (Guildf). 2012, 53, 5232–5241. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer Chemical Structure | Abbreviation |
|---|---|
 | KF |
 | DVS |
 | HDDA |
 | 1-MI |
| Formulation | KF:DVS:HDDA Molar Ratio | Weight Fractions | |||
|---|---|---|---|---|---|
| KF | DVS | HDDA | 1-MI | ||
| DVS-HDDA 1-0 | 1:1:0 | 0.51 | 0.49 | 0 | 0.02 |
| DVS-HDDA 3-1 | 4:3:1 | 0.46 | 0.33 | 0.21 | 0.02 |
| DVS-HDDA 1-1 | 2:1:1 | 0.42 | 0.20 | 0.38 | 0.02 |
| DVS-HDDA 1-3 | 4:1:3 | 0.39 | 0.09 | 0.52 | 0.02 |
| DVS-HDDA 0-1 | 1:0:1 | 0.36 | 0 | 0.64 | 0.02 |
| Functionality | T (°C) | x (%) | t (min) c |
|---|---|---|---|
| 3 a | 35 | 92 | 300 |
| 80 | 98 | 180 | |
| 6 b | 35 | 77 | 240 |
| 80 | 90 | 90 |
| f | 3 | 6 |
| Tg0 (K) | 195.2 | 184.8 |
| Tg∞ (K) | 325.3 | 443.4 |
| ΔCp0 (J/g·K) | 0.619 | 0.666 |
| ΔCp∞ (J/g·K) | 0.396 | 0.243 |
| λ | 0.640 | 0.365 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konuray, O.; Fernández-Francos, X.; Ramis, X.; Serra, À. Acetoacetate Based Thermosets Prepared by Dual-Michael Addition Reactions. Polymers 2019, 11, 1408. https://doi.org/10.3390/polym11091408
Konuray O, Fernández-Francos X, Ramis X, Serra À. Acetoacetate Based Thermosets Prepared by Dual-Michael Addition Reactions. Polymers. 2019; 11(9):1408. https://doi.org/10.3390/polym11091408
Chicago/Turabian StyleKonuray, Osman, Xavier Fernández-Francos, Xavier Ramis, and Àngels Serra. 2019. "Acetoacetate Based Thermosets Prepared by Dual-Michael Addition Reactions" Polymers 11, no. 9: 1408. https://doi.org/10.3390/polym11091408
APA StyleKonuray, O., Fernández-Francos, X., Ramis, X., & Serra, À. (2019). Acetoacetate Based Thermosets Prepared by Dual-Michael Addition Reactions. Polymers, 11(9), 1408. https://doi.org/10.3390/polym11091408