A Coarse-Grained Force Field for Silica–Polybutadiene Interfaces and Nanocomposites



Abstract
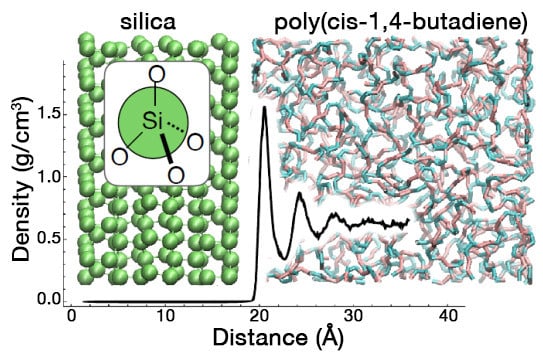
:
1. Introduction
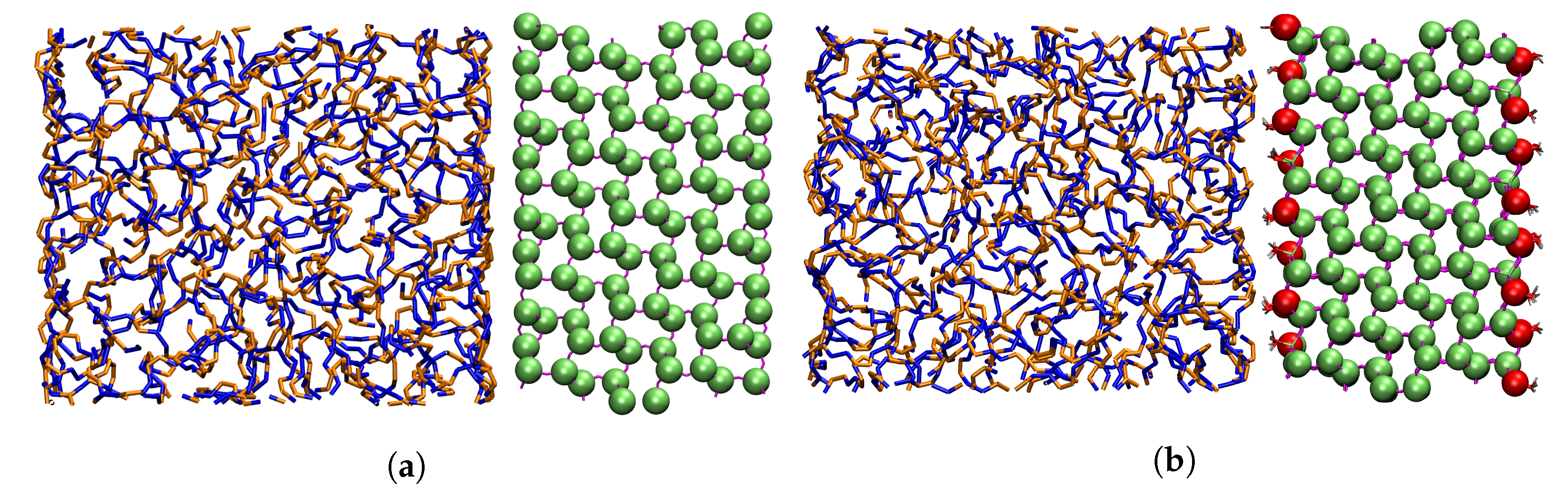
2. Models
3. Methods
3.1. Equilibration
3.2. Boltzmann Inversion
3.3. Dynamical Mechanical Analysis
4. Results
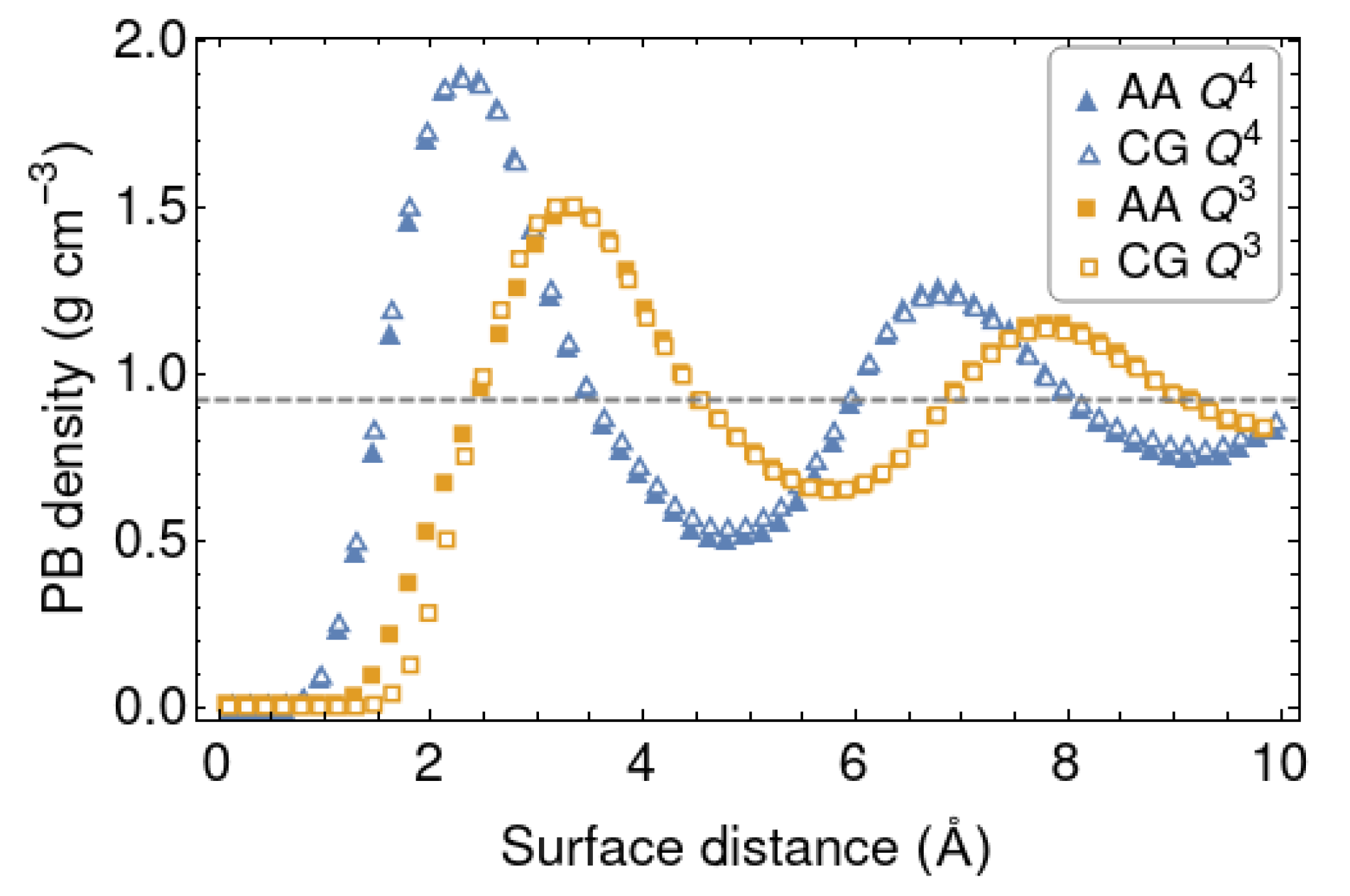
4.1. Force Field Parameters and Transferability
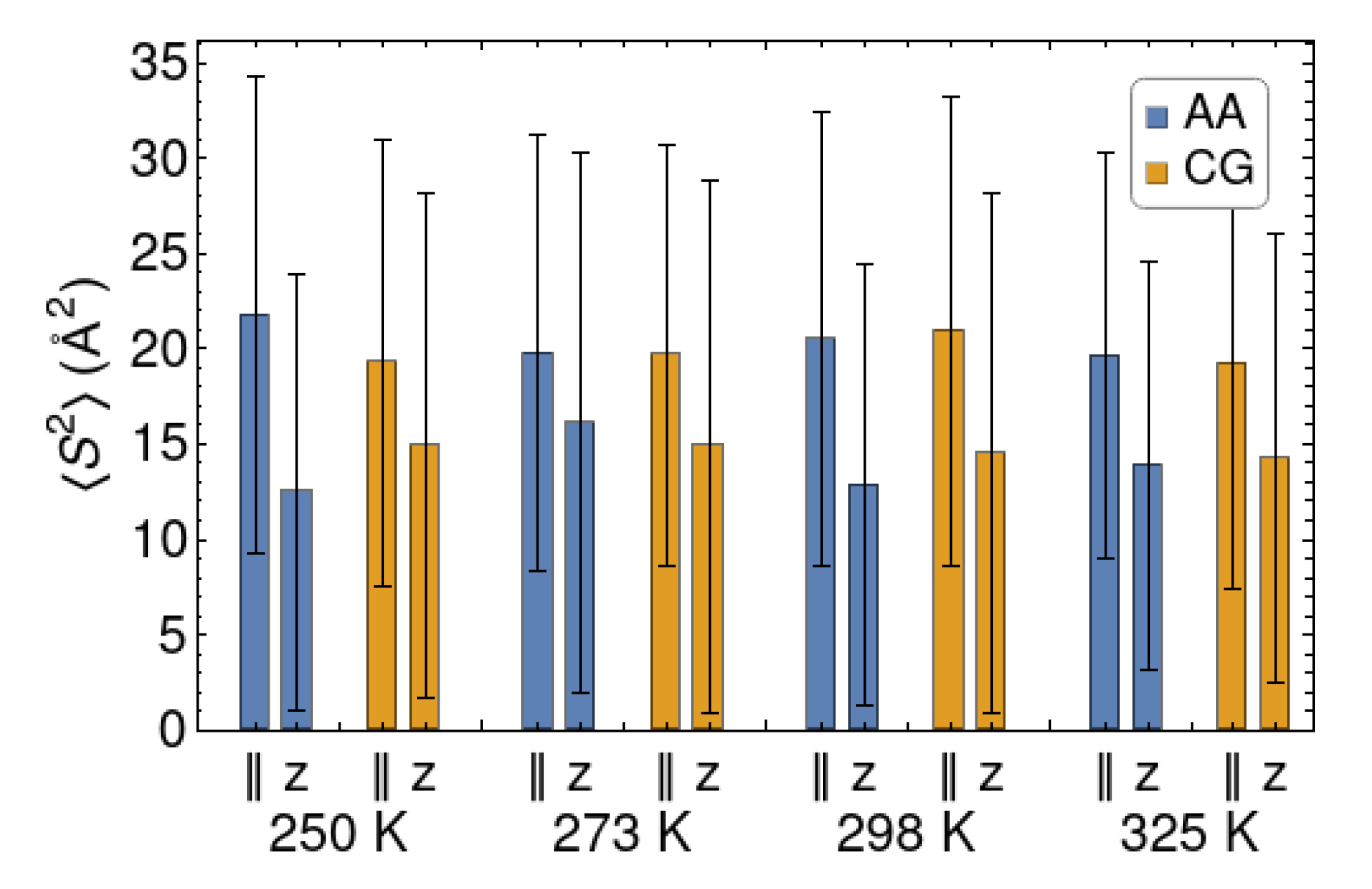
4.2. Dynamical Mechanical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vaia, R.A.; Giannelis, E.P. Polymer Nanocomposites: Status and Opportunities. MRS Bull. 2001, 26, 394–401. [Google Scholar] [CrossRef]
- Balazs, A.C.; Emrick, T.; Russell, T.P. Nanoparticle Polymer Composites: Where Two Small Worlds Meet. Science 2006, 314, 1107–1110. [Google Scholar] [CrossRef]
- Kumar, S.K.; Benicewicz, B.C.; Vaia, R.A.; Winey, K.I. Are Polymer Nanocomposites Practical for Applications? Macromolecules 2017, 50, 714–731. [Google Scholar] [CrossRef]
- Jancar, J.; Douglas, J.; Starr, F.; Kumar, S.; Cassagnau, P.; Lesser, A.; Sternstein, S.; Buehler, M. Current issues in research on structure–property relationships in polymer nanocomposites. Polymer 2010, 51, 3321–3343. [Google Scholar] [CrossRef]
- Lin, C.C.; Parrish, E.; Composto, R.J. Macromolecule and Particle Dynamics in Confined Media. Macromolecules 2016, 49, 5755–5772. [Google Scholar] [CrossRef]
- Rittigstein, P.; Priestley, R.D.; Broadbelt, L.J.; Torkelson, J.M. Model polymer nanocomposites provide an understanding of confinement effects in real nanocomposites. Nat. Mater. 2007, 6, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Torquato, S. Random Heterogeneous Materials; Interdisciplinary Applied Mathematics; Springer: New York, NY, USA, 2002; Volume 16. [Google Scholar] [CrossRef]
- Gent, A.N. Engineering with Rubber. How to Design Rubber Components; Hanser: Munich, Germany, 2012. [Google Scholar] [CrossRef]
- Diani, J.; Fayolle, B.; Gilormini, P. A review on the Mullins effect. Eur. Polym. J. 2009, 45, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Medalia, A.I. Effect of carbon black on ultimate properties of rubber vulcanizates. Rubber Chem. Technol. 1987, 60, 45–61. [Google Scholar] [CrossRef]
- Vilgis, T.A.; Heinrich, G.; Klüppel, M. Reinforcement of Polymer Nano-Composites: Theory, Experiments and Applications; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar] [CrossRef]
- Kraus, G. Reinforcement of Elastomers; Interscience: New York, NY, USA, 1965. [Google Scholar] [CrossRef]
- Heinrich, G.; Klüppel, M. Recent advances in the theory of filler networking in elastomers. In Filled Elastomers Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2002; pp. 1–44. [Google Scholar] [CrossRef]
- Wang, M.J. Effect of polymer–filler and filler–filler interactions on dynamic properties of filled vulcanizates. Rubber Chem. Technol. 1998, 71, 520–589. [Google Scholar] [CrossRef]
- Göritz, D.; Raab, H.; Fröhlich, J.; Maier, P.G. Surface structure of carbon black and reinforcement. Rubber Chem. Technol. 1999, 72, 929–945. [Google Scholar] [CrossRef]
- Raos, G. Application of the Christensen-Lo Model to the Reinforcement of Elastomers by Fractal Fillers. Macromol. Theory Simul. 2003, 12, 17–23. [Google Scholar] [CrossRef]
- Allegra, G.; Raos, G.; Vacatello, M. Theories and simulations of polymer-based nanocomposites: From chain statistics to reinforcement. Prog. Polym. Sci. 2008, 33, 683–731. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, H.; Wu, Y. Computational Thermomechanical Properties of Silica–Epoxy Nanocomposites by Molecular Dynamic Simulation. Polymers 2017, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Hager, J.; Hentschke, R.; Hojdis, N.W.; Karimi-Varzaneh, H.A. Computer Simulation of Particle–Particle Interaction in a Model Polymer Nanocomposite. Macromolecules 2015, 48, 9039–9049. [Google Scholar] [CrossRef]
- Pavlov, A.S.; Khalatur, P.G. Filler reinforcement in cross-linked elastomer nanocomposites: Insights from fully atomistic molecular dynamics simulation. Soft Matter 2016, 12, 5402–5419. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.S.; Khalatur, P.G. Fully atomistic molecular dynamics simulation of nanosilica-filled crosslinked polybutadiene. Chem. Phys. Lett. 2016, 653, 90–95. [Google Scholar] [CrossRef]
- Pfaller, S.; Possart, G.; Steinmann, P.; Rahimi, M.; Müller-Plathe, F.; Böhm, M.C. Investigation of interphase effects in silica–polystyrene nanocomposites based on a hybrid molecular-dynamics–finite-element simulation framework. Phys. Rev. E 2016, 93, 052505. [Google Scholar] [CrossRef]
- Meyer, J.; Hentschke, R.; Hager, J.; Hojdis, N.W.; Karimi-Varzaneh, H.A. Molecular Simulation of Viscous Dissipation due to Cyclic Deformation of a Silica–Silica Contact in Filled Rubber. Macromolecules 2017, 50, 6679–6689. [Google Scholar] [CrossRef]
- Maurel, G.; Goujon, F.; Schnell, B.; Malfreyt, P. Multiscale Modeling of the Polymer–Silica Surface Interaction: From Atomistic to Mesoscopic Simulations. J. Phys. Chem. C 2015, 119, 4817–4826. [Google Scholar] [CrossRef]
- Guseva, D.V.; Komarov, P.V.; Lyulin, A.V. Molecular-dynamics simulations of thin polyisoprene films confined between amorphous silica substrates. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [Green Version]
- Guseva, D.V.; Komarov, P.V.; Lyulin, A.V. Computational synthesis, structure, and glass transition of (1,4) Cis-polyisoprene-based nanocomposite by multiscale modeling. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 473–485. [Google Scholar] [CrossRef]
- Skountzos, E.N.; Anastassiou, A.; Mavrantzas, V.G.; Theodorou, D.N. Determination of the Mechanical Properties of a Poly(methyl methacrylate) Nanocomposite with Functionalized Graphene Sheets through Detailed Atomistic Simulations. Macromolecules 2014, 47, 8072–8088. [Google Scholar] [CrossRef]
- Hu, Y.; Ding, J. Effects of morphologies of carbon nanofillers on the interfacial and deformation behavior of polymer nanocomposites—A molecular dynamics study. Carbon 2016, 107, 510–524. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, J.; Wu, Y.; Zhang, L.; Wang, Z.; Li, Y. Molecular insights into the effect of graphene packing on mechanical behaviors of graphene reinforced cis-1,4-polybutadiene polymer nanocomposites. Phys. Chem. Chem. Phys. 2017, 19, 22417–22433. [Google Scholar] [CrossRef] [PubMed]
- Rouhi, S.; Alizadeh, Y.; Ansari, R. On the elastic properties of single-walled carbon nanotubes/poly(ethylene oxide) nanocomposites using molecular dynamics simulations. J. Mol. Model. 2016, 22, 41. [Google Scholar] [CrossRef]
- Lin, F.; Xiang, Y.; Shen, H.S. Temperature dependent mechanical properties of graphene reinforced polymer nanocomposites—A molecular dynamics simulation. Compos. Part B Eng. 2017, 111, 261–269. [Google Scholar] [CrossRef]
- Pandey, Y.N.; Papakonstantopoulos, G.J.; Doxastakis, M. Polymer/Nanoparticle Interactions: Bridging the Gap. Macromolecules 2013, 46, 5097–5106. [Google Scholar] [CrossRef]
- Baeza, G.P.; Genix, A.C.; Degrandcourt, C.; Petitjean, L.; Gummel, J.; Couty, M.; Oberdisse, J. Multiscale Filler Structure in Simplified Industrial Nanocomposite Silica/SBR Systems Studied by SAXS and TEM. Macromolecules 2012, 46, 317–329. [Google Scholar] [CrossRef]
- Gersappe, D. Molecular Mechanisms of Failure in Polymer Nanocomposites. Phys. Rev. Lett. 2002, 89, 058301. [Google Scholar] [CrossRef]
- Hagita, K.; Morita, H.; Takano, H. Molecular dynamics simulation study of a fracture of filler–filled polymer nanocomposites. Polymer 2016, 99, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zheng, Z.; Davris, T.; Li, F.; Liu, J.; Wu, Y.; Zhang, L.; Lyulin, A.V. Influence of Morphology on the Mechanical Properties of Polymer Nanocomposites Filled with Uniform or Patchy Nanoparticles. Langmuir 2016, 32, 8473–8483. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Z.; Wen, S.; Yang, Q.; Zhang, L.; Zhong, C.; Liu, L. Molecular simulation study of role of polymer–particle interactions in the strain-dependent viscoelasticity of elastomers (Payne effect). J. Chem. Phys. 2014, 141, 104901. [Google Scholar] [CrossRef] [PubMed]
- Yagyu, H. Simulations of the effects of filler aggregation and filler-rubber bond on the elongation behavior of filled cross-linked rubber by coarse-grained molecular dynamics. Soft Mater. 2017, 15, 263–271. [Google Scholar] [CrossRef]
- Molinari, N.; Sutton, A.; Mostofi, A. Mechanisms of reinforcement in polymer nanocomposites. Phys. Chem. Chem. Phys. 2018, 20, 23085–23094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquini, M.; Raos, G. Tunable interaction potentials and morphology of polymer–nanoparticle blends. J. Chem. Phys. 2020, 152, 174902. [Google Scholar] [CrossRef]
- Raos, G.; Moreno, M.; Elli, S. Computational Experiments on Filled Rubber Viscoelasticity: What Is the Role of Particle–particle Interactions? Macromolecules 2006, 39, 6744–6751. [Google Scholar] [CrossRef]
- Raos, G.; Casalegno, M. Nonequilibrium simulations of filled polymer networks: Searching for the origins of reinforcement and nonlinearity. J. Chem. Phys. 2011, 134, 054902. [Google Scholar] [CrossRef]
- Groot, R.D. Electrostatic interactions in dissipative particle dynamics—simulation of polyelectrolytes and anionic surfactants. J. Chem. Phys. 2003, 118, 11265–11277. [Google Scholar] [CrossRef]
- Buglakov, A.; Ivanov, V.; Komarov, P.; Zherenkova, L.; Chiu, Y.T. A Study of Films Based on Acrylic Copolymers: Mesoscopic Simulation. Polym. Sci. Ser. A 2020, 62, 307–319. [Google Scholar] [CrossRef]
- Mann, B.A.; Holm, C.; Kremer, K. Swelling of polyelectrolyte networks. J. Chem. Phys. 2005, 122, 154903. [Google Scholar] [CrossRef] [PubMed]
- Yelash, L.; Müller, M.; Paul, W.; Binder, K. How well can coarse-grained models of real polymers describe their structure? the case of polybutadiene. J. Chem. Theory Comput. 2006, 2, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; Leone, G. Recent advances in the polymerization of butadiene over the last decade. Polyolefins J. 2014, 1, 43–60. [Google Scholar]
- Ozawa, Y.; Takata, T. Synthesis and property of end-functionalized poly(cis-1,4-butadiene) and its application to rubber compound. J. Appl. Polym. Sci. 2019, 136, 1–8. [Google Scholar] [CrossRef]
- Israelachvili, J. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Yethiraj, A. Molecular modeling of polymers at surfaces. Chem. Eng. J. 1999, 74, 109–115. [Google Scholar] [CrossRef]
- Froltsov, V.A.; Klüppel, M.; Raos, G. Molecular dynamics simulation of rupture in glassy polymer bridges within filler aggregates. Phys. Rev. E 2012, 86, 041801. [Google Scholar] [CrossRef]
- Batistakis, C.; Michels, M.; Lyulin, A.V. Confinement-induced stiffening of thin elastomer films: Linear and nonlinear mechanics vs local dynamics. Macromolecules 2014, 47, 4690–4703. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, J.; Ding, M.; Shi, T. Effects of Polymer–Wall Interactions on Entanglements and Dynamics of Confined Polymer Films. J. Phys. Chem. B 2017, 121, 1448–1454. [Google Scholar] [CrossRef]
- Smith, G.D.; Paul, W. United Atom Force Field for Molecular Dynamics Simulations of 1,4-Polybutadiene Based on Quantum Chemistry Calculations on Model Molecules. J. Phys. Chem. A 1998, 102, 1200–1208. [Google Scholar] [CrossRef]
- He, L.; Sewell, T.D.; Thompson, D.L. Molecular dynamics simulations of shock waves in cis-1,4-polybutadiene melts. J. Appl. Phys. 2013, 114, 163517. [Google Scholar] [CrossRef]
- David, A.; De Nicola, A.; Tartaglino, U.; Milano, G.; Raos, G. Viscoelasticity of Short Polymer Liquids from Atomistic Simulations. J. Electrochem. Soc. 2019, 166, B3246–B3256. [Google Scholar] [CrossRef]
- Izvekov, S.; Voth, G.A. Modeling real dynamics in the coarse-grained representation of condensed phase systems. J. Chem. Phys. 2006. [Google Scholar] [CrossRef] [PubMed]
- Meinel, M.K.; Müller-Plathe, F. Loss of Molecular Roughness upon Coarse-Graining Predicts the Artificially Accelerated Mobility of Coarse-Grained Molecular Simulation Models. J. Chem. Theory Comput. 2020, 16, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Belton, D.J.; Deschaume, O.; Perry, C.C. An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances. FEBS J. 2012, 279, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.S.; Puddu, V.; Berry, R.J.; Varshney, V.; Patwardhan, S.V.; Perry, C.C.; Heinz, H. Force field and a surface model database for silica to simulate interfacial properties in atomic resolution. Chem. Mater. 2014, 26, 2647–2658. [Google Scholar] [CrossRef]
- Rahimi, M.; Iriarte-Carretero, I.; Ghanbari, A.; Böhm, M.C.; Müller-Plathe, F. Mechanical behavior and interphase structure in a silica–polystyrene nanocomposite under uniaxial deformation. Nanotechnology 2012, 23, 305702. [Google Scholar] [CrossRef]
- Liu, S.; Böhm, M.C.; Müller-Plathe, F. Role of the interfacial area for structure and dynamics in polymer nanocomposites: Molecular dynamics simulations of polystyrene with silica nanoparticles of different shapes. Mater. Res. Express 2016, 3, 105301. [Google Scholar] [CrossRef]
- Pastore, R.; Raos, G. Glassy dynamics of a polymer monolayer on a heterogeneous disordered substrate. Soft Matter 2015, 11, 8083–8091. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Li, F.; Liu, J.; Pastore, R.; Raos, G.; Wu, Y.; Zhang, L. Effects of chemically heterogeneous nanoparticles on polymer dynamics: Insights from molecular dynamics simulations. Soft Matter 2018, 14. [Google Scholar] [CrossRef]
- Pastore, R.; David, A.; Casalegno, M.; Greco, F.; Raos, G. Influence of wall heterogeneity on nanoscopically confined polymers. Phys. Chem. Chem. Phys. 2019, 21, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Mark, J.E. Physical Properties of Polymers Handbook; Springer: New York, NY, USA, 2007; Volume 1076. [Google Scholar]
- Sandia National Laboratories. LAMMPS, Large-Scale Atomic/Molecular Massively Parallel Simulator. Available online: https://lammps.sandia.gov/ (accessed on 29 June 2020).
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Reith, D.; Pütz, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24, 1624–1636. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Kott, S.J. Liquid structuring at solid interfaces as probed by direct force measurements: The transition from simple to complex liquids and polymer fluids. J. Chem. Phys. 1988, 88, 7162–7166. [Google Scholar] [CrossRef]
- Ruths, M.; Granick, S. Tribology of Confined Fomblin-Z Perfluoropolyalkyl Ethers: Role of Chain-End Chemical Functionality. J. Phys. Chem. B 1999, 103, 8711–8721. [Google Scholar] [CrossRef]
- Sun, G.; Butt, H.J. Adhesion between Solid Surfaces in Polymer Melts: Bridging of Single Chains. Macromolecules 2004, 37, 6086–6089. [Google Scholar] [CrossRef]
- Shen, J.; Lin, X.; Liu, J.; Li, X. Effects of Cross-Link Density and Distribution on Static and Dynamic Properties of Chemically Cross-Linked Polymers. Macromolecules 2019, 52, 121–134. [Google Scholar] [CrossRef]
- Thompson, A.P.; Plimpton, S.J.; Mattson, W. General formulation of pressure and stress tensor for arbitrary many-body interaction potentials under periodic boundary conditions. J. Chem. Phys. 2009, 131, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Thompson, T.; Wang, S.Q.; von Meerwall, E.D.; Halasa, A. Investigating Linear and Nonlinear Viscoelastic Behavior Using Model Silica–particle-Filled Polybutadiene. Macromolecules 2005, 38, 8816–8824. [Google Scholar] [CrossRef]
- Wang, W.; Hou, G.; Zheng, Z.; Wang, L.; Liu, J.; Wu, Y.; Zhang, L.; Lyulin, A.V. Designing polymer nanocomposites with a semi-interpenetrating or interpenetrating network structure: Toward enhanced mechanical properties. Phys. Chem. Chem. Phys. 2017, 19, 15808–15820. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
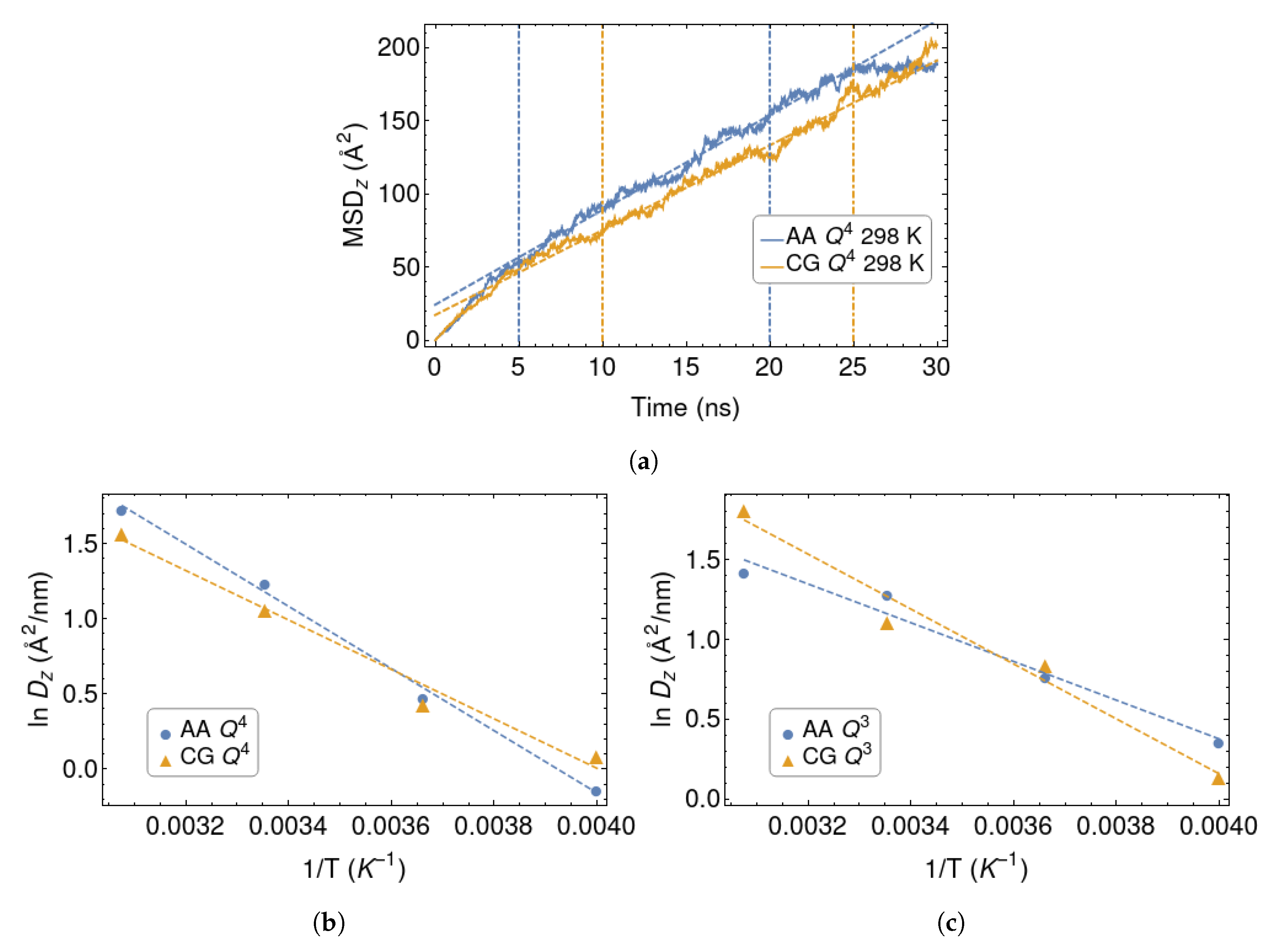{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH | CH | Q3 | Q4 | |||||
|---|---|---|---|---|---|---|---|---|
| CH | 0.0936 | 4.500 | ||||||
| CH | 0.1015 | 4.257 | 0.1000 | 3.800 | ||||
| Q3 | 0.0967 | 5.065 | 0.1000 | 4.654 | 0.1000 | 5.700 | ||
| Q4 | 0.1676 | 4.450 | 0.1732 | 4.089 | 0.1732 | 5.008 | 0.3000 | 4.400 |
| Silica Surface Model | Ea (kcal/mol) |
|---|---|
| AA | 4.11 |
| CG | 3.27 |
| AA | 2.41 |
| CG | 3.41 |
| T (K) | Surface | (ps) | (ps) | |
|---|---|---|---|---|
| 325 | AA Q | 2269 | 0.488 | 4751 |
| CG Q | 2994 | 0.497 | 6055 | |
| AA Q | 2536 | 0.517 | 4777 | |
| CG Q | 2736 | 0.506 | 5354 | |
| 298 | AA Q | 3457 | 0.520 | 6448 |
| CG Q | 2620 | 0.535 | 4660 | |
| AA Q | 3446 | 0.590 | 5301 | |
| CG Q | 3788 | 0.562 | 6242 | |
| 273 | AA Q | 5463 | 0.593 | 8348 |
| CG Q | 5490 | 0.562 | 9046 | |
| AA Q | 6187 | 0.545 | 10,684 | |
| CG Q | 5420 | 0.550 | 9227 | |
| 250 | AA Q | 7176 | 0.524 | 13,211 |
| CG Q | 7336 | 0.531 | 13,209 | |
| AA Q | 12,037 | 0.705 | 15,134 | |
| CG Q | 10,335 | 0.654 | 14,025 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, A.; Pasquini, M.; Tartaglino, U.; Raos, G. A Coarse-Grained Force Field for Silica–Polybutadiene Interfaces and Nanocomposites. Polymers 2020, 12, 1484. https://doi.org/10.3390/polym12071484
David A, Pasquini M, Tartaglino U, Raos G. A Coarse-Grained Force Field for Silica–Polybutadiene Interfaces and Nanocomposites. Polymers. 2020; 12(7):1484. https://doi.org/10.3390/polym12071484
Chicago/Turabian StyleDavid, Alessio, Marta Pasquini, Ugo Tartaglino, and Guido Raos. 2020. "A Coarse-Grained Force Field for Silica–Polybutadiene Interfaces and Nanocomposites" Polymers 12, no. 7: 1484. https://doi.org/10.3390/polym12071484
APA StyleDavid, A., Pasquini, M., Tartaglino, U., & Raos, G. (2020). A Coarse-Grained Force Field for Silica–Polybutadiene Interfaces and Nanocomposites. Polymers, 12(7), 1484. https://doi.org/10.3390/polym12071484