
Self-Initiated Butyl Acrylate Polymerizations in Bulk and in Solution Monitored By In-Line Techniques


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Polymerization with In-Line DSC Measurement
2.4. Polymerization with In-Line FT-NIR Measurement
3. Results and Discussion
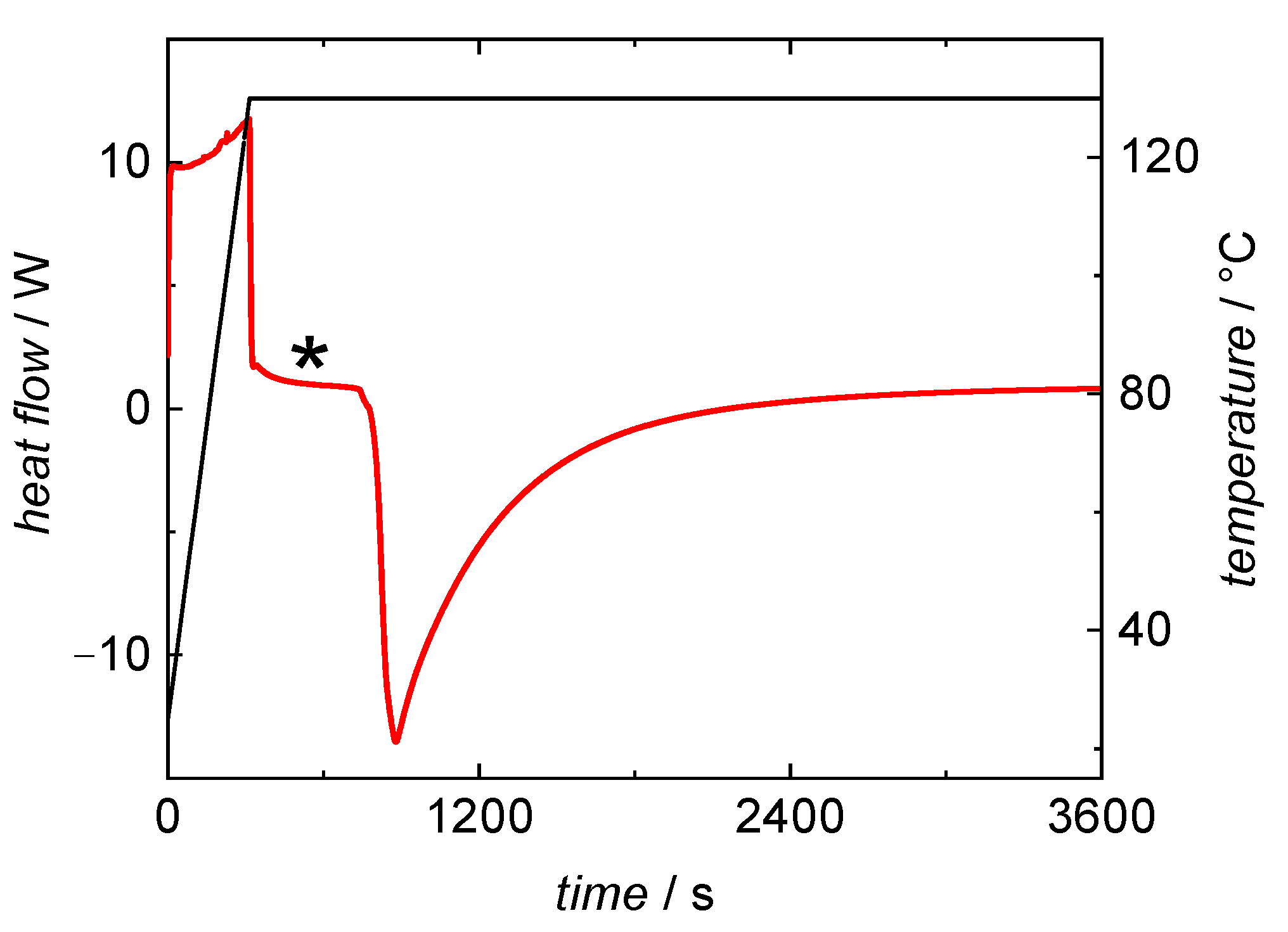
3.1. BA Polymerizations with In-Line DSC Monitoring
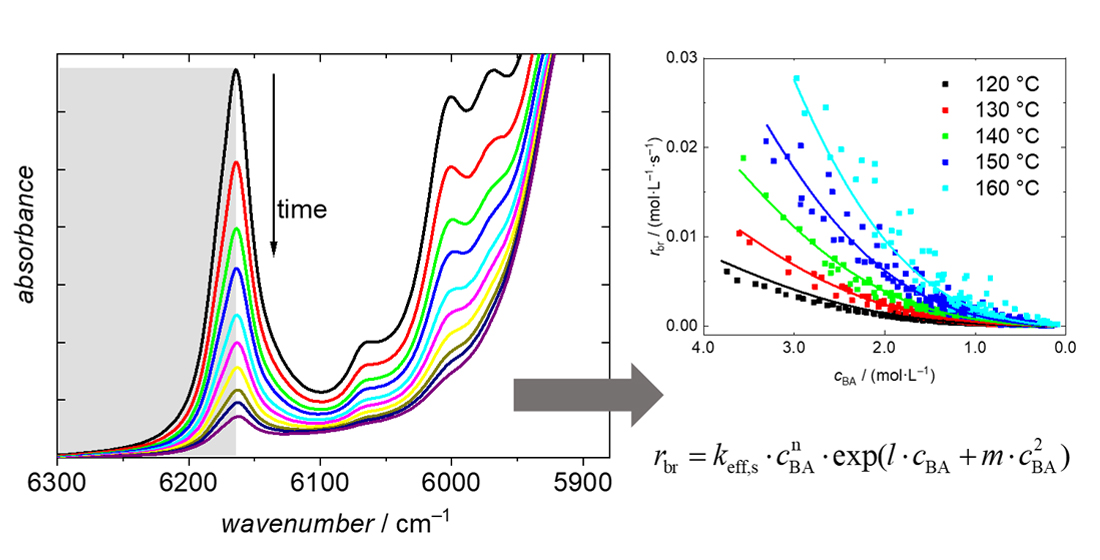
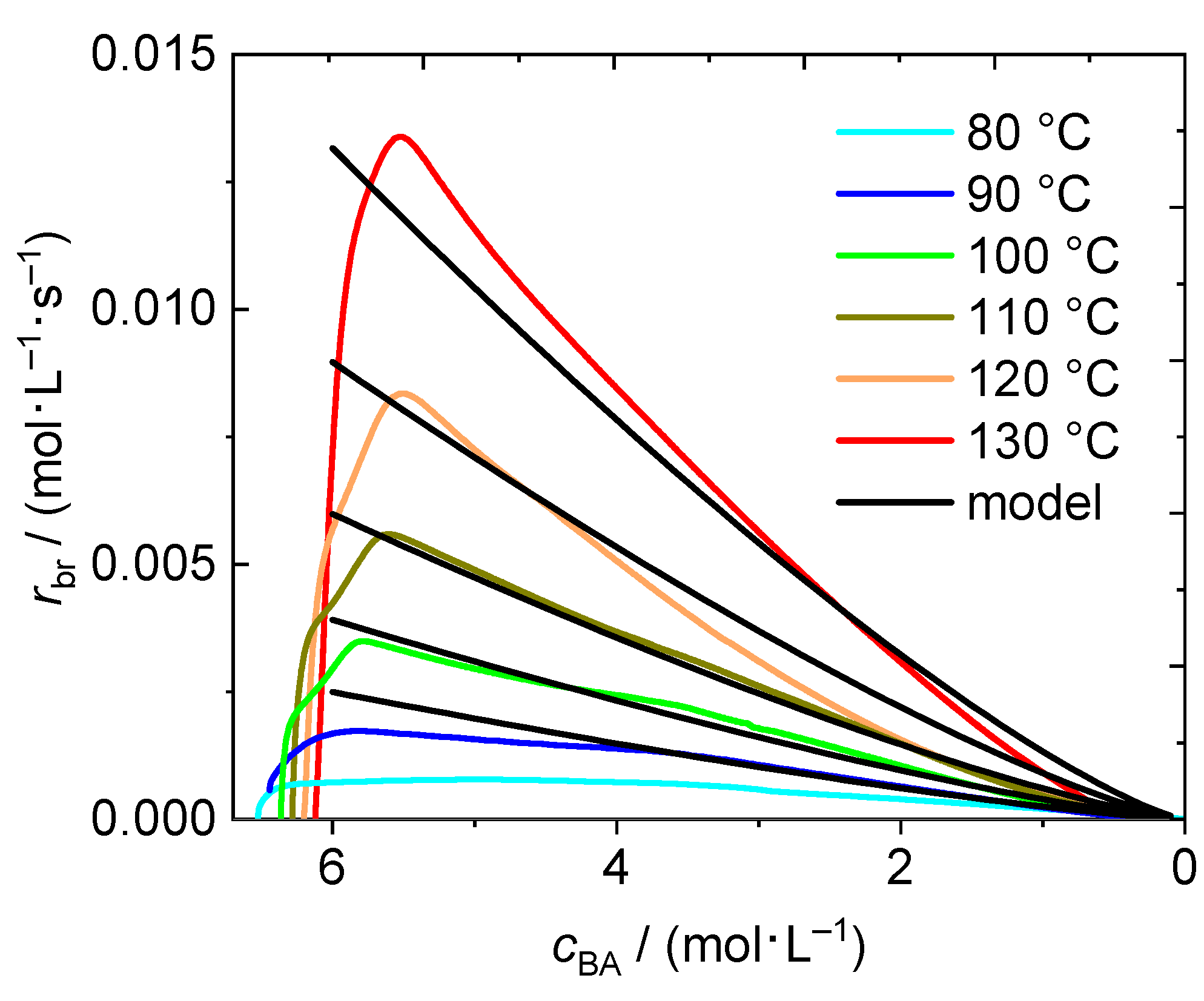
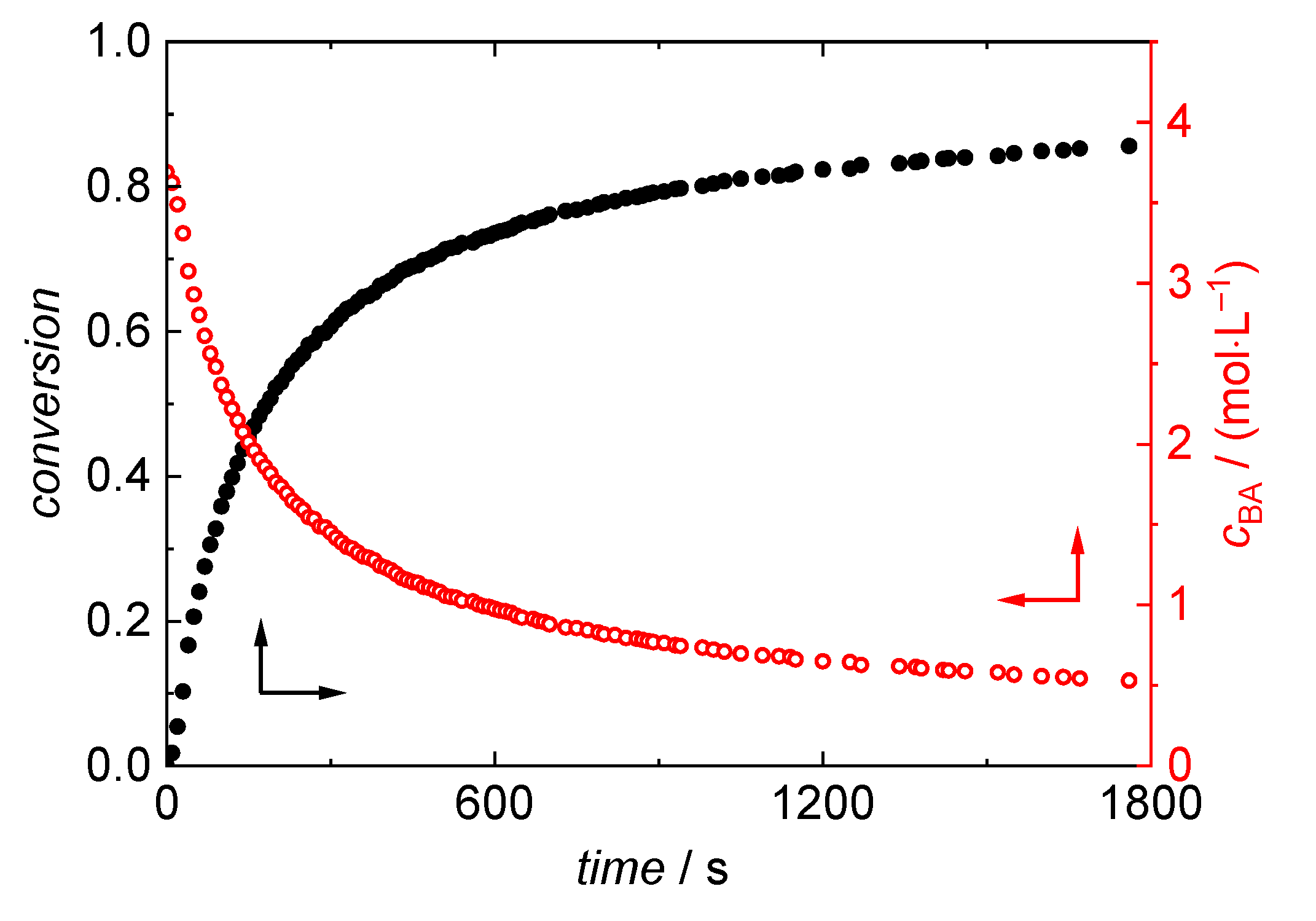
3.2. BA Polymerizations with In-Line FT-NIR Monitoring

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballard, N.; Asua, J.M. Radical polymerization of acrylic monomers: An overview. Prog. Polym. Sci. 2018, 79, 40–60. [Google Scholar] [CrossRef]
- Beuermann, S.; Paquet, D.A., Jr.; McMinn, J.H.; Hutchinson, R.A. Determination of Free-Radical Propagation Rate Coefficients of Butyl, 2-Ethylhexyl and Dodecyl Acrylates by Pulsed-Laser Polymerization. Macromolecules 1996, 29, 4206–4215. [Google Scholar] [CrossRef]
- Ahmad, N.M.; Heatley, F.; Lovell, P.A. Chain transfer to polymer in free-radical solution polymerization of n-butyl acrylate studied by NMR spectroscopy. Macromolecules 1998, 31, 2822–2827. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Leiza, J.R.; Schoonbrood, H.C.D.; Asua, J.M. A decrease in effective acrylate propagation rate constants caused by intramolecular chain transfer. Macromolecules 2000, 33, 4–7. [Google Scholar] [CrossRef]
- van Herk, A.M. Historic account of the development in the understanding of the propagation kinetics of acrylate radical polymerizations. Macromol. Rapid Commun. 2009, 30, 1964–1968. [Google Scholar] [CrossRef]
- Junkers, T.; Barner-Kowollik, C. The role of mid-chain radicals in acrylate free radical polymerization: Branching and scission. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7585–7605. [Google Scholar] [CrossRef]
- Willemse, R.X.E.; van Herk, A.M.; Panchenko, E.; Junkers, T.; Buback, M. PLP-ESR monitoring of Midchain Radicals in n-Butyl Acrylate Polymerization. Macromolecules 2005, 38, 5098–5103. [Google Scholar] [CrossRef]
- Azukizawa, M.; Yamada, B.; Hill, D.J.T.; Pomery, P.J. Radical polymerization of phenyl acrylate as studied by ESR spectroscopy: Concurrence of propagating and mid-chain radicals. Macromol. Chem. Phys. 2000, 201, 774–781. [Google Scholar] [CrossRef]
- Ballard, N.; Veloso, A.; Asua, J.M. Mid-Chain Radical Migration in the Radical Polymerization of n-Butyl Acrylate. Polymers 2018, 10, 765. [Google Scholar] [CrossRef] [Green Version]
- Vandenbergh, J.; Junkers, T. Macromonomers from AGET activation of poly(n-butyl acrylate) precursors: Radical transfer pathways and midchain radical migration. Macromolecules 2012, 45, 6850–6856. [Google Scholar] [CrossRef]
- Vir, A.B.; Marien, Y.W.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. From n-butyl acrylate Arrhenius parameters for backbiting and tertiary propagation to β-scission via stepwise pulsed laser polymerization. Polym. Chem. 2019, 10, 4116–4125. [Google Scholar] [CrossRef]
- Drache, M.; Stehle, M.; Mätzig, J.; Brandl, K.; Jungbluth, M.; Namyslo, J.; Schmidt, A.; Beuermann, S. Identification of β scission products from free radical polymerizations of butyl acrylate at high temperature. Polym. Chem. 2019, 10, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Van Steenberge, P.H.M.; Vandenbergh, J.; Junkers, T.; D’hooge, D.R.; Marin, G.B. Kinetic Monte Carlo Generation of Complete Electron Spray Ionization Mass Spectra for Acrylate Macromonomer Synthesis. Macromolecules 2017, 50, 2625–2636. [Google Scholar] [CrossRef]
- Peck, A.N.F.; Hutchinson, R.A. Secondary reactions in the high-temperature free radical polymerization of butyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Rantow, F.S.; Soroush, M.; Grady, M.C.; Kalfas, G.A. Spontaneous polymerization and chain microstructure evolution in high-temperature solution polymerization of n-butyl acrylate. Polymer 2006, 47, 1423–1435. [Google Scholar] [CrossRef]
- Riazi, H.; Shamsabadi, A.A.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Kalfas, G.A.; Petkovska, V.I.; Bruni, C.; Grady, M.C.; Soroush, M. Experimental study of the spontaneous thermal homopolymerization of methyl and n-butyl acrylate. J. Appl. Polym. Sci. 2010, 118, 1898–1909. [Google Scholar] [CrossRef]
- Shamsabadi, A.A.; Moghadam, N.; Srinivasan, S.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling. Processes 2016, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Riazi, H.; Shamsabadi, A.A.; Grady, M.C.; Rappe, A.M.; Soroush, M. Experimental and theoretical Study of the Self-Initiation Reaction of Methyl Acrylate in Free-Radical Polymerization. Ind. Eng. Chem. Res. 2018, 57, 532–539. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational Study of the Self-Initiation Mechanism in Thermal Polymerization of Methyl Acrylate. J. Phys. Chem. A 2009, 113, 10787–10794. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Tao, J.; Grady, M.C.; Soroush, M.; Rappe, A.M. Modeling Spin-Forbidden Monomer Self-Initiation Reactions in Spontaneous Free-Radical Polymerization of Acrylates and Methacrylates. J. Phys. Chem. A 2014, 118, 9310–9318. [Google Scholar] [CrossRef]
- Laki, S.; Shamsabadi, A.A.; Riazi, H.; Grady, M.C.; Rappe, A.M.; Soroush, M. Experimental and Mechanistic Modeling Study of Self-Initiated High-Temperature Polymerization of Ethyl Acrylate. Ind. Eng. Chem. Res. 2020, 59, 2621–2630. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Self-Initiation Mechanism in Spontaneous Thermal Polymerization of Ethyl and n-Butyl Acrylate: A Theoretical Study. J. Phys. Chem. A 2010, 114, 7975–7983. [Google Scholar] [CrossRef] [Green Version]
- Hamzehlou, S.; Ballard, N.; Reyes, Y.; Aguirre, A.; Asua, J.; Leiza, J. Analyzing the discrepancies in the activation energies of the backbiting and β-scission reactions in the radical polymerization of n-butyl acrylate. Polym. Chem. 2016, 7, 2069–2077. [Google Scholar] [CrossRef]
- Buback, M. Spectroscopy of fluid phases—The study of reactions and equilibrium up to high conversion. Angew. Chem. Int. Ed. Engl. 1991, 30, 641–653. [Google Scholar] [CrossRef]
- Haven, J.J.; Junkers, T. Online Monitoring of Polymerizations: Current Status. Eur. J. Org. Chem. 2017, 2017, 6474–6482. [Google Scholar] [CrossRef]
- Vrijsen, J.H.; Thomlinson, I.A.; Levere, M.E.; Lyall, C.L.; Davidson, M.G.; Hintermair, U.; Junkers, T. Online tracing of molecular weight evolution during radical polymerization via high-resolution FlowNMR spectroscopy. Polym. Chem. 2020, 11, 3546–3550. [Google Scholar] [CrossRef]
- Buback, M.; Hippler, H.; Schweer, J.; Vögele, H.-P. Time-resolved study of laser induced high pressure ethylene polymerization. Die Makromol. Chem. Rapid Commun. 1986, 7, 261–265. [Google Scholar] [CrossRef]
- Buback, M.; Kowollik, C. Termination Kinetics in free-radical Bulk Terpolymerization—The Systems Methyl acrylate—Butyl acrylate—Dodecyl acrylate and Methyl methacrylate—Butyl methacrylate—Dodecyl methacrylate. Macromol. Chem. Phys. 1999, 200, 1765–1770. [Google Scholar] [CrossRef]
- Drawe, P.; Kattner, H.; Buback, M. Kinetics and Modeling of the Radical Polymerization of Trimethylaminoethyl methacrylate chloride in Aqueous Solution. Macromol. Chem. Phys. 2016, 217, 2755–2764. [Google Scholar] [CrossRef]
- Möller, E.; Beuermann, S. Homogeneous phase copolymerizations of vinylidene fluoride and hexafluoropropene in supercritical carbon dioxide. Macromol. React. Eng. 2011, 5, 8–21. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Hesse, P.; Junkers, T.; Lacík, I. Free-Radical Polymerization Kinetics of 2-Acrylamido-2-methylpropanesulfonic acid in Aqueous Solution. Macromolecules 2006, 39, 509–516. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Schmaltz, C. Termination rate coefficients of butyl acrylate free-radical homopolymerization in supercritical CO2 and in bulk. Ind. Eng. Chem. Res. 1999, 38, 3338–3344. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Russell, G.T. Kinetics of free radical solution polymerization of methyl methacrylate over extended conversion range. Macromol. Chem. Phys. 1995, 196, 2493–2516. [Google Scholar] [CrossRef]
- Achilias, D.S.; Siafaka, P.I. Polymerization Kinetics of Poly(2-Hydroxyethyl Methacrylate) Hydrogels and Nanocomposite Materials. Processes 2017, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-W.; Sun, Y.-M.; Huang, W.-F. Curing Kinetics of the Synthesis of Poly(2-hydroxyethylmethacrylate) (PHEMA) with Ethylene GlycolDimethacrylate (EGDMA) as a Crosslinking Agent. J. Polym. Sci. Part A 1997, 35, 1873–1889. [Google Scholar] [CrossRef]
- Achilias, D.S.; Tsagkalias, I.S. Investigation of radical polymerization kinetics of poly(ethylene glycol)methacrylate hydrogels via DSC and mechanistic or isoconversional models. J. Therm. Anal. Calorim. 2018, 134, 1307–1315. [Google Scholar] [CrossRef]
- Lee, E.J.; Park, H.J.; Kim, S.M.; Lee, K.J. Effect of Azo and Peroxide Initiators on a Kinetic Study of Methyl Methacrylate Free Radical Polymerization by DSC. Macromol. Res. 2018, 26, 322–331. [Google Scholar] [CrossRef]
- Achilias, D.S. Investigation of radical polymerization kinetics using DSX and mechanistic or isoconversional methods. J. Therm. Anal. Calorim. 2014, 116, 1379–1386. [Google Scholar] [CrossRef]
- Victoria-Valenzuela, D.; Herrera-Ordonez, J.; Arcos-Casarrubias, A.; Vazquez-Torres, H. Kinetics of Bulk Free-Radical Polymerization of Butyl Methacrylate Isomers Studied by Reaction Calorimetry. Macromol. React. Eng. 2018, 12, 1700046. [Google Scholar] [CrossRef]
- Achilias, D.I.; Verros, G.D. Modeling of Diffusion-Controlled Reactions in Free Radical Solution and Bulk Polymerization: Model Validation by DSC Experiments. J. Appl. Polym. Sci. 2010, 116, 1842–1856. [Google Scholar] [CrossRef]
- Buback, M.; Kurz, C.H.; Schmaltz, C. Pressure dependence of propagation rate coefficients in free-radical homopolymerizations of methyl acrylate and dodecyl acrylate. Macromol. Chem. Phys. 1998, 199, 1721–1727. [Google Scholar] [CrossRef]
- Ogo, Y.; Yokawa, M. Effect of pressure on the propagation and termination rate constants of radical polymerization. Die Makromol. Chem. 1977, 178, 453–464. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; El-Rezzi, V.; Jürgens, M.; Nelke, D. Influence of Monomer Structure on the Propagation Kinetics of Acrylate and Methacrylate Homopolymerization studied via PLP-SEC in Fluid CO2. Macromol. Chem. Phys. 2004, 205, 876–883. [Google Scholar] [CrossRef]
- McCurdy, K.G.; Laidler, K.J. Thermochemical studies of some acrylate and methacrylate polymerizations in emulsion systems. Can. J. Chem. 1964, 42, 818–824. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing Home Page. Available online: https://www.r-project.org (accessed on 16 March 2021).
- Junkers, T. Novel Single Pulse–Pulsed Laser Polymerization Methods for the Determination of Chain-Length Dependent Termination Kinetics in Conventional and Controlled Free-Radical Polymerization; Sierke Verlag: Göttingen, Germany, 2006. [Google Scholar]
- Degener, B. Laserinduzierte Radikalische Polymerisation von Butylacrylat in Einem Weiten Umsatzbereich. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 1992. [Google Scholar]
- Jungbluth, M.; Drache, M. Investigation into the chain transfer to solvents in butyl acrylate polymerizations. Unpublished Results.
- Zammit, M.D.; Davis, T.P.; Willett, G.D.; O’Driscoll, K.F. The Effect of Solvent on the Homo-Propagation Rate Coefficients of Styrene and Methyl Methacrylate. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 2311–2321. [Google Scholar] [CrossRef]
- Kamachi, M. Influence of solvent on free-radical polymerization of vinyl compounds. Adv. Polym. Sci. 1981, 38, 55–87. [Google Scholar] [CrossRef]
- Olaj, O.F.; Schnöll-Bitai, I. Solvent Effects on the Rate Constant of Chain Propagation in Free Radical Polymerization. Mon. Chem. 1999, 130, 731–740. [Google Scholar] [CrossRef]
- Beuermann, S. Solvent influence on propagation kinetics in radical polymerizations studied by pulsed laser initiated polymerization. Macromol. Rapid Commun. 2009, 30, 1066–1088. [Google Scholar] [CrossRef]
- Beuermann, S. Impact of H-bonding on the propagation kinetics in butyl methacrylate radical polymerizations. Macromolecules 2004, 37, 1037–1041. [Google Scholar] [CrossRef]
- Beuermann, S.; Nelke, D. The influence of hydrogen bonding on the propagation rate coefficient in free-radical polymerizations of hydroxypropyl methacrylate. Macromol. Chem. Phys. 2003, 204, 460–470. [Google Scholar] [CrossRef]
- Hutchinson, R.A.; Paquet, D.A., Jr.; Beuermann, S.; McMinn, J.H. Investigation of Methacrylate Free-Radical Depropagation Kinetics by Pulsed-Laser Polymerization. Ind. Eng. Chem. Res. 1998, 37, 3567–3574. [Google Scholar] [CrossRef]
- Reichhardt, C. Solvents and Solvent Effects in Organic Chemistry, 2nd ed.; VCH Verlagsgesellschaft mbH: Weinheim, Germany, 1990. [Google Scholar]
- Jeličić, A.; Yasin, M.; Beuermann, S. Toward the Description and Prediction of Solvent Induced Variations in Methacrylate Propagation Rate Coefficients on the Basis of Solvatochromic Parameters. Macromol. React. Eng. 2009, 5, 232–242. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook Second Edition; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
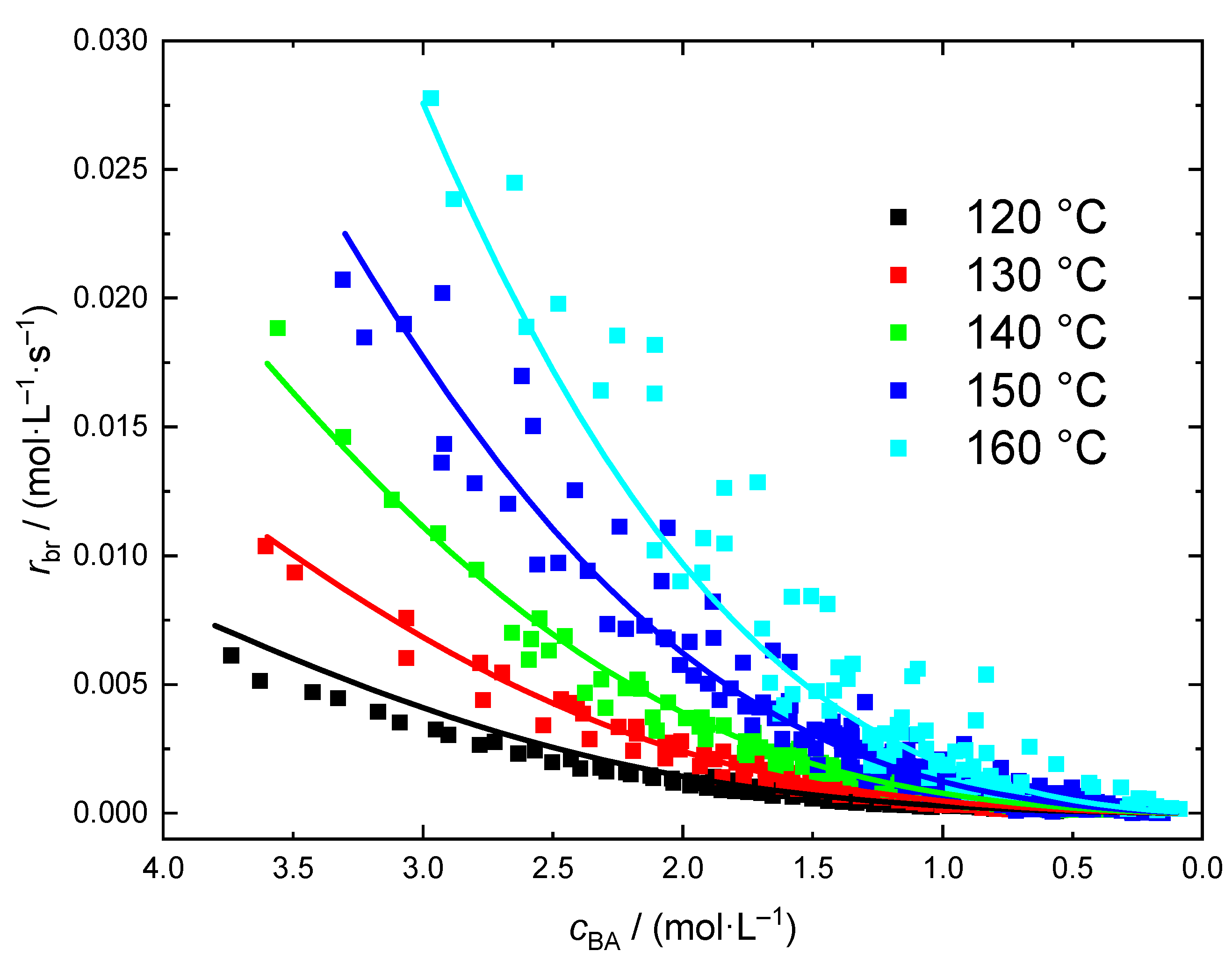{kind=link}
{kind=link}
| Coefficient | Value | Error |
|---|---|---|
| a0 (lnA) | 8.47 | 0.20 |
| a1 (Ea)/(kJ·mol−1) | 50.59 | 0.61 |
| a2 (n) | 1.28 | 0.03 |
| T/°C | x | cBA,res/(mol·L−1) | t/s |
|---|---|---|---|
| 120 | 0.82 | 0.78 | 3600 |
| 130 | 0.89 | 0.41 | 3600 |
| 140 | 0.92 | 0.31 | 3600 |
| 150 | 0.86 | 0.53 | 1800 |
| 160 | 0.82 | 0.67 | 600 |
| 170 | 0.83 | 0.62 | 600 |
| 180 | 0.86 | 0.51 | 600 |
| Coefficient | Value | Error |
|---|---|---|
| a0 (lnA) | 11.55 | 0.51 |
| a1 (Ea)/(kJ·mol–1) | 67.52 | 1.80 |
| a3 (l)/(L·mol–1) | 1.04 | 0.08 |
| a4 (m)/(L2·mol–2) | −0.10 | 0.02 |
| Solvent | δD | δP | δH |
|---|---|---|---|
| toluene | 18.0 | 1.4 | 2.0 |
| xylene | 17.6 | 1.0 | 3.1 |
| 1,4 dioxane | 19.0 | 1.8 | 7.4 |
| acetone | 15.5 | 10.4 | 7.0 |
| 2-butanone | 16.0 | 9.0 | 5.1 |
| Coefficient | Value | Error | |
|---|---|---|---|
| 2-octanone | a01 | −10.40 | 0.16 |
| a3/(kJ·mol−1) | 2.22 | 0.19 | |
| a4/(kJ·mol−1) | −0.38 | 0.05 | |
| toluene, xylene, mesitylene | a01 | −10.36 | 0.18 |
| a3/(kJ·mol–1) | 1.85 | 0.19 | |
| a4/(kJ·mol−1) | −0.29 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mätzig, J.; Drache, M.; Beuermann, S. Self-Initiated Butyl Acrylate Polymerizations in Bulk and in Solution Monitored By In-Line Techniques. Polymers 2021, 13, 2021. https://doi.org/10.3390/polym13122021
Mätzig J, Drache M, Beuermann S. Self-Initiated Butyl Acrylate Polymerizations in Bulk and in Solution Monitored By In-Line Techniques. Polymers. 2021; 13(12):2021. https://doi.org/10.3390/polym13122021
Chicago/Turabian StyleMätzig, Jonas, Marco Drache, and Sabine Beuermann. 2021. "Self-Initiated Butyl Acrylate Polymerizations in Bulk and in Solution Monitored By In-Line Techniques" Polymers 13, no. 12: 2021. https://doi.org/10.3390/polym13122021
APA StyleMätzig, J., Drache, M., & Beuermann, S. (2021). Self-Initiated Butyl Acrylate Polymerizations in Bulk and in Solution Monitored By In-Line Techniques. Polymers, 13(12), 2021. https://doi.org/10.3390/polym13122021