
High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of GO Aqueous Dispersion
2.3. Preparation of GO Ethanol Dispersion by Solvent Replacement
2.4. Preparation of GO/PA66 Salt Nanocomposites by In Situ Precipitation
2.5. Fabrication of GO/PA66 Nanocomposite Fibers by In Situ Polymerization
2.6. Characterizations
3. Results and Discussion
3.1. Formation of GO/PA66 Salt Nanocomposites
3.2. In Situ Polymerization of GO/PA66 Salt Nanocomposites
3.3. Properties of GO/PA66 Nanocomposite Fibers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spina, R.; Cavalcante, B. Evaluation of grinding of unfilled and glass fiber reinforced polyamide 6,6. Polymers 2020, 12, 2288. [Google Scholar] [CrossRef]
- Cakal Sarac, E.; Haghighi Poudeh, L.; Berktas, I.; Saner Okan, B. Scalable fabrication of high-performance graphene/polyamide 66 nanocomposites with controllable surface chemistry by melt compounding. J. Appl. Polym. Sci. 2021, 138, 1–12. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, X.; Zhang, X.; Liu, H.; Zhang, H.; Zhang, X. Polyamide 66 and amino-functionalized multi-walled carbon nanotube composites and their melt-spun fibers. J. Mater. Sci. 2019, 54, 11056–11068. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, J.; Yang, H.; Lang, J.; Yang, H. Comparative study on the flame-retardant properties and mechanical properties of PA66 with different dicyclohexyl hypophosphite acid metal salts. Polymers 2019, 11, 1956. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Liao, J.; Dong, J.; Wu, J.; Qiu, H.; Zhou, X.; Li, J.; Jiang, D.; He, T.C.; Quan, Z. An effective treatment of experimental osteomyelitis using the antimicrobial titanium/silver-containing nHP66 (nano-hydroxyapatite/polyamide-66) nanoscaffold biomaterials. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirvakili, S.M.; Hunter, I.W. Multidirectional Artificial Muscles from Nylon. Adv. Mater. 2017, 29, 1–7. [Google Scholar] [CrossRef]
- Haines, C.S.; Lima, M.D.; Li, N.; Spinks, G.M.; Foroughi, J.; Madden, J.D.W.; Kim, S.H.; Fang, S.; De Andrade, M.J.; Göktepe, F.; et al. Artificial muscles from fishing line and sewing thread. Science 2014, 343, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Li, S.M.; Hsiao, S.T.; Liao, W.H.; Chen, P.H.; Yang, S.Y.; Tien, H.W.; Ma, C.C.M.; Hu, C.C. Integration of tailored reduced graphene oxide nanosheets and electrospun polyamide-66 nanofabrics for a flexible supercapacitor with high-volume- and high-area-specific capacitance. Carbon 2014, 73, 87–98. [Google Scholar] [CrossRef]
- Oh, J.Y.; Choi, Y.S.; Yang, S.J.; Kim, J.; Choi, H.S.; Choi, G.D.; Yun, C.H.; Lee, B.K.; Park, C.R. Effect of microstructure and morphological properties of carbon nanotubes on the length reduction during melt processing. Compos. Sci. Technol. 2015, 112, 42–49. [Google Scholar] [CrossRef]
- Dericiler, K.; Sadeghi, H.M.; Yagci, Y.E.; Sas, H.S.; Okan, B.S. Experimental and numerical investigation of flow and alignment behavior of waste tire-derived graphene nanoplatelets in PA66 matrix during melt-mixing and injection. Polymers 2021, 13, 949. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.G.; Lee, J.E.; Hwang, S.H.; Han, J.H.; Chae, H.G.; Park, Y. Bin Enhancement in mechanical properties of polyamide 66-carbon fiber composites containing graphene oxide-carbon nanotube hybrid nanofillers synthesized through in situ interfacial polymerization. Compos. Part A Appl. Sci. Manuf. 2020, 135, 105938. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Orlita, M.; Faugeras, C.; Plochocka, P.; Neugebauer, P.; Martinez, G.; Maude, D.K.; Barra, A.L.; Sprinkle, M.; Berger, C.; De Heer, W.A.; et al. Approaching the dirac point in high-mobility multilayer epitaxial graphene. Phys. Rev. Lett. 2008, 101, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Young, R.J.; Liu, M.; Kinloch, I.A.; Li, S.; Zhao, X.; Vallés, C.; Papageorgiou, D.G. The mechanics of reinforcement of polymers by graphene nanoplatelets. Compos. Sci. Technol. 2018, 154, 110–116. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; Zhou, J.; Li, B. Thermal Conductivity of Polymers and Their Nanocomposites. Adv. Mater. 2018, 30, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Petkovich, N.D.; Liu, K.; Qian, Y.; Macosko, C.W.; Stein, A. Unsaturated polyester resin toughening with very low loadings of GO derivatives. Polymer 2017, 110, 149–157. [Google Scholar] [CrossRef]
- Park, Y.T.; Qian, Y.; Chan, C.; Suh, T.; Nejhad, M.G.; Macosko, C.W.; Stein, A. Epoxy toughening with low graphene loading. Adv. Funct. Mater. 2015, 25, 575–585. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Gao, X.F.; Liu, H.H.; Zhang, J.; Zhang, X.X. Green fabrication of functionalized graphene via one-step method and its reinforcement for polyamide 66 fibers. Mater. Chem. Phys. 2020, 240, 122288. [Google Scholar] [CrossRef]
- Papadopoulou, E.L.; Pignatelli, F.; Marras, S.; Marini, L.; Davis, A.; Athanassiou, A.; Bayer, I.S. Nylon 6,6/graphene nanoplatelet composite films obtained from a new solvent. RSC Adv. 2016, 6, 6823–6831. [Google Scholar] [CrossRef]
- Cho, B.G.; Lee, S.; Hwang, S.H.; Han, J.H.; Chae, H.G.; Park, Y. Bin Influence of hybrid graphene oxide-carbon nanotube as a nano-filler on the interfacial interaction in nylon composites prepared by in situ interfacial polymerization. Carbon 2018, 140, 324–337. [Google Scholar] [CrossRef]
- Ji, X.; Xu, Y.; Zhang, W.; Cui, L.; Liu, J. Review of functionalization, structure and properties of graphene/polymer composite fibers. Compos. Part A Appl. Sci. Manuf. 2016, 87, 29–45. [Google Scholar] [CrossRef]
- Dallinger, A.; Kindlhofer, P.; Greco, F.; Coclite, A.M. Multiresponsive Soft Actuators Based on a Thermoresponsive Hydrogel and Embedded Laser-Induced Graphene. ACS Appl. Polym. Mater. 2021, 3, 1809–1818. [Google Scholar] [CrossRef]
- Cataldi, P.; Steiner, P.; Raine, T.; Lin, K.; Kocabas, C.; Young, R.J.; Bissett, M.; Kinloch, I.A.; Papageorgiou, D.G. Multifunctional Biocomposites Based on Polyhydroxyalkanoate and Graphene/Carbon Nanofiber Hybrids for Electrical and Thermal Applications. ACS Appl. Polym. Mater. 2020, 2, 3525–3534. [Google Scholar] [CrossRef]
- Cseri, L.; Baugh, J.; Alabi, A.; AlHajaj, A.; Zou, L.; Dryfe, R.A.W.; Budd, P.M.; Szekely, G. Graphene oxide-polybenzimidazolium nanocomposite anion exchange membranes for electrodialysis. J. Mater. Chem. A 2018, 6, 24728–24739. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Yu, B.; Yang, T.; Wu, Y.; Yu, H.; Huang, T. In situ polymerization of nylon 66/reduced graphene oxide nanocomposites. J. Nanomater. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef]
- Hong, B.J.; Compton, O.C.; An, Z.; Eryazici, I.; Nguyen, S.T. Successful stabilization of graphene oxide in electrolyte solutions: Enhancement of biofunctionalization and cellular uptake. ACS Nano 2012, 6, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Dhayal, V.; Hashmi, S.Z.; Kumar, U.; Choudhary, B.L.; Kuznetsov, A.E.; Dalela, S.; Kumar, S.; Kaya, S.; Dolia, S.N.; Alvi, P.A. Spectroscopic studies, molecular structure optimization and investigation of structural and electrical properties of novel and biodegradable Chitosan-GO polymer nanocomposites. J. Mater. Sci. 2020, 55, 14829–14847. [Google Scholar] [CrossRef]
- Alammar, A.; Park, S.H.; Ibrahim, I.; Deepak, A.; Holtzl, T.; Dumée, L.F.; Lim, H.N.; Szekely, G. Architecting neonicotinoid-scavenging nanocomposite hydrogels for environmental remediation. Appl. Mater. Today 2020, 21, 100878. [Google Scholar] [CrossRef]
- Kinloch, I.A.; Suhr, J.; Lou, J.; Young, R.J.; Ajayan, P.M. Composites with carbon nanotubes and graphene: An outlook. Science 2018, 362, 547–553. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Shen, L.; Zhang, X.; Wang, Y.; Bao, N.; Kung, H.H. An efficient and eco-friendly solution-chemical route for preparation of ultrastable reduced graphene oxide suspensions. AIChE J. 2014, 60, 2757–2764. [Google Scholar] [CrossRef]
- Li, C.; Guo, Y.; Shen, L.; Ji, C.; Bao, N. Scalable concentration process of graphene oxide dispersions via cross-flow membrane filtration. Chem. Eng. Sci. 2019, 200, 127–137. [Google Scholar] [CrossRef]
- Yoon, K.Y.; An, S.J.; Chen, Y.; Lee, J.H.; Bryant, S.L.; Ruoff, R.S.; Huh, C.; Johnston, K.P. Graphene oxide nanoplatelet dispersions in concentrated NaCl and stabilization of oil/water emulsions. J. Colloid Interface Sci. 2013, 403, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Lu, H.; Zhuang, Z.; Wang, X.P.; Fang, Q.F. Nano-hydroxyapatite/poly(L-lactic acid) composite synthesized by a modified in situ precipitation: Preparation and properties. J. Mater. Sci. Mater. Med. 2010, 21, 3077–3083. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Zhang, X.; Gao, X.; Liu, H.; Zhang, X. Fabrication of high-strength PET fibers modified with graphene oxide of varying lateral size. J. Mater. Sci. 2020, 55, 8940–8953. [Google Scholar] [CrossRef]
- Min, C.; Nie, P.; Song, H.J.; Zhang, Z.; Zhao, K. Study of tribological properties of polyimide/graphene oxide nanocomposite films under seawater-lubricated condition. Tribol. Int. 2014, 80, 131–140. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Li, G.; Zhao, X.-Y.; Jiang, J.-M. High solubility, low-dielectric constant, and optical transparency of novel polyimides derived from 3,3′,5,5′-tetramethyl-4,4′-diaminodiphenyl-4″-isopropyltoluene. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 3309–3317. [Google Scholar] [CrossRef]
- Zangmeister, R.A.; Morris, T.A.; Tarlov, M.J. Characterization of polydopamine thin films deposited at short times by autoxidation of dopamine. Langmuir 2013, 29, 8619–8628. [Google Scholar] [CrossRef]
- Sheng, X.; Mo, R.; Ma, Y.; Zhang, X.; Zhang, L.; Wu, H. Waterborne Epoxy Resin/Polydopamine Modified Zirconium Phosphate Nanocomposite for Anticorrosive Coating. Ind. Eng. Chem. Res. 2019, 58, 16571–16580. [Google Scholar] [CrossRef]
- Wan, Y.J.; Tang, L.C.; Gong, L.X.; Yan, D.; Li, Y.B.; Wu, L.B.; Jiang, J.X.; Lai, G.Q. Grafting of epoxy chains onto graphene oxide for epoxy composites with improved mechanical and thermal properties. Carbon 2014, 69, 467–480. [Google Scholar] [CrossRef]
- Ramesh, C.; Keller, A.; Eltink, S.J.E.A. Studies on the crystallization and melting of nylon-6,6: 1. The dependence of the Brill transition on the crystallization temperature. Polymer 1994, 35, 2483–2487. [Google Scholar] [CrossRef]
- Zhang, G.; Yan, D. Crystallization kinetics and melting behavior of nylon 10,10 in nylon 10,10-montmorillonite nanocomposites. J. Appl. Polym. Sci. 2003, 88, 2181–2188. [Google Scholar] [CrossRef]
- Garcia, D.; Starkweather, H.W. Hydrogen bonding in nylon 66 and model compounds. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 537–555. [Google Scholar] [CrossRef]
- Morimune-Moriya, S.; Yada, S.; Kuroki, N.; Ito, S.; Hashimoto, T.; Nishino, T. Strong reinforcement effects of nanodiamond on mechanical and thermal properties of polyamide 66. Compos. Sci. Technol. 2020, 199, 108356. [Google Scholar] [CrossRef]
- Basavaraj, E.; Ramaraj, B. A study on mechanical, thermal, and wear characteristics of nylon 66/molybdenum disulfide composites reinforced with glass fibers. Polym. Compos. 2012, 33, 1570–1577. [Google Scholar] [CrossRef]
- Abdallah, W.; Yilmazer, U. Polyamide 66 nanocomposites based on organoclays treated with thermally stable phosphonium salts. J. Appl. Polym. Sci. 2013, 127, 772–783. [Google Scholar] [CrossRef]
- Zhou, K.; Jiang, S.; Shi, Y.; Liu, J.; Wang, B.; Hu, Y.; Gui, Z. Multigram-scale fabrication of organic modified MoS2nanosheets dispersed in polystyrene with improved thermal stability, fire resistance, and smoke suppression properties. RSC Adv. 2014, 4, 40170–40180. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
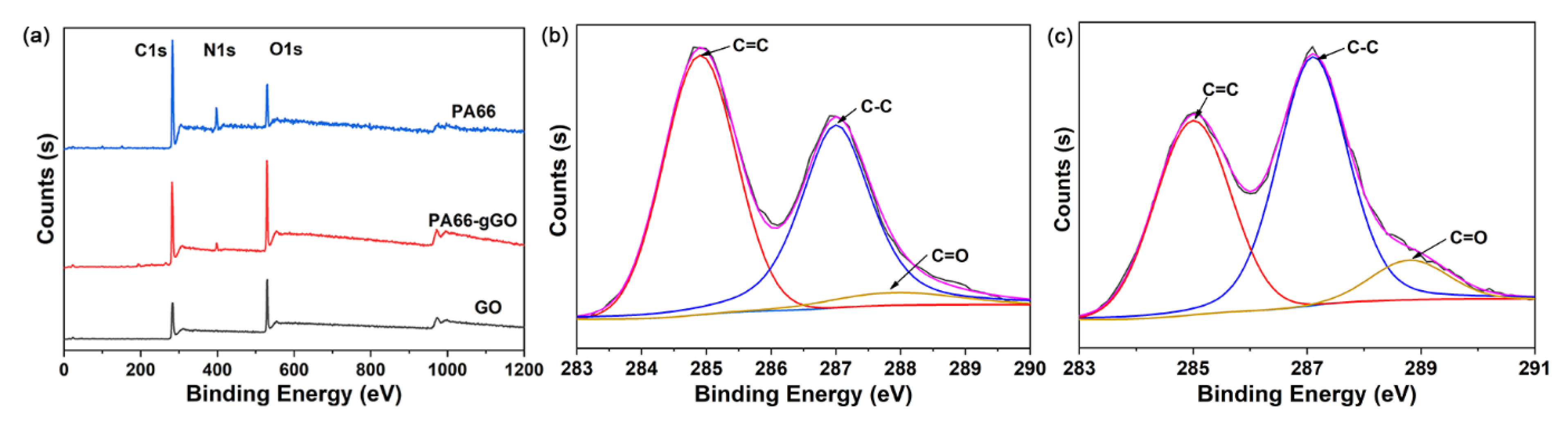{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tm (°C) | Tc (°C) | ΔHc (J/g) | Xc (%) |
|---|---|---|---|---|
| PA66-GO-0 | 260.5 | 228.4 | 53.86 | 28.5 |
| PA66-GO-0.1 | 261.1 | 228.8 | 48.15 | 25.6 |
| PA66-GO-0.2 | 261.3 | 231.1 | 50.59 | 26.9 |
| PA66-GO-0.3 | 261.8 | 234.0 | 49.62 | 26.3 |
| PA66-GO-0.4 | 262.1 | 234.3 | 52.63 | 27.9 |
| PA66-GO-0.5 | 262.5 | 234.5 | 55.09 | 29.2 |
| Samples | E’ (MPa) | Tan δ | Tg (°C) |
|---|---|---|---|
| PA66-GO-0 | 1345 | 0.120 | 43.2 |
| PA66-GO-0.1 | 1475 | 0.110 | 51.6 |
| PA66-GO-0.2 | 1691 | 0.081 | 56.1 |
| PA66-GO-0.3 | 1900 | 0.076 | 57.5 |
| PA66-GO-0.4 | 1317 | 0.073 | 58.4 |
| PA66-GO-0.5 | 970 | 0.072 | 59.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, A.; Wu, J.; Shen, L.; Zhang, X.; Bao, N. High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers 2021, 13, 1688. https://doi.org/10.3390/polym13111688
Gu A, Wu J, Shen L, Zhang X, Bao N. High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers. 2021; 13(11):1688. https://doi.org/10.3390/polym13111688
Chicago/Turabian StyleGu, Ao, Jian Wu, Liming Shen, Xiaoyan Zhang, and Ningzhong Bao. 2021. "High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization" Polymers 13, no. 11: 1688. https://doi.org/10.3390/polym13111688
APA StyleGu, A., Wu, J., Shen, L., Zhang, X., & Bao, N. (2021). High-Strength GO/PA66 Nanocomposite Fibers via In Situ Precipitation and Polymerization. Polymers, 13(11), 1688. https://doi.org/10.3390/polym13111688