
Unravelling Constant pH Molecular Dynamics in Oligopeptides with Explicit Solvation Model




Abstract
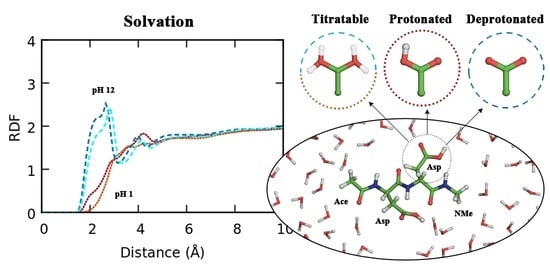
:
1. Introduction
2. Materials and Methods
2.1. Capped Tripeptides
2.2. Oligopeptides
2.3. Preparation of the Input Peptide Structures
2.4. All-Atom Conventional and Constant pH Molecular Dynamics Simulations
2.5. Energetic and Conformational Analysis
3. Results
3.1. Capped Tripeptides with Explicit Water Molecules
3.1.1. Conformational Sampling Disagrees in Deprotonated Forms of Amino Acids with Several Protonation States
3.1.2. Energy Contributions Reveal Deficiencies in the Reproduction of Electrostatics’ Interactions
3.2. Titratable Aspartic Acids in Adjacent and Terminal Positions in Oligopeptide
3.2.1. Position of the Titratable Amino Acids Modulates the Conformational Sampling
3.2.2. Terminal Titratable Residues Describe Correctly the Conformational Properties
3.2.3. Different Description on Electrostatics and Dihedral Energies Causes Deviations in Conformational Sampling and Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamelberg, D.; Mongan, J.; Mccammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.C.; Tsai, C.-C.; Ellis, C.R.; Shen, J. Proton-Coupled Conformational Allostery Modulates the Inhibitor Selectivity for β-Secretase. J. Phys. Chem. Lett. 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 12, 5933–5944. [Google Scholar] [CrossRef]
- Huang, Y.; Yue, Z.; Tsai, C.-C.; Henderson, J.A.; Shen, J. Predicting Catalytic Proton Donors and Nucleophiles in Enzymes: How Adding Dynamics Helps Elucidate the Structure−Function Relationships. J. Phys. Chem. Lett. 2018, 9, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Kraml, J.; Kahler, U.; Kamenik, A.S.; Liedl, K.R. Catalytic Site pK a Values of Aspartic, Cysteine, and Serine Proteases: Constant pH MD Simulations. J. Chem. Inf. Model. 2020, 60, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.B.P.S.; Vila-Viç, D.; Rocchia, W.; Machuqueiro, M. PypKa: A Flexible Python Module for Poisson—Boltzmann-Based pKa Calculations. J. Chem. Inf. Model. 2020, 60, 4442–4448. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H+ + 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Barazorda-Ccahuana, H.L.; Gómez, B.; Mas, F.; Madurga, S. Effect of pH on the Supramolecular Structure of Helicobacter pylori Urease by Molecular Dynamics Simulations. Polymers 2020, 12, 2713. [Google Scholar] [CrossRef] [PubMed]
- Beroza, P.; Fredkin, D.R.; Okamura, M.Y.; Feher, G. Protonation of interacting residues in a protein by a Monte Carlo method: Application to lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 1991, 88, 5804–5808. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.E.; Pettitt, B.M. Molecular Dynamics At a Constant pH. Int. J. High Perform. Comput. Appl. 1994, 8, 47–53. [Google Scholar] [CrossRef]
- Baptista, A.M.; Martel, P.J.; Petersen, S.B. Simulation of protein conformational freedom as a function of pH: Constant-pH molecular dynamics using implicit titration. Proteins Struct. Funct. Bioinform. 1997, 27, 523–544. [Google Scholar] [CrossRef]
- Lee, M.S.; Salsbury, F.R.; Brooks, C.L. Constant-pH Molecular Dynamics Using Continuous Titration Coordinates. Proteins Struct. Funct. Bioinform. 2004, 56, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Tegeler, F.; Groenhof, G.; Grubmüller, H. Constant pH Molecular Dynamics in Explicit Solvent with λ-Dynamics. J. Chem. Theory Comput. 2011, 7, 1962–1978. [Google Scholar] [CrossRef]
- Wallace, J.A.; Shen, J.K. Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J. Chem. Theory Comput. 2011, 7, 2617–2629. [Google Scholar] [CrossRef]
- Goh, G.B.; Hulbert, B.S.; Zhou, H.; Brooks III, C.L. Constant pH Molecular Dynamics of Proteins in Explicit Solvent with Proton Tautomerism. Proteins 2014, 82, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Baptista, A.M.; Teixeira, V.H.; Soares, C.M.; Nio, A.; Baptista, M.; Udio, C.; Soares, M. Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys. 2002, 117, 926. [Google Scholar] [CrossRef]
- Bürgi, R.; Kollman, P.A.; Van Gunsteren, W.F. Simulating proteins at constant pH: An approach combining molecular dynamics and Monte Carlo simulation. Proteins Struct. Funct. Genet. 2002, 47, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Swails, J.M.; York, D.M.; Roitberg, A.E. Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput. 2014, 10, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Mongan, J.; Case, D.A. Biomolecular simulations at constant pH. Curr. Opin. Struct. Biol. 2005, 15, 157–163. [Google Scholar] [CrossRef]
- Williams, S.L.; Blachly, P.G.; Mccammon, J.A. Measuring the successes and deficiencies of constant pH molecular dynamics: A blind prediction study. Proteins Struct. Funct. Bioinform. 2011, 79, 3381–3388. [Google Scholar] [CrossRef]
- Machuqueiro, M.; Baptista, A.M. Is the prediction of pKa values by constant-pH molecular dynamics being hindered by inherited problems? Proteins Struct. Funct. Bioinform. 2011, 79, 3437–3447. [Google Scholar] [CrossRef]
- Wallace, J.A.; Wang, Y.; Shi, C.; Pastoor, K.J.; Nguyen, B.-L.; Xia, K.; Shen, J.K. Toward accurate prediction of pKa values for internal protein residues: The importance of conformational relaxation and desolvation energy. Proteins Struct. Funct. Bioinform. 2011, 79, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Morrow, B.H.; Shi, C.; Shen, J.K. Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. [Google Scholar] [CrossRef]
- Dobrev, P.; Phani, S.; Vemulapalli, B.; Nath, N.; Griesinger, C.; Grubmü, H. Probing the Accuracy of Explicit Solvent Constant pH Molecular Dynamics Simulations for Peptides. J. Chem. Theory Comput. 2020, 16, 2561–2569. [Google Scholar] [CrossRef]
- Itoh, S.G.; Damjanović, A.; Brooks, B.R. pH replica-exchange method based on discrete protonation states. Proteins Struct. Funct. Bioinform. 2011, 79, 3420–3436. [Google Scholar] [CrossRef] [Green Version]
- Machuqueiro, M.; Baptista, A.M. Constant-pH Molecular Dynamics with Ionic Strength Effects: Protonation-Conformation Coupling in Decalysine. J. Phys. Chem. B 2006, 110, 2927–2933. [Google Scholar] [CrossRef]
- Williams, S.L.; De Oliveira, C.A.F.; Mccammon, J.A. Coupling Constant pH Molecular Dynamics with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2010, 6, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wallace, J.A.; Yue, Z.; Shen, J.K. Introducing Titratable Water to All-Atom Molecular Dynamics at Constant pH. Biophys. J. 2013, 105, L15–L17. [Google Scholar] [CrossRef] [Green Version]
- Yeager, A.V.; Swails, J.M.; Miller, B.R. Improved Accuracy for Constant pH-REMD Simulations through Modification of Carboxylate Effective Radii. J. Chem. Theory Comput. 2017, 13, 4624–4635. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Privat, C.; Madurga, S.; Mas, F.; Rubio-Martínez, J. On the Use of the Discrete Constant pH Molecular Dynamics to Describe the Conformational Space of Peptides. Polymers 2021, 13, 99. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 1902. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000. [Google Scholar] [CrossRef] [Green Version]
- Perez, J.J.; Santos Tomas, M.; Rubio-Martinez, J. Assessment of the Sampling Performance of Multiple-Copy Dynamics versus a Unique Trajectory. J. Chem. Inf. Model. 2016, 56, 1950–1962. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capped Tripeptides | ||||||
|---|---|---|---|---|---|---|
| Residue | cMD | cpHMD | PS | Intrinsic pKa | ||
| pH 1 | pH 12 | pH 14 | ||||
| ASP | ✓ | d- | 4.0 | |||
| ASH | ✓ | pn | ||||
| AS4 | ✓ | ✓ | t | |||
| GLU | ✓ | d- | 4.4 | |||
| GLH | ✓ | pn | ||||
| GL4 | ✓ | ✓ | t | |||
| HIE | ✓ | dn | 6.6 | |||
| HID | ✓ | dn | ||||
| HIP | ✓ | ✓ | ✓ | p+/t | ||
| CYM | ✓ | d- | 8.5 | |||
| CYS | ✓ | ✓ | ✓ | pn/t | ||
| TYR | ✓ | ✓ | pn/t | 9.6 | ||
| LYN | ✓ | dn | 10.4 | |||
| LYS | ✓ | ✓ | ✓ | p+/t | ||
| DA8D | ||||||
| pH 1 | pH 10 | |||||
| ASP | ✓ | d- | 4.0 | |||
| ASH | ✓ | pn | ||||
| AS4 | ✓ | ✓ | t | |||
| A4D2A4 | ||||||
| pH 1 | pH 10 | |||||
| ASP | ✓ | d- | 4.0 | |||
| ASH | ✓ | pn | ||||
| AS4 | ✓ | ✓ | t | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Privat, C.; Madurga, S.; Mas, F.; Rubio-Martinez, J. Unravelling Constant pH Molecular Dynamics in Oligopeptides with Explicit Solvation Model. Polymers 2021, 13, 3311. https://doi.org/10.3390/polym13193311
Privat C, Madurga S, Mas F, Rubio-Martinez J. Unravelling Constant pH Molecular Dynamics in Oligopeptides with Explicit Solvation Model. Polymers. 2021; 13(19):3311. https://doi.org/10.3390/polym13193311
Chicago/Turabian StylePrivat, Cristian, Sergio Madurga, Francesc Mas, and Jaime Rubio-Martinez. 2021. "Unravelling Constant pH Molecular Dynamics in Oligopeptides with Explicit Solvation Model" Polymers 13, no. 19: 3311. https://doi.org/10.3390/polym13193311
APA StylePrivat, C., Madurga, S., Mas, F., & Rubio-Martinez, J. (2021). Unravelling Constant pH Molecular Dynamics in Oligopeptides with Explicit Solvation Model. Polymers, 13(19), 3311. https://doi.org/10.3390/polym13193311