Influence of Calcium Binding on Conformations and Motions of Anionic Polyamino Acids. Effect of Side Chain Length


Abstract
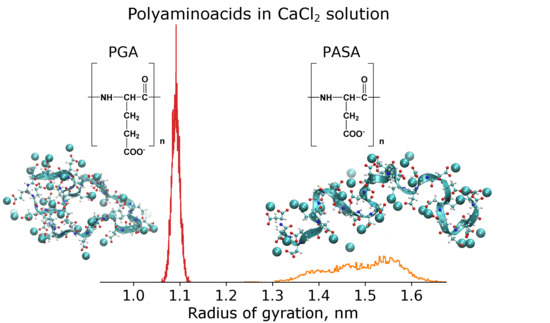
:
1. Introduction
2. Model Description
3. MD Simulation
3.1. MD Parameters
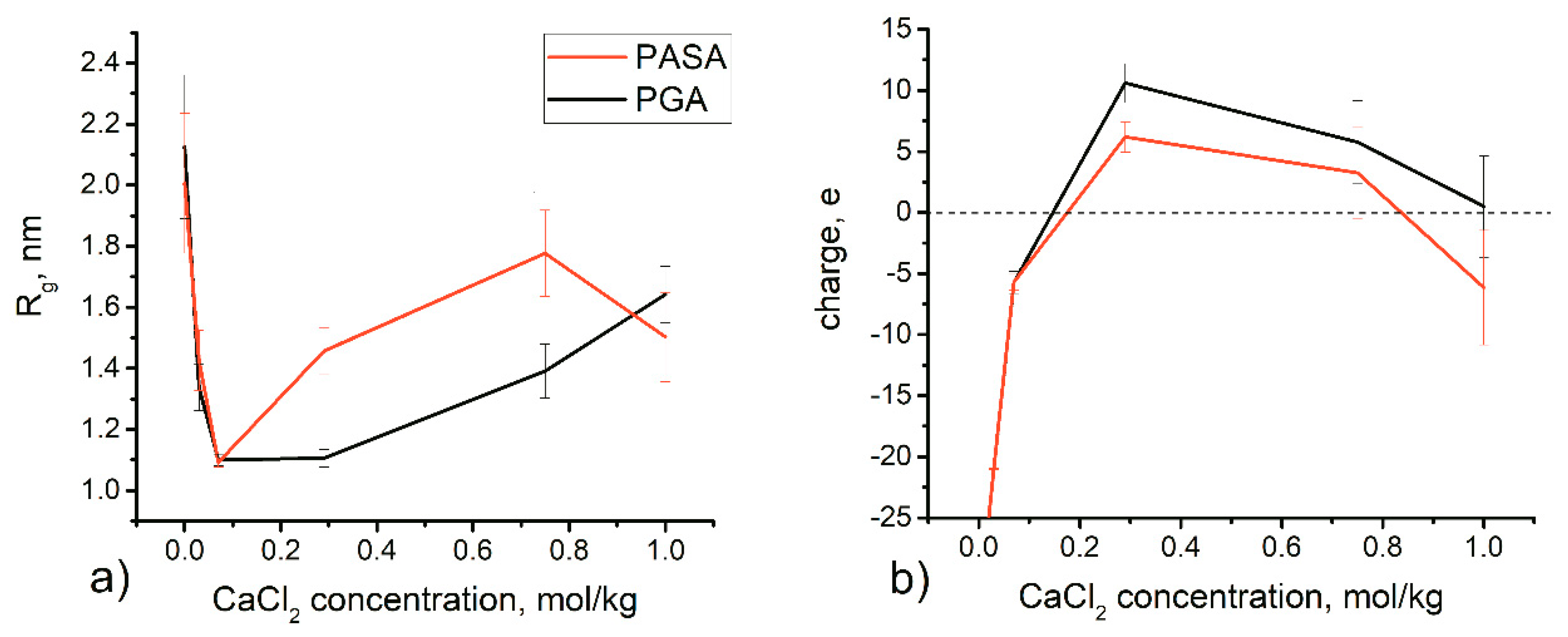
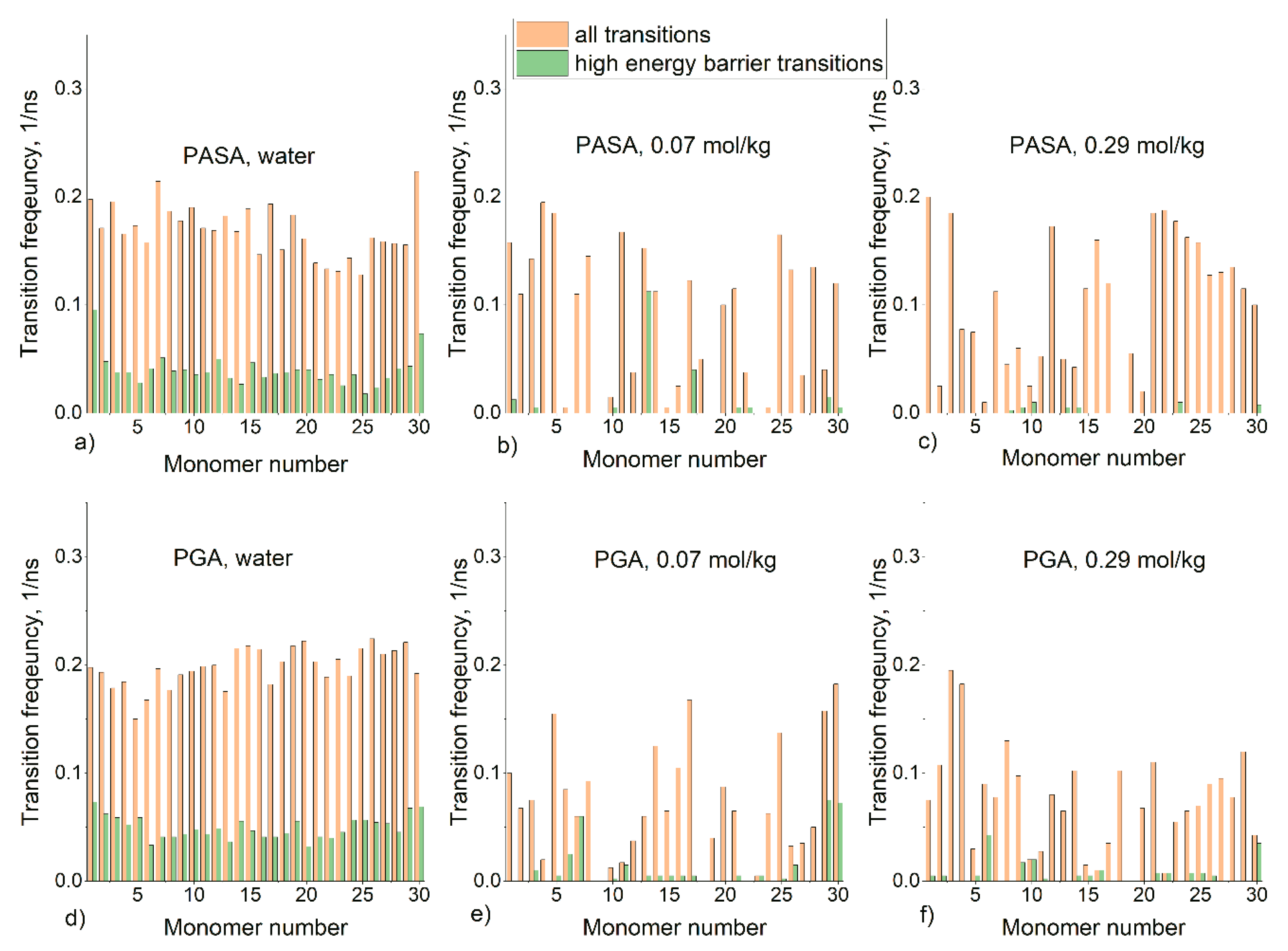
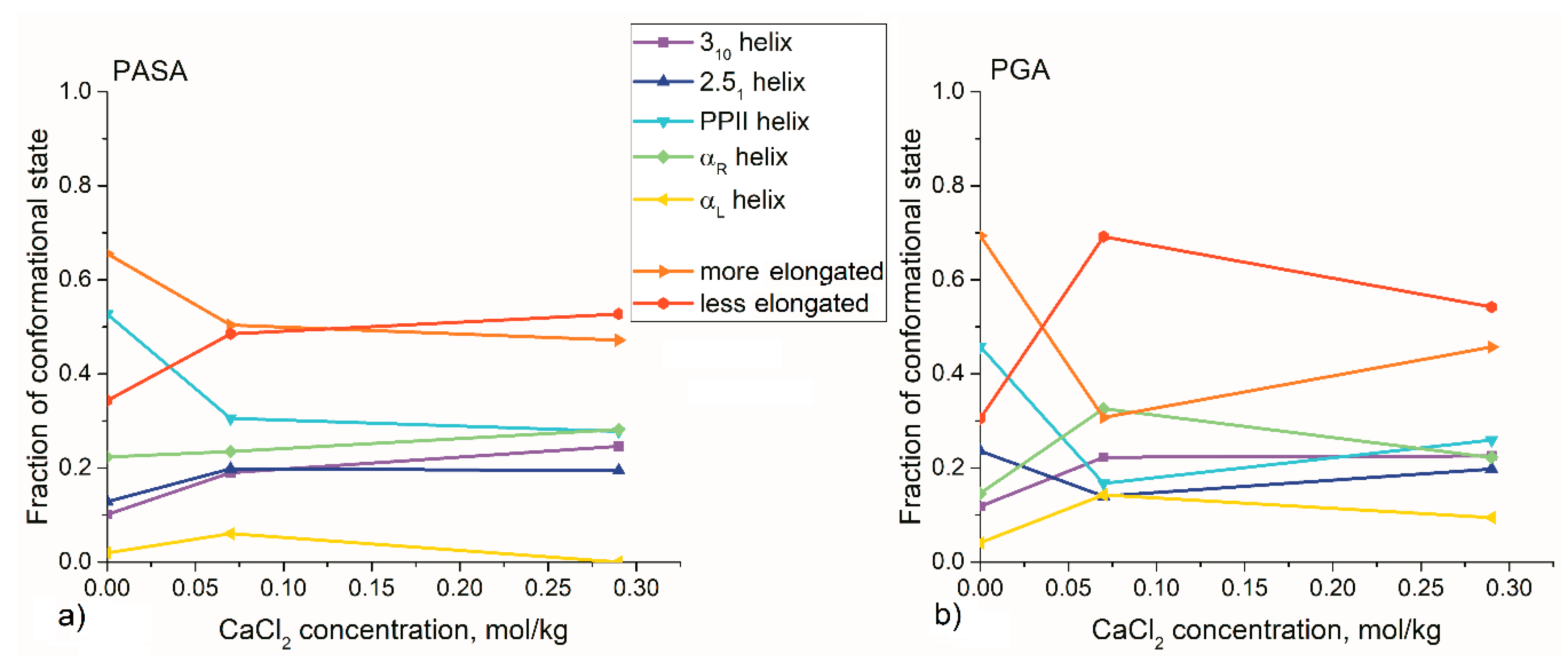
3.2. Results from Classical Unbiased MD Simulations
4. Hamiltonian Replica Exchange Simulation
4.1. Hamiltonian Replica Exchange Parameters
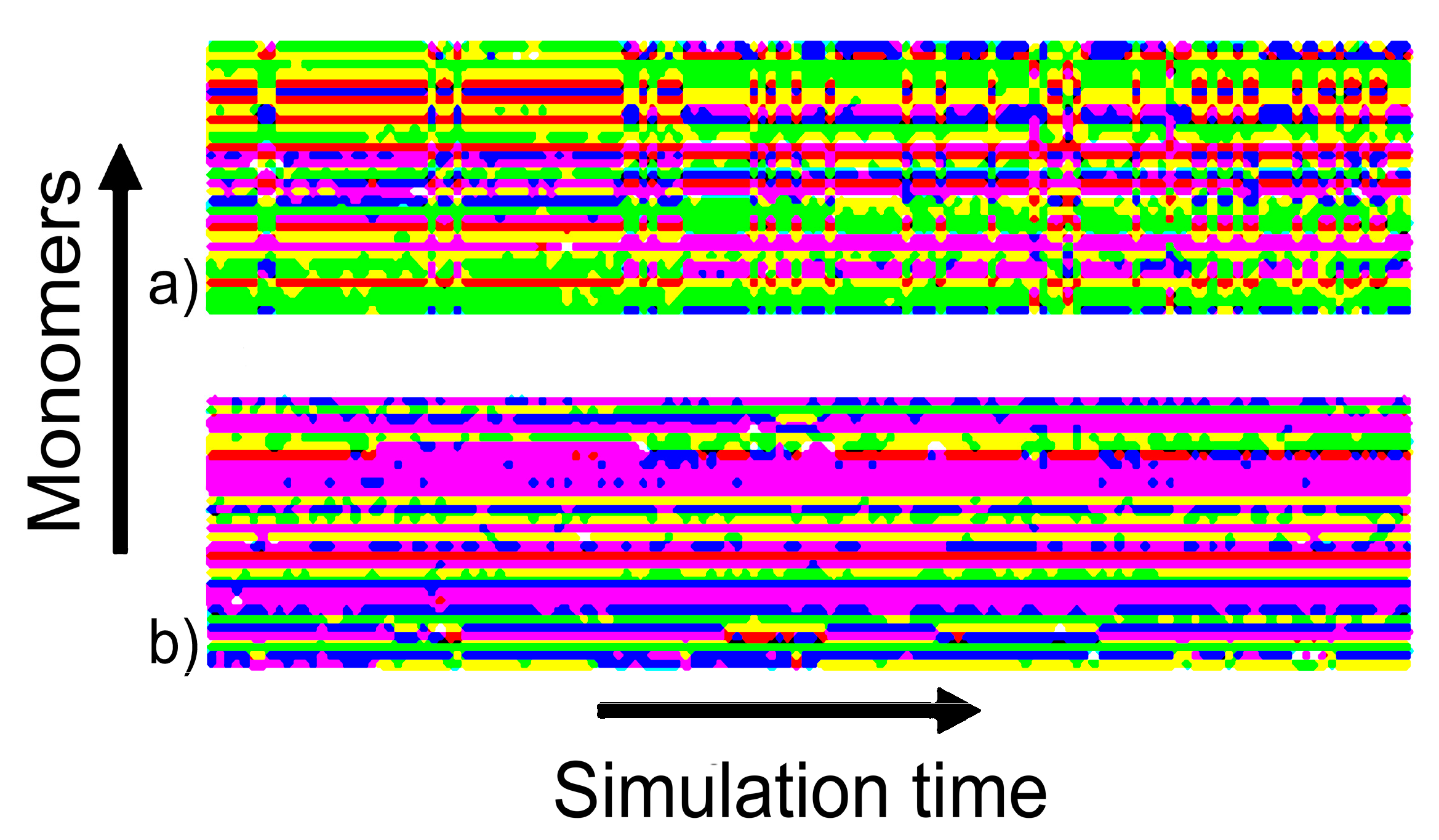
4.2. Analysis of Effectiveness of HRE Simulation
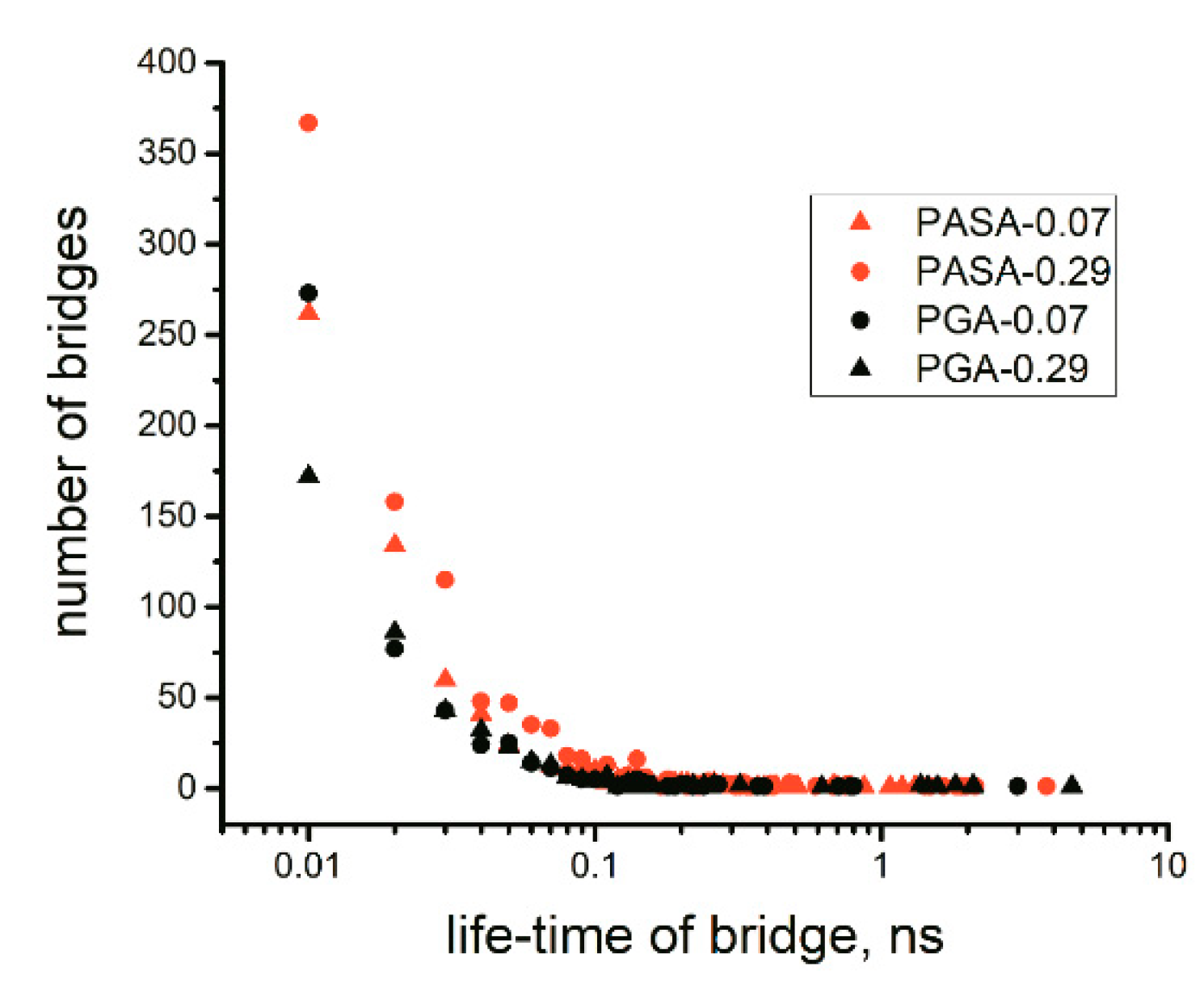
4.2.1. Lifetime of Calcium Bridges
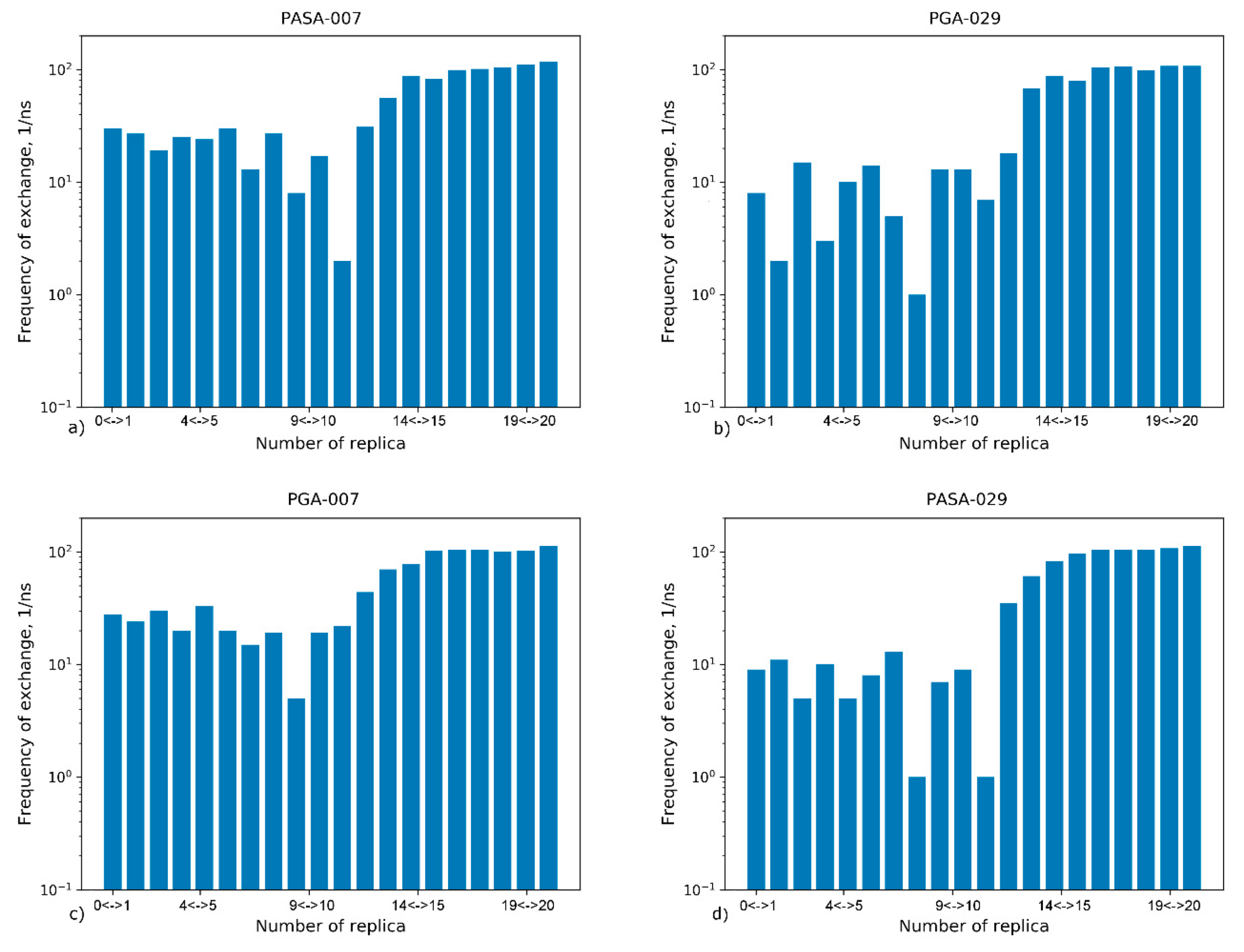
4.2.2. Exchange Frequencies between Replicas
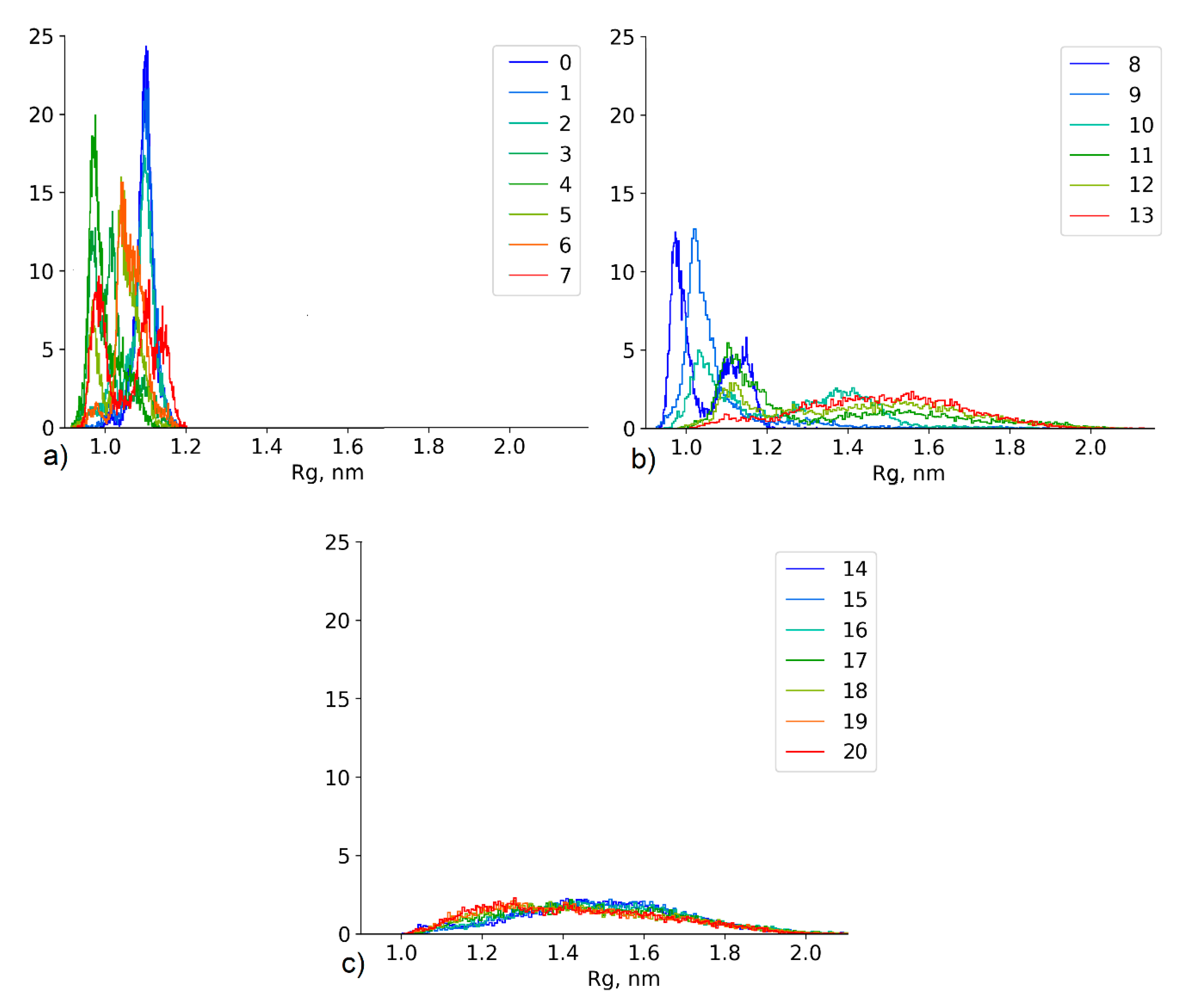
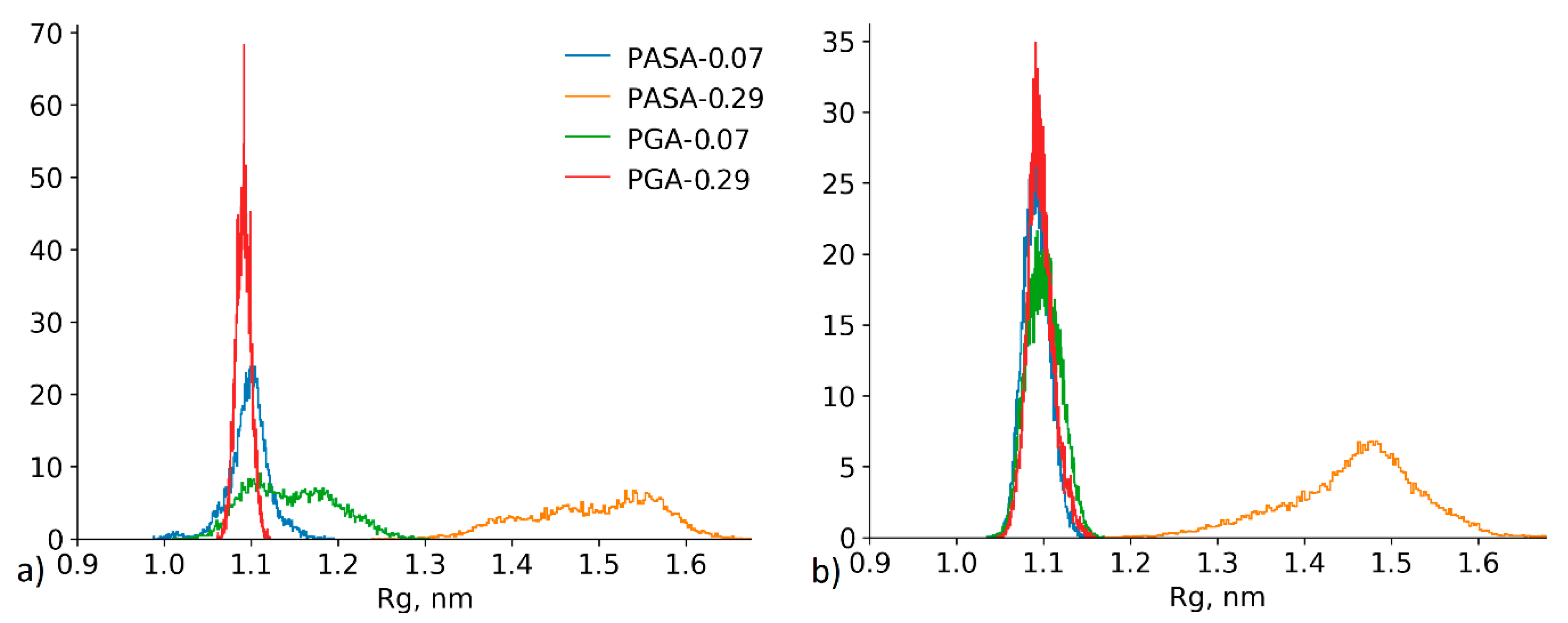
4.2.3. Effect of the Conformational State Exchange on the Gyration Radius Distribution
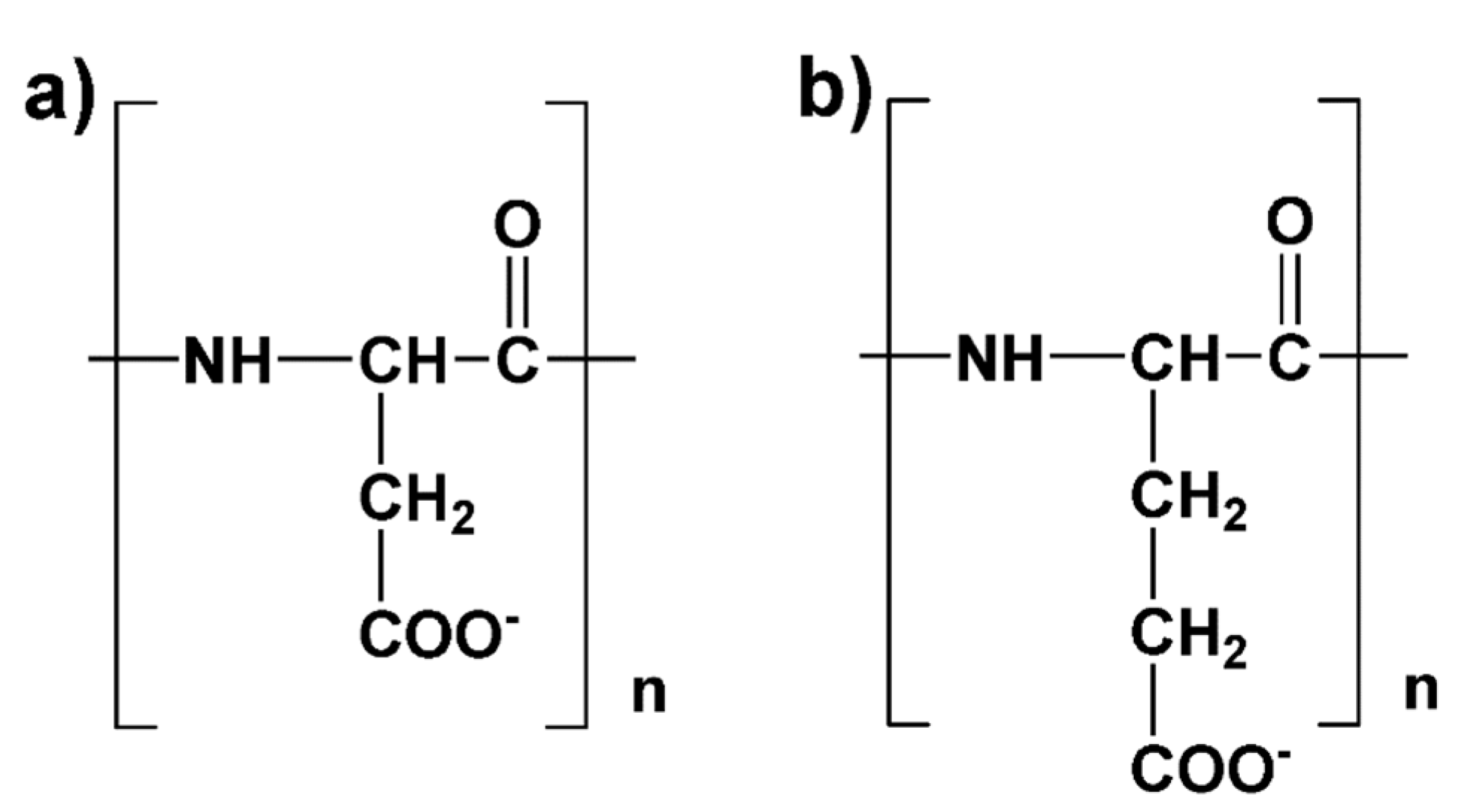
4.3. Results of HRE Simulation
4.3.1. Chain Conformations
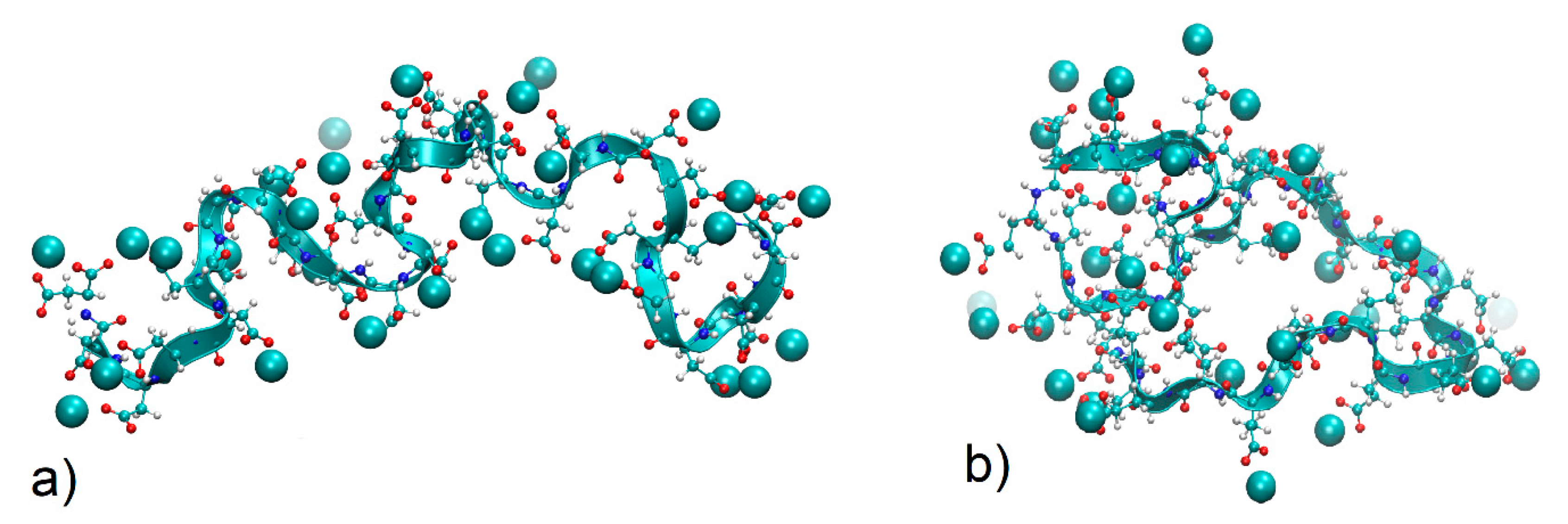
4.3.2. Local Conformations
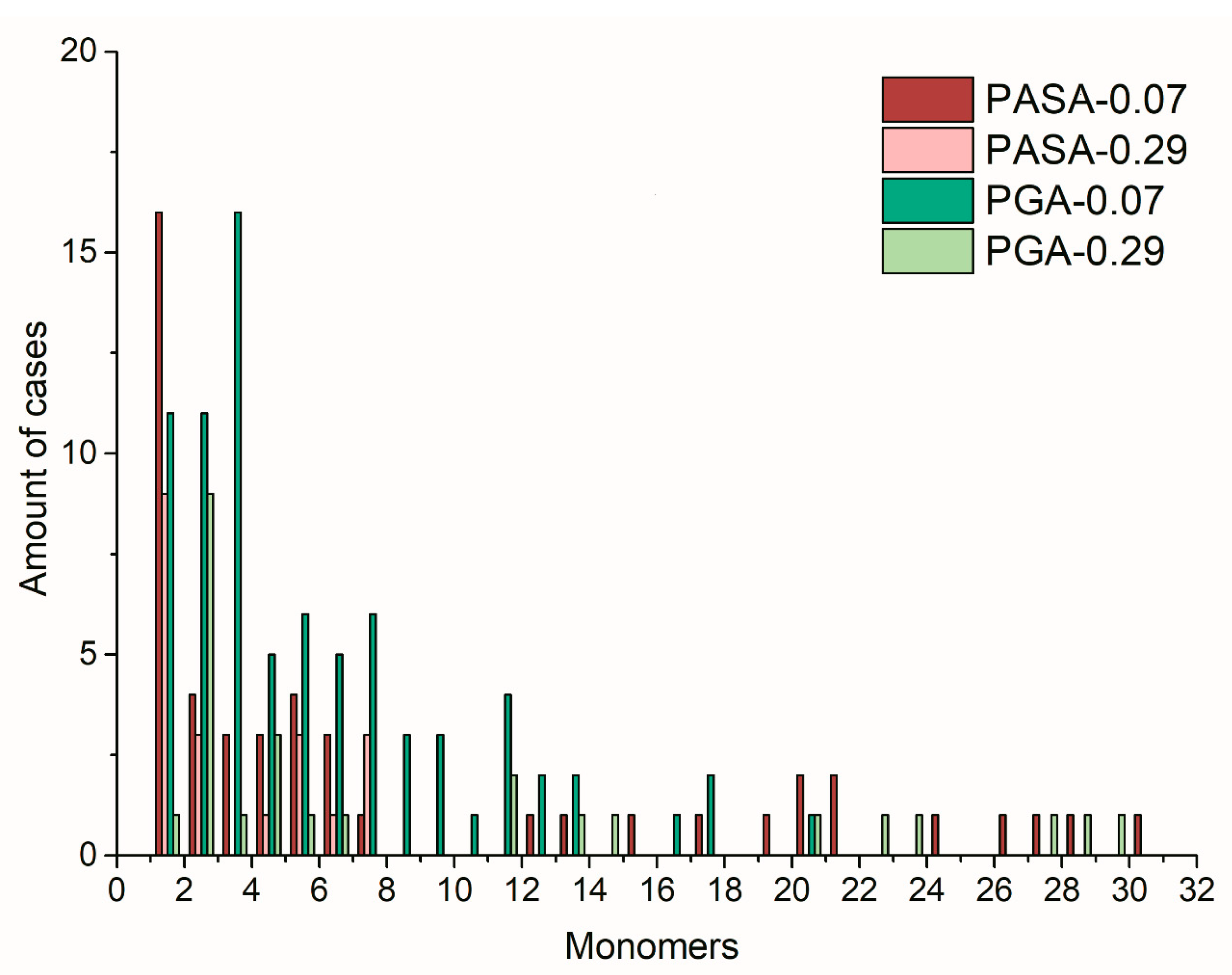
4.3.3. Distribution of Calcium Ions Adsorbed by PASA and PGA Chains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wilts, E.M.; Herzberger, J.; Long, T.E. Addressing water scarcity: Cationic polyelectrolytes in water treatment and purification. Polym. Int. 2018, 67, 799–814. [Google Scholar] [CrossRef]
- Migahed, M.A.; Rashwan, S.M.; Kamel, M.M.; Habib, R.E. Synthesized polyaspartic acid derivatives as corrosion and scale inhibitors in desalination operations. Cogent Eng. 2017, 4, 1366255. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, X.; Carbone, E.J.; Nelson, C.; Kan, H.M.; Lo, K.W.H. Poly aspartic acid peptide-linked PLGA based nanoscale particles: Potential for bone-targeting drug delivery applications. Int. J. Pharm. 2014, 475, 547–557. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.G.; De Koker, S.; Sukhorukov, G.B.; Kreft, O.; Parak, W.J.; Skirtach, A.G.; Demeester, J.; De Smedt, S.C.; Hennink, W.E. Polyelectrolyte microcapsules for biomedical applications. Soft Matter 2009, 5, 282–291. [Google Scholar] [CrossRef]
- Lankalapalli, S.; Kolapalli, V.R.M. Polyelectrolyte complexes: A review of their applicability in drug delivery technology. Indian J. Pharm. Sci. 2009, 71, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattari, S.; Dadkhah Tehrani, A.; Adeli, M. pH-Responsive Hybrid Hydrogels as Antibacterial and Drug Delivery Systems. Polymers 2018, 10, 660. [Google Scholar] [CrossRef] [Green Version]
- Gwak, G.; Jung, B.; Han, S.; Hong, S. Evaluation of poly (aspartic acid sodium salt) as a draw solute for forward osmosis. Water Res. 2015, 80, 294–305. [Google Scholar] [CrossRef]
- Thula, T.T.; Svedlund, F.; Rodriguez, D.E.; Podschun, J.; Pendi, L.; Gower, L.B. Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents. Polymers 2011, 3, 10–35. [Google Scholar] [CrossRef] [Green Version]
- Quan, B.D.; Sone, E.D. The effect of polyaspartate chain length on mediating biomimetic remineralization of collagenous tissues. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [Green Version]
- Cantaert, B.; Beniash, E.; Meldrum, F.C. The role of poly(aspartic acid) in the precipitation of calcium phosphate in confinement. J. Mater. Chem. B 2013, 1, 6586–6595. [Google Scholar] [CrossRef]
- Yao, C.-H.; Yang, S.-P.; Chen, Y.-S.; Chen, K.-Y. Electrospun Poly(γ–glutamic acid)/β–Tricalcium Phosphate Composite Fibrous Mats for Bone Regeneration. Polymers 2019, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picker, A.; Kellermeier, M.; Seto, J.; Gebauer, D.; Cölfen, H. The multiple effects of amino acids on the early stages of calcium carbonate crystallization. Z. Krist. 2012, 227, 744–757. [Google Scholar] [CrossRef] [Green Version]
- Bewernitz, M.A.; Gebauer, D.; Long, J.; Cölfen, H.; Gower, L.B. A metastable liquid precursor phase of calcium carbonate and its interactions with polyaspartate. Faraday Discuss. 2012, 159, 291–312. [Google Scholar] [CrossRef] [Green Version]
- Averianov, I.V.; Stepanova, M.A.; Gofman, I.V.; Nikolaeva, A.L.; Korzhikov-Vlakh, V.A.; Karttunen, M.; Korzhikova-Vlakh, E.G. Chemical modification of nanocrystalline cellulose for improved interfacial compatibility with poly(lactic acid). Mendeleev Commun. 2019, 29, 220–222. [Google Scholar] [CrossRef]
- Rezania, A.; Healy, K.E. The effect of peptide surface density on mineralization of a matrix deposited by osteogenic cells. J. Biomed. Mater. Res. 2000, 52, 595–600. [Google Scholar] [CrossRef]
- Cao, L.; Wang, L.; Fan, L.; Xiao, W.; Lin, B.; Xu, Y.; Liang, J.; Cao, B. RGDC Peptide-Induced Biomimetic Calcium Phosphate Coating Formed on AZ31 Magnesium Alloy. Materials 2017, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.A.; Ebrahimpour, A.; Perez, L.; Smesko, S.A.; Nancollas, G.H. The dual role of polyelectrolytes and proteins as mineralization promoters and inhibitors of calcium oxalate monohydrate. Calcif. Tissue Int. 1989, 45, 122–128. [Google Scholar] [CrossRef]
- Öner, M.; Doǧan, Ö. Inhibitory effect of polyelectrolytes on crystallization kinetics of hydroxyapatite. Prog. Cryst. Growth Charact. Mater. 2005, 50, 39–51. [Google Scholar] [CrossRef]
- Bar-Yosef Ofir, P.; Govrin-Lippman, R.; Garti, N.; Füredi-Milhofer, H. The Influence of Polyelectrolytes on the Formation and Phase Transformation of Amorphous Calcium Phosphate. Cryst. Growth Des. 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Hunter, G.K.; OYoung, J.; Grohe, B.; Karttunen, M.; Goldberg, H.A. The flexible polyelectrolyte hypothesis of protein-biomineral interaction. Langmuir 2010, 26, 18639–18646. [Google Scholar] [CrossRef]
- Muthukumar, M. 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50, 9528–9560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.D.; Olvera De La Cruz, M. Manipulation of Confined Polyelectrolyte Conformations through Dielectric Mismatch. ACS Nano 2019, 13, 9298–9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.-F.; Hsiao, P.-Y. Effect of chain stiffness on ion distributions around a polyelectrolyte in multivalent salt solutions. J. Chem. Phys. 2010, 132, 024905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, Q.; Zhao, H.; Shu, X.; Zhang, Q.; Yang, Y.; Liu, J.; Wu, S. Molecular dynamics study of the effects of calcium ions on the conformational properties of comb-like poly(acrylic acid-co-methyl allyl. Comput. Mater. Sci. 2015, 109, 90–96. [Google Scholar] [CrossRef]
- Chung, Y.T.; Huang, C.I. Ion condensation behavior and dynamics of water molecules surrounding the sodium poly(methacrylic acid) chain in water: A molecular dynamics study. J. Chem. Phys. 2012, 136, 124903. [Google Scholar] [CrossRef]
- Staňo, R.; Nová, L.; Uhlík, F.; Košovan, P. Multivalent counterions accumulate in star-like polyelectrolytes and collapse the polymer in spite of increasing its ionization. Soft Matter 2020, 16, 1047–1055. [Google Scholar] [CrossRef]
- Head, J.H. Partial Optimization of Adsorbates on Clusters: Oxygen on Al(111); Springer: Boston, MA, USA, 1992; pp. 415–422. [Google Scholar]
- Kundagrami, A.; Muthukumar, M. Theory of competitive counterion adsorption on flexible polyelectrolytes: Divalent salts. J. Chem. Phys. 2008, 128, 244901. [Google Scholar] [CrossRef] [Green Version]
- Grohe, B.; Hug, S.; Langdon, A.; Jalkanen, J.; Rogers, K.A.; Goldberg, H.A.; Karttunen, M.; Hunter, G.K. Mimicking the biomolecular control of calcium oxalate monohydrate crystal growth: Effect of contiguous glutamic acids. Langmuir 2012, 28, 12182–12190. [Google Scholar] [CrossRef]
- Huang, C.I.; Olvera De La Cruz, M. Polyelectrolytes in multivalent salt solutions: Monomolecular versus multimolecular aggregation. Macromolecules 2002, 35, 976–986. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Borisov, O.V.; Birshtein, T.M. Polyelectrolyte brush interaction with multivalent ions. Macromolecules 1999, 32, 8189–8196. [Google Scholar] [CrossRef]
- Yu, J.; Jackson, N.E.; Xu, X.; Brettmann, B.K.; Ruths, M.; De Pablo, J.J.; Tirrell, M. Multivalent ions induce lateral structural inhomogeneities in polyelectrolyte brushes. Sci. Adv. 2017, 3, eaao1497. [Google Scholar] [CrossRef] [PubMed]
- Brettmann, B.K.; Laugel, N.; Hoffmann, N.; Pincus, P.; Tirrell, M. Bridging contributions to polyelectrolyte brush collapse in multivalent salt solutions. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 284–291. [Google Scholar] [CrossRef]
- Levin, Y. Electrostatic correlations: From plasma to biology. Rep. Prog. Phys. 2002, 65, 1577. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte solutions. III. An analysis based on the mayer ionic solution theory. J. Chem. Phys. 1969, 51, 3249–3252. [Google Scholar] [CrossRef]
- Ghelichi, M.; Eikerling, M.H. Conformational Properties of Comb-Like Polyelectrolytes: A Coarse-Grained MD Study. J. Phys. Chem. B 2016, 120, 2859–2867. [Google Scholar] [CrossRef]
- Kuo, H.T.; Liu, S.L.; Chiu, W.C.; Fang, C.J.; Chang, H.C.; Wang, W.R.; Yang, P.A.; Li, J.H.; Huang, S.J.; Huang, S.L.; et al. Effect of charged amino acid side chain length on lateral cross-strand interactions between carboxylate- and guanidinium-containing residues in a β-hairpin. Amino Acids 2015, 47, 885–898. [Google Scholar] [CrossRef]
- Meuzelaar, H.; Vreede, J.; Woutersen, S. Influence of Glu/Arg, Asp/Arg, and Glu/Lys Salt Bridges on α-Helical Stability and Folding Kinetics. Biophys. J. 2016, 110, 2328–2341. [Google Scholar] [CrossRef] [Green Version]
- Duboué-Dijon, E.; Delcroix, P.; Martinez-Seara, H.; Hladílková, J.; Coufal, P.; Křížek, T.; Jungwirth, P. Binding of Divalent Cations to Insulin: Capillary Electrophoresis and Molecular Simulations. J. Phys. Chem. B 2018, 122, 5640–5648. [Google Scholar] [CrossRef] [Green Version]
- Lins, L.; Nasir, M.N.; Laurent, P.; Deleu, M.; Flore, C.; Ongena, M. Analysis of calcium-induced effects on the conformation of fengycin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 110, 450–457. [Google Scholar] [CrossRef]
- Kawasaki, H.; Soma, N.; Kretsinger, R.H. Molecular Dynamics Study of the Changes in Conformation of Calmodulin with Calcium Binding and/or Target Recognition. Sci. Rep. 2019, 9, 10688. [Google Scholar] [CrossRef]
- Cisneros, A.; Karttunen, M.; Ren, P.; Sagui, C. Classical Electrostatics for Biomolecular Simulations. Chem. Rev. 2014, 114, 779–814. [Google Scholar] [CrossRef] [Green Version]
- Karttunen, M.; Rottler, J.; Vattulainen, I.; Sagui, C. Chapter 2 Electrostatics in Biomolecular Simulations: Where Are We Now and Where Are We Heading? Curr. Top. Membr. 2008, 60, 49–89. [Google Scholar] [CrossRef]
- Wong-ekkabut, J.; Karttunen, M. The good, the bad and the user in soft matter simulations. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.; Gilmer, J.; Mayes, H.B.; Mobley, D.L.; Monroe, J.I.; Prasad, S.; Zuckerman, D.M. Best Practices for Foundations in Molecular Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catte, A.; Girych, M.; Javanainen, M.; Loison, C.; Melcr, J.; Miettinen, M.S.; Monticelli, L.; Määttä, J.; Oganesyan, V.S.; Ollila, O.H.S.; et al. Molecular electrometer and binding of cations to phospholipid bilayers. Phys. Chem. Chem. Phys. 2016, 18, 32560–32569. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Aksimentiev, A. New tricks for old dogs: Improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef]
- Leontyev, I.V.; Stuchebrukhov, A.A. Electronic Continuum Model for Molecular Dynamics Simulations of Biological Molecules. J. Chem. Theory Comput. 2010, 6, 1498–1508. [Google Scholar] [CrossRef]
- Melcr, J.; Martinez-Seara, H.; Nencini, R.; Kolafa, J.; Jungwirth, P.; Ollila, O.H.S. Accurate Binding of Sodium and Calcium to a POPC Bilayer by Effective Inclusion of Electronic Polarization. J. Phys. Chem. B 2018, 122, 4546–4557. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Lopes, P.E.M.; Guvench, O.; MacKerell, A.D. Current status of protein force fields for molecular dynamics simulations. In Molecular Modeling of Proteins, 2nd ed.; Kukol, A., Ed.; Humana Press: New York, NY, USA, 2014; pp. 47–71. ISBN 9781493914654. [Google Scholar]
- Lemkul, J.A.; Huang, J.; Roux, B.; MacKerell, A.D. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116, 4983–5013. [Google Scholar] [CrossRef]
- Baker, C.M. Polarizable force fields for molecular dynamics simulations of biomolecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 241–254. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.-P.; Ren, P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Bedrov, D.; Piquemal, J.P.; Borodin, O.; MacKerell, A.D.; Roux, B.; Schröder, C. Molecular Dynamics Simulations of Ionic Liquids and Electrolytes Using Polarizable Force Fields. Chem. Rev. 2019, 119, 7940–7995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, A.T.; Hughes, Z.E.; Walsh, T.R. Improving the description of interactions between Ca2+ and protein carboxylate groups, including γ-carboxyglutamic acid: Revised CHARMM22∗ parameters. RSC Adv. 2015, 5, 67820–67828. [Google Scholar] [CrossRef] [Green Version]
- Tolmachev, D.A.; Boyko, O.S.; Lukasheva, N.V.; Martinez-Seara, H.; Karttunen, M. Overbinding and Qualitative and Quantitative Changes Caused by Simple Na+ and K+ Ions in Polyelectrolyte Simulations: Comparison of Force Fields with and without NBFIX and ECC Corrections. J. Chem. Theory Comput. 2020, 16, 677–687. [Google Scholar] [CrossRef]
- Lesk, A.M. Introduction to Bioinformatics; Oxford University Press: Oxford, UK, 2019; ISBN 9780198794141. [Google Scholar]
- Schnabel, W. Main-Chain Scission of Linear Polymers as Studied by Pulse Radiolysis and Flash Photolysis. In Crosslinking and Scission in Polymers; Springer: Dordrecht, The Netherlands, 1990; pp. 37–56. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef]
- Nguyen, P.H. Replica exchange simulation method using temperature and solvent viscosity. J. Chem. Phys. 2010, 132, 144109. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Galvelis, R.; Sugita, Y. Replica state exchange metadynamics for improving the convergence of free energy estimates. J. Comput. Chem. 2015, 36, 1446–1455. [Google Scholar] [CrossRef]
- Dama, J.F.; Rotskoff, G.; Parrinello, M.; Voth, G.A. Transition-tempered metadynamics: Robust, convergent metadynamics via on-the-fly transition barrier estimation. J. Chem. Theory Comput. 2014, 10, 3626–3633. [Google Scholar] [CrossRef] [PubMed]
- Do, T.N.; Choy, W.Y.; Karttunen, M. Binding of Disordered Peptides to Kelch: Insights from Enhanced Sampling Simulations. J. Chem. Theory Comput. 2016, 12, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.; Bussi, G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2013, 16, 163–199. [Google Scholar] [CrossRef] [Green Version]
- Camilloni, C.; Pietrucci, F. Advanced simulation techniques for the thermodynamic and kinetic characterization of biological systems. Adv. Phys. 2018, 3, 885–916. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200–212. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 70902. [Google Scholar] [CrossRef] [Green Version]
- Meli, M.; Colombo, G. A hamiltonian replica exchange molecular dynamics (MD) method for the study of folding, based on the analysis of the stabilization determinants of proteins. Int. J. Mol. Sci. 2013, 14, 12157–12169. [Google Scholar] [CrossRef] [Green Version]
- Laio, A.; Rodriguez-Fortea, A.; Gervasio, F.L.; Ceccarelli, M.; Parrinello, M. Assessing the accuracy of metadynamics. J. Phys. Chem. B 2005, 109, 6714–6721. [Google Scholar] [CrossRef]
- Kahlen, J.; Peter, C.; Donadio, D. Molecular simulation of oligo-glutamates in a calcium-rich aqueous solution: Insights into peptide-induced polymorph selection. CrystEngComm 2015, 17, 6863–6867. [Google Scholar] [CrossRef] [Green Version]
- Lemke, T.; Peter, C.; Kukharenko, O. Efficient Sampling and Characterization of Free Energy Landscapes of Ion-Peptide Systems. J. Chem. Theory Comput. 2018, 14, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Aksimentiev, A. Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar] [CrossRef]
- Venable, R.M.; Luo, Y.; Gawrisch, K.; Roux, B.; Pastor, R.W. Simulations of anionic lipid membranes: Development of interaction-specific ion parameters and validation using NMR data. J. Phys. Chem. B 2013, 117, 10183–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laage, D.; Stirnemann, G. Effect of Ions on Water Dynamics in Dilute and Concentrated Aqueous Salt Solutions. J. Phys. Chem. B 2019, 123, 3312–3324. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wilson, J.; Aksimentiev, A. Improved model of hydrated calcium ion for molecular dynamics simulations using classical biomolecular force fields. Biopolymers 2016, 105, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [Green Version]
- Daniele, P.G.; Foti, C.; Gianguzza, A.; Prenesti, E.; Sammartano, S. Weak alkali and alkaline earth metal complexes of low molecular weight ligands in aqueous solution. Co-ord. Chem. Rev. 2008, 252, 1093–1107. [Google Scholar] [CrossRef]
- Prorok, M.; Castellino, F.J. Thermodynamics of binding of calcium, magnesium, and zinc to the N- methyl-D-aspartate receptor ion channel peptidic inhibitors, conantokin-G and conantokin-T. J. Biol. Chem. 1998, 273, 19573–19578. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.S.; Ray, J. Counterion condensation revisited. J. Biomol. Struct. Dyn. 1998, 16, 461–476. [Google Scholar] [CrossRef]
- Kubitzki, M.B.; De Groot, B.L. Molecular dynamics simulations using temperature-enhanced essential dynamics replica exchange. Biophys. J. 2007, 92, 4262–4270. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, J.; Muniyat, M.I.; Gaalswyk, K. On-line Optimization of Hamiltonian Replica Exchange Simulations. bioRxiv 2017, 228262. [Google Scholar] [CrossRef] [Green Version]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Bulo, R.E.; Donadio, D.; Laio, A.; Molnar, F.; Rieger, J.; Parrinello, M. “Site binding” of Ca2+ ions to polyacrylates in water: A molecular dynamics study of coiling and aggregation. Macromolecules 2007, 40, 3437–3442. [Google Scholar] [CrossRef]
- Ostermeir, K.; Zacharias, M. Hamiltonian replica-exchange simulations with adaptive biasing of peptide backbone and side chain dihedral angles. J. Comput. Chem. 2014, 35, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Hritz, J.; Oostenbrink, C. Optimization of replica exchange molecular dynamics by fast mimicking. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olander, D.S.; Holtzer, A. The Stability of the Polyglutamic Acid α Helix. J. Am. Chem. Soc. 1968, 90, 4549–4560. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, M.; Holtzer, A. The Helix-Coil Transition in Solutions of Polyglutamic Acid. J. Am. Chem. Soc. 1964, 86, 538–543. [Google Scholar] [CrossRef]
- Raiteri, P.; Demichelis, R.; Gale, J.D.; Kellermeier, M.; Gebauer, D.; Quigley, D.; Wright, L.B.; Walsh, T.R. Exploring the influence of organic species on pre- and post-nucleation calcium carbonate. Faraday Discuss. 2012, 159, 61–85. [Google Scholar] [CrossRef] [Green Version]
- Fischer, V.; Landfester, K.; Muñoz-Espí, R. Stabilization of calcium oxalate metastable phases by oligo(L-glutamic acid): Effect of peptide chain length. Cryst. Growth Des. 2011, 11, 1880–1890. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaCl2 Concentration, mol/kg | Number of Ca2+ Ions | Number of Cl− Ions |
|---|---|---|
| 0 (water) | 0 | 0 |
| 0.03 | 6 | 12 |
| 0.07 | 14 | 28 |
| 0.29 | 58 | 116 |
| 0.75 | 145 | 290 |
| 1 | 190 | 380 |
| Replica | T, K | α | σ, nm | ε, kJ mol−1 |
|---|---|---|---|---|
| 0 | 300 | 0 | 0.273239 | 0.50208 |
| 1 | 300.75 | −0.05 | 0.275077 | 0.500581 |
| 2 | 301.5 | −0.1 | 0.276915 | 0.499083 |
| 3 | 302.25 | −0.15 | 0.278753 | 0.497584 |
| 4 | 303 | −0.2 | 0.280591 | 0.496085 |
| 5 | 303.75 | −0.25 | 0.282429 | 0.494587 |
| 6 | 304.5 | −0.3 | 0.284267 | 0.493088 |
| 7 | 305.25 | −0.35 | 0.286105 | 0.491589 |
| 8 | 306 | −0.4 | 0.287943 | 0.49009 |
| 9 | 306.75 | −0.45 | 0.289781 | 0.488592 |
| 10 | 307.5 | −0.5 | 0.291619 | 0.487093 |
| 11 | 308.25 | −0.55 | 0.293457 | 0.485594 |
| 12 | 309 | −0.6 | 0.295295 | 0.484096 |
| 13 | 309.75 | −0.65 | 0.297134 | 0.482597 |
| 14 | 310.5 | −0.7 | 0.298972 | 0.481098 |
| 15 | 311.25 | −0.75 | 0.30081 | 0.4796 |
| 16 | 312 | −0.8 | 0.302648 | 0.478101 |
| 17 | 312.75 | −0.85 | 0.304486 | 0.476602 |
| 18 | 313.5 | −0.9 | 0.306324 | 0.475103 |
| 19 | 314.25 | −0.95 | 0.308162 | 0.473605 |
| 20 | 315 | −1 | 0.31 | 0.472106 |
| PASA-0.07 mol/kg | PASA-0.29 mol/kg | PGA-0.07 mol/kg | PGA-0.29 mol/kg | |
|---|---|---|---|---|
| Rx, nm | 0.866 ± 0.037 | 1.391 ± 0.085 | 0.912 ± 0.075 | 0.845 ± 0.012 |
| Ry, nm | 0.607 ± 0.030 | 0.452 ± 0.031 | 0.557 ± 0.034 | 0.563 ± 0.156 |
| Rz, nm | 0.289 ± 0.032 | 0.292 ± 0.046 | 0.409 ± 0.043 | 0.399 ± 0.016 |
| (Ry + Rz)/2, nm | 0.578 ± 0.022 | 0.842 ± 0.032 | 0.660 ± 0.34 | 0.622 ± 0.008 |
| B | 0.481 ± 0.039 | 0.731 ± 0.030 | 0.466 ± 0.062 | 0.430 ± 0.014 |
| k2 | 0.082 ± 0.014 | 0.233 ± 0.037 | 0.059 ± 0.020 | 0.047 ± 0.005 |
| P | −0.169 ± 0.266 | 0.911 ± 0.056 | 0.603 ± 0.293 | 0.435 ± 0.154 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolmachev, D.; Lukasheva, N.; Mamistvalov, G.; Karttunen, M. Influence of Calcium Binding on Conformations and Motions of Anionic Polyamino Acids. Effect of Side Chain Length. Polymers 2020, 12, 1279. https://doi.org/10.3390/polym12061279
Tolmachev D, Lukasheva N, Mamistvalov G, Karttunen M. Influence of Calcium Binding on Conformations and Motions of Anionic Polyamino Acids. Effect of Side Chain Length. Polymers. 2020; 12(6):1279. https://doi.org/10.3390/polym12061279
Chicago/Turabian StyleTolmachev, Dmitry, Natalia Lukasheva, George Mamistvalov, and Mikko Karttunen. 2020. "Influence of Calcium Binding on Conformations and Motions of Anionic Polyamino Acids. Effect of Side Chain Length" Polymers 12, no. 6: 1279. https://doi.org/10.3390/polym12061279
APA StyleTolmachev, D., Lukasheva, N., Mamistvalov, G., & Karttunen, M. (2020). Influence of Calcium Binding on Conformations and Motions of Anionic Polyamino Acids. Effect of Side Chain Length. Polymers, 12(6), 1279. https://doi.org/10.3390/polym12061279