
Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations

 ,
,  ,
,  and
and 
Abstract
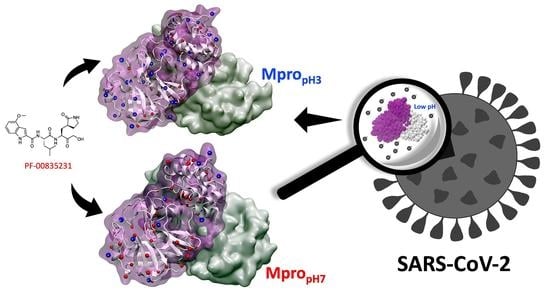
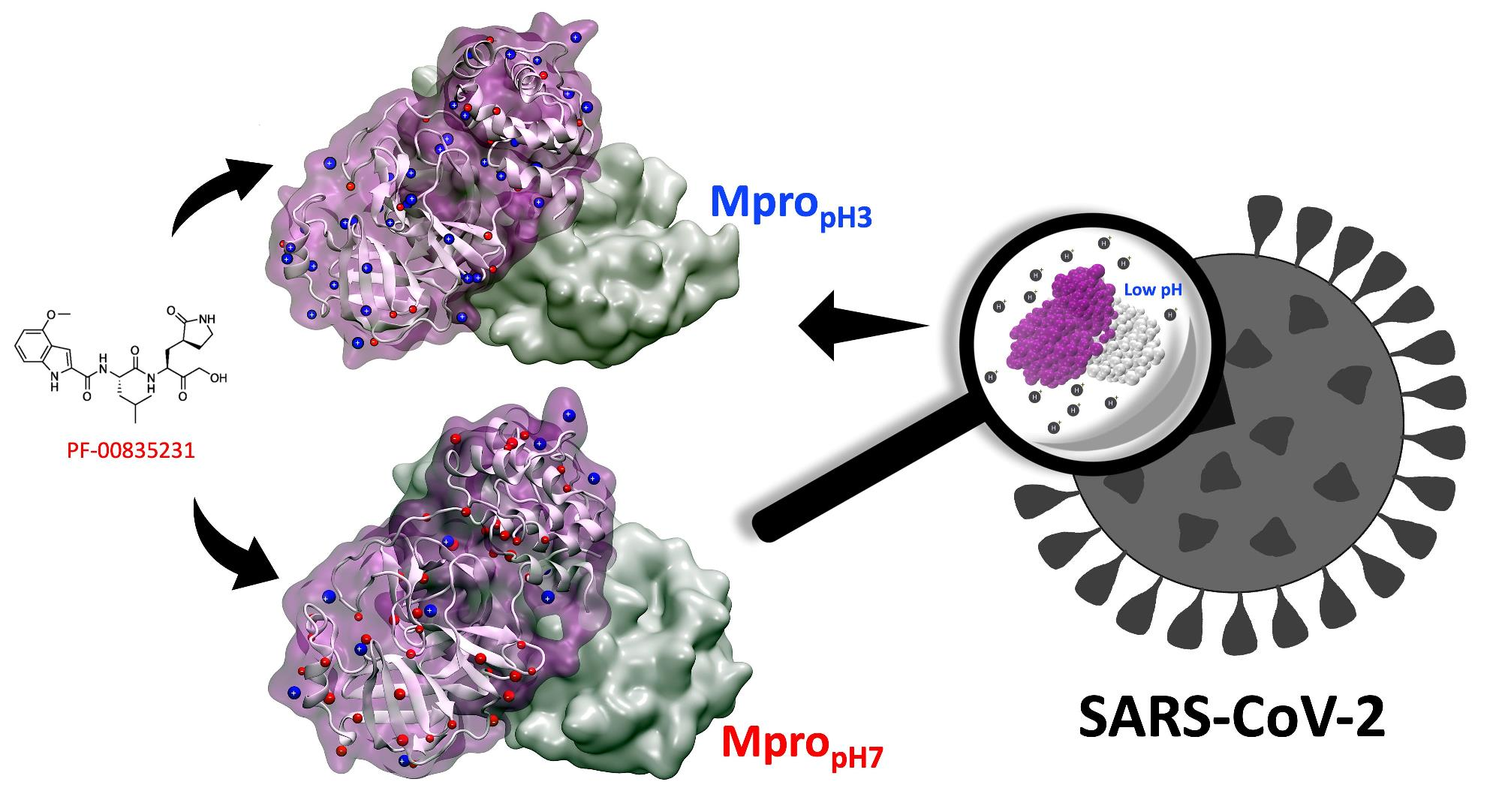
:
1. Introduction
2. Computational Methods
2.1. Protonation/Deprotonation States by SGCMC
2.2. Molecular Dynamics Simulation Details
2.3. Molecular Docking
2.4. Simulation Data Analysis
3. Results and Discussion
3.1. Protonation/Deprotonation States of SARS-CoV-2 Mpro
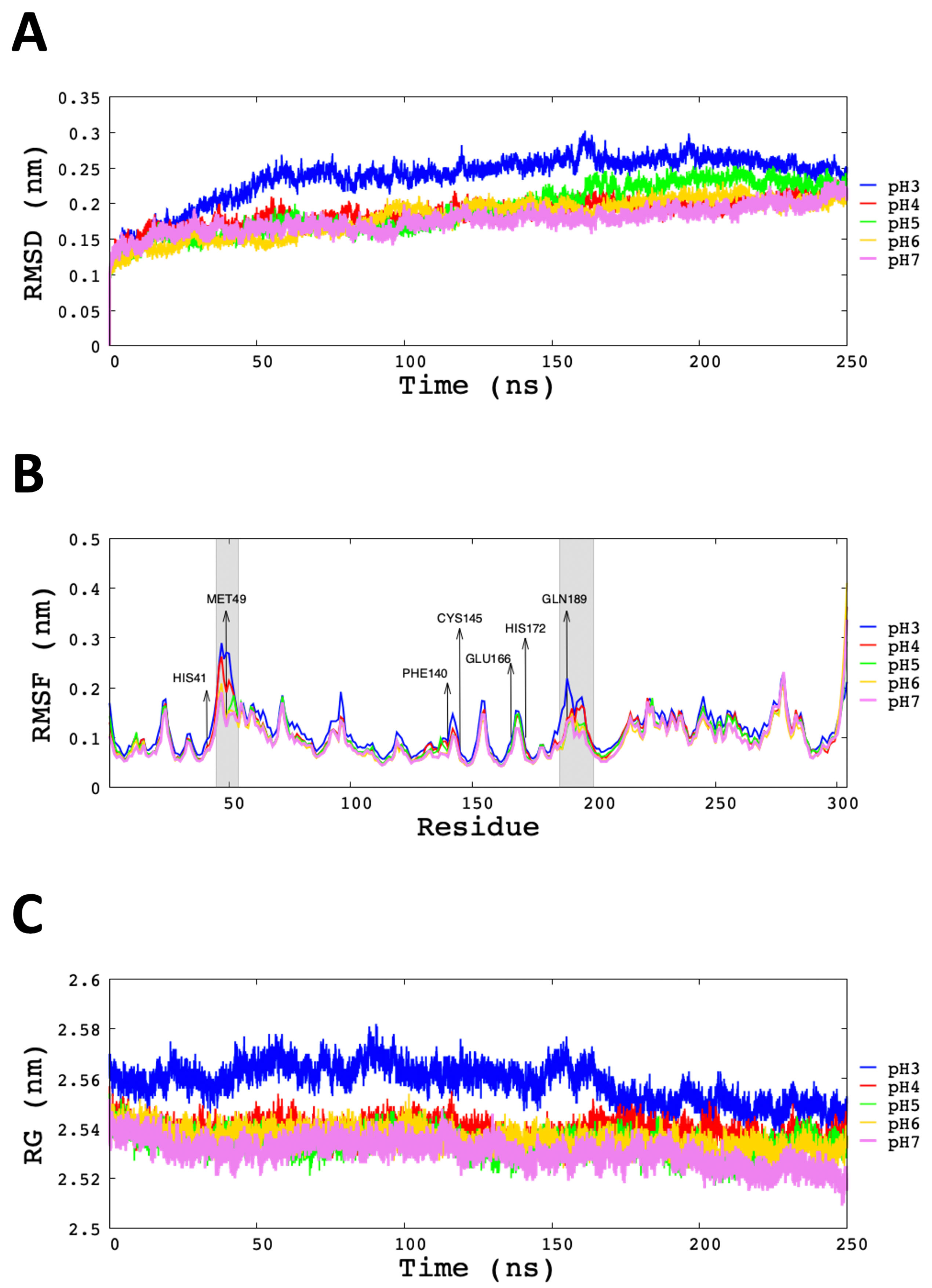
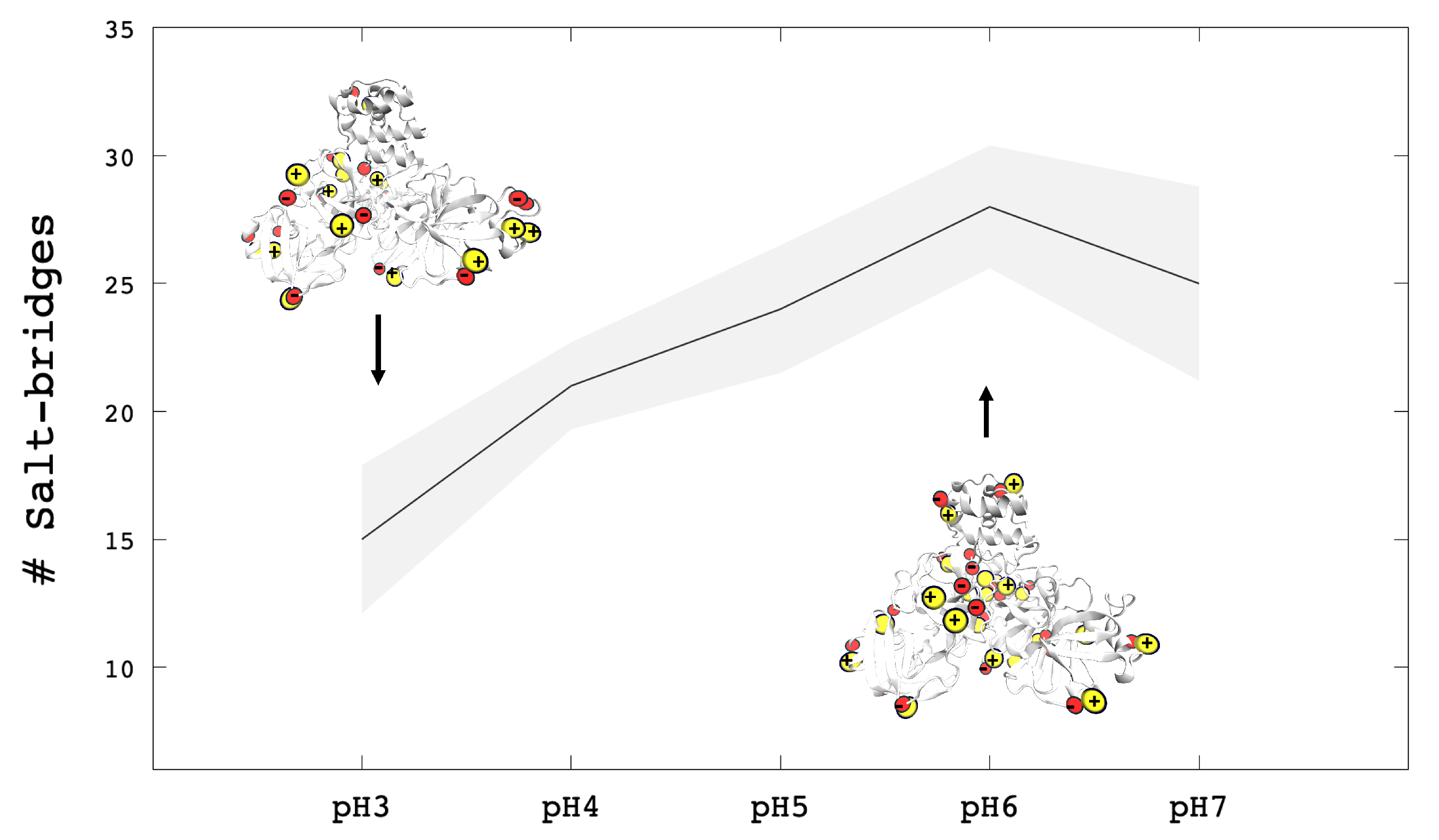
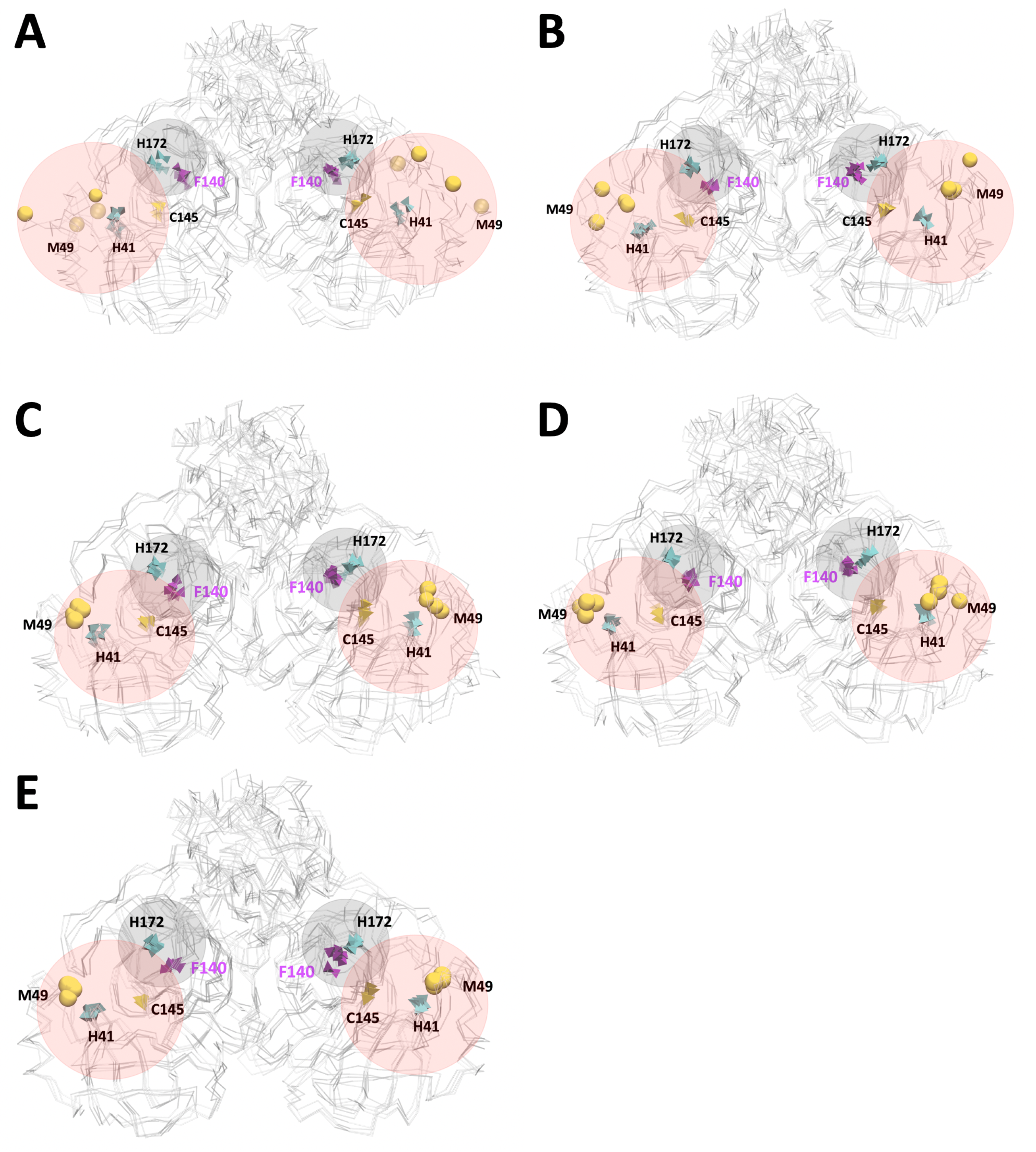
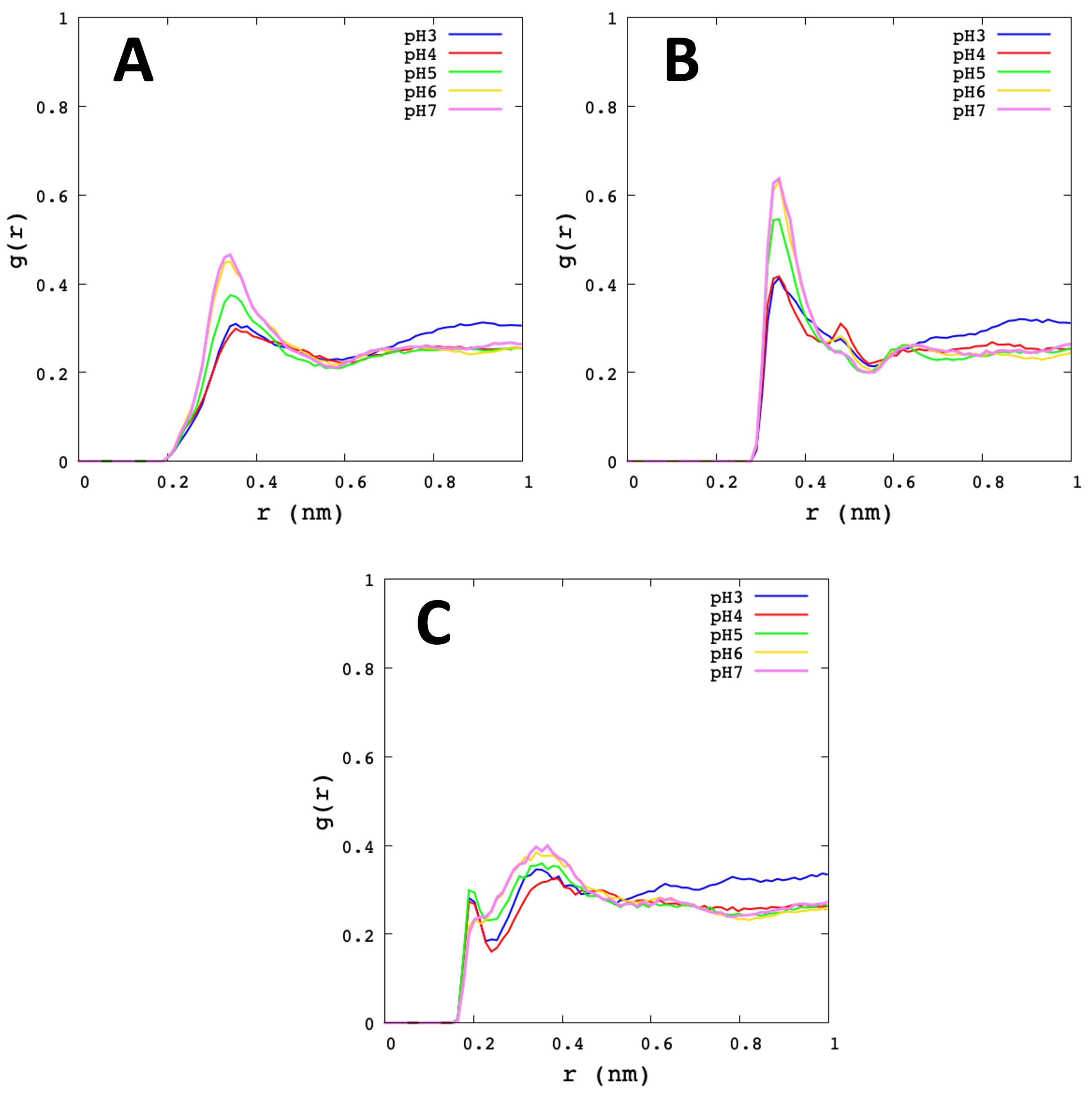
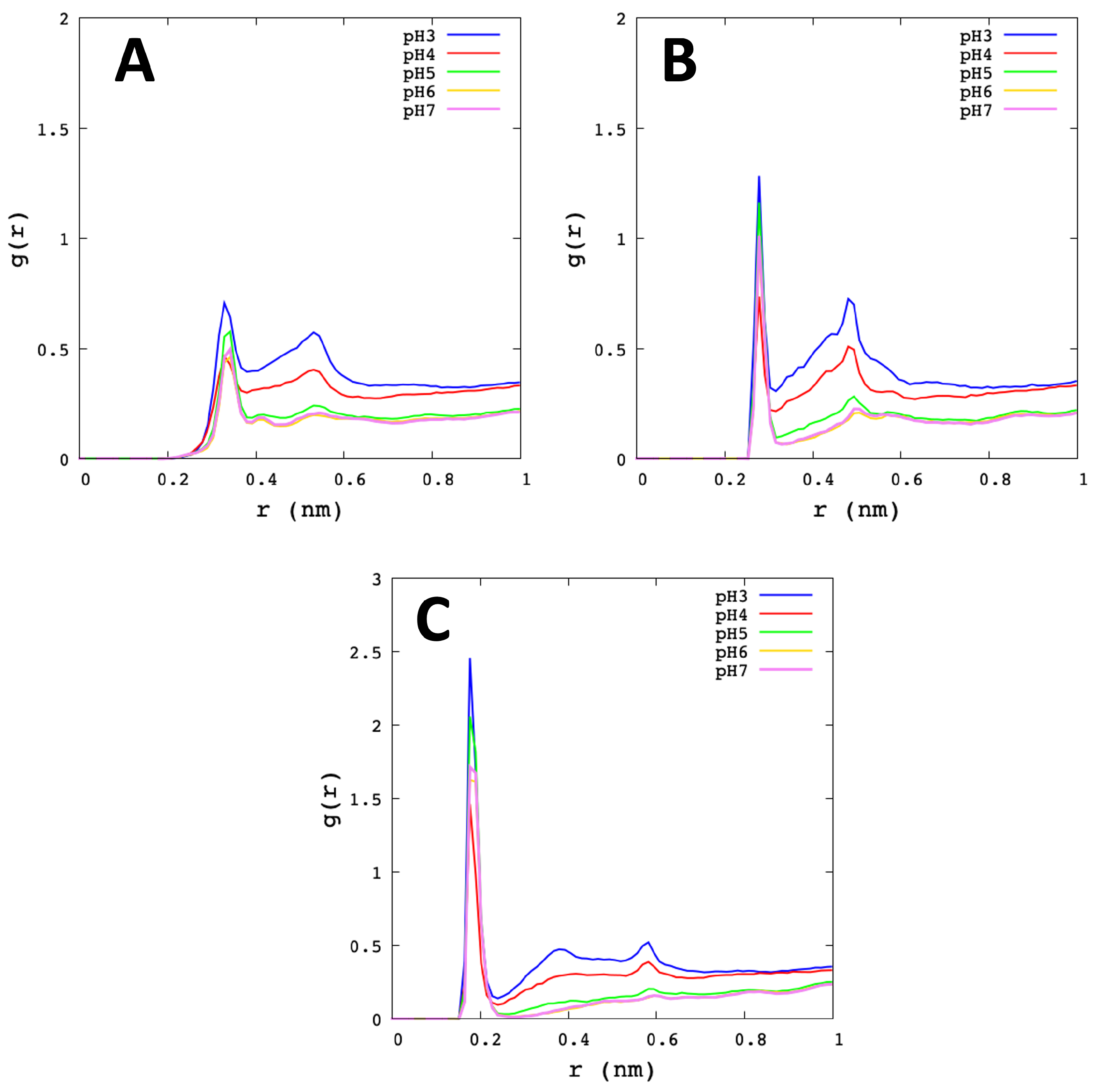
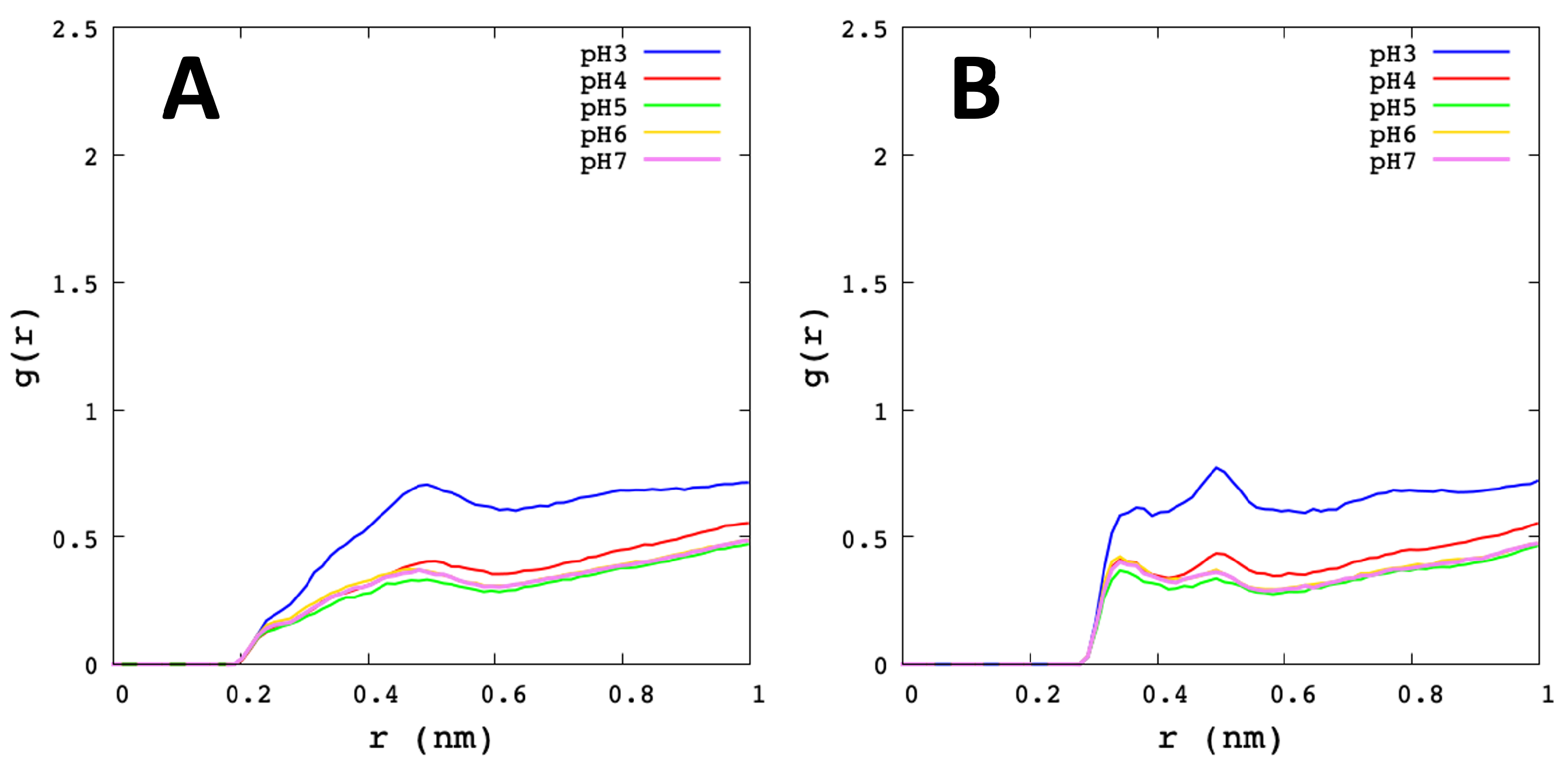
3.2. Analysis of Molecular Dynamic Simulation
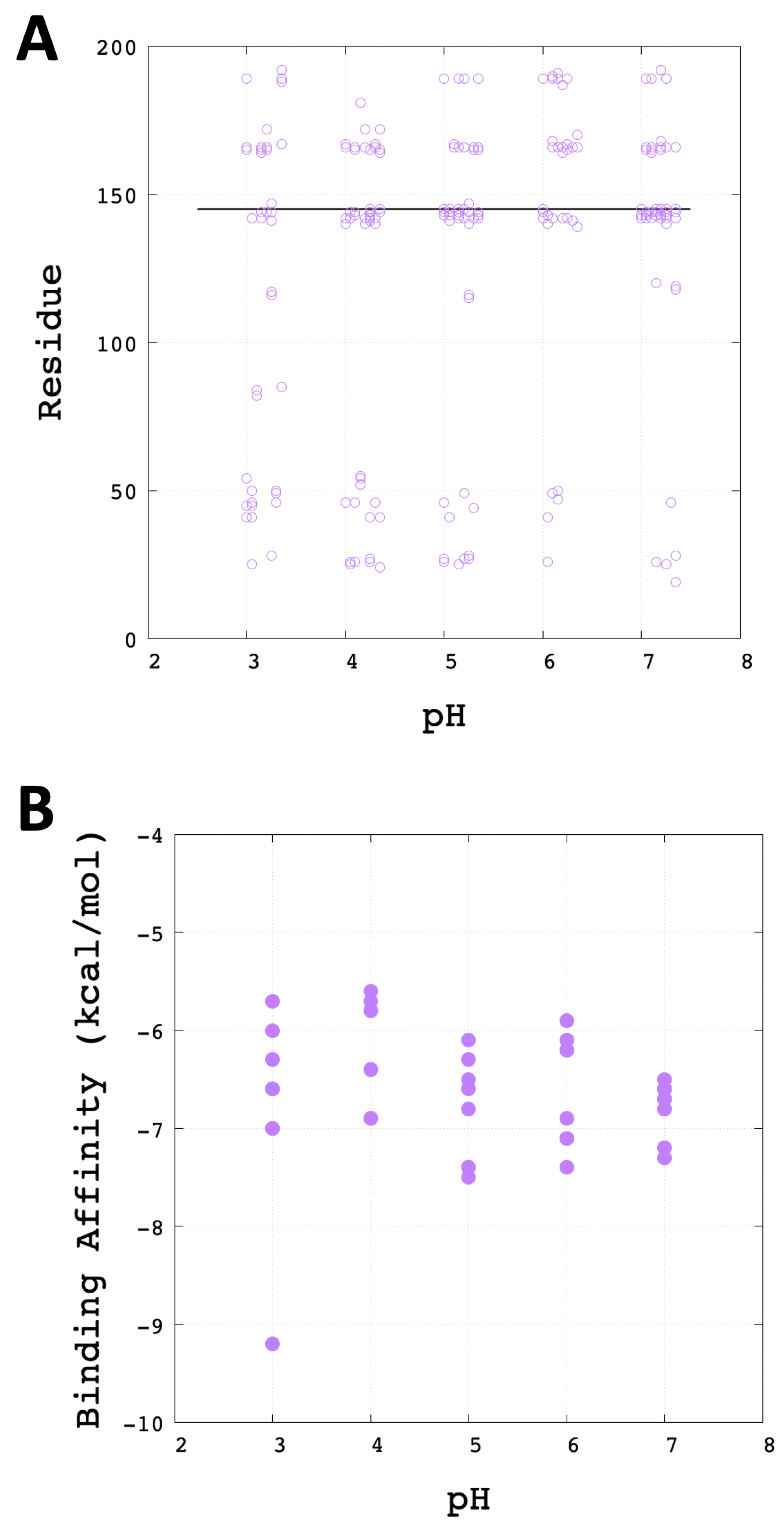
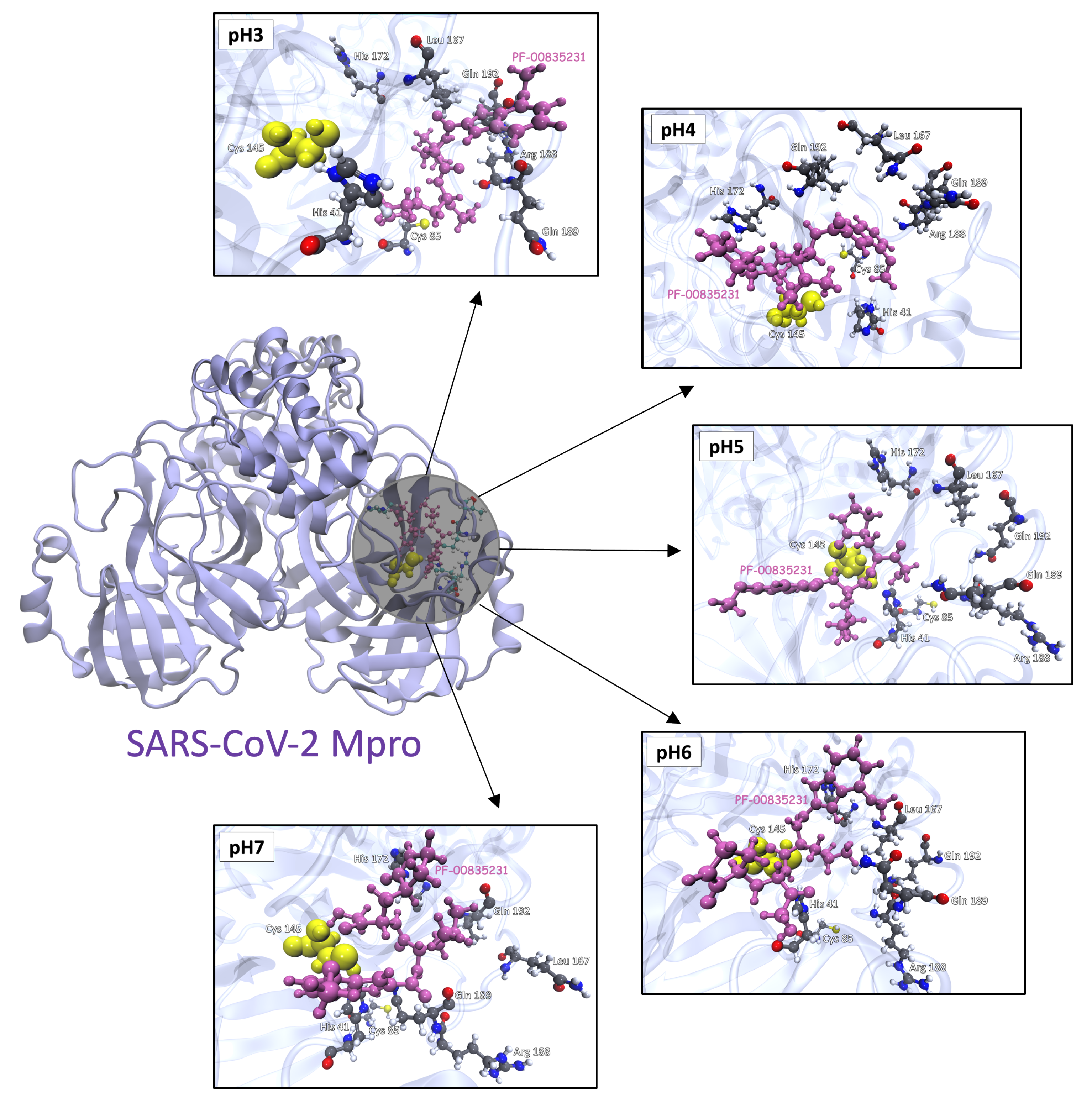
3.3. Molecular Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| Mpro | Main Protease |
| SGCMC | Semi-Grand Canonical Monte Carlo |
| MD | Molecular Dynamics |
| RMSD | Root-Mean Squared Deviation |
| RMSF | Root-Mean Squared Fluctuation |
| RG | Radius of Gyration |
| HB | Number of Hydrogen Bonds |
| SASA | Solvent Accessible Surface Area |
| RDF | Radial Distribution Function |
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. [Google Scholar]
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-mediated entry via the endosome of human coronavirus 229E. J. Virol. 2009, 83, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Shen, H.M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724. [Google Scholar] [CrossRef]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sieben, C.; Ludwig, K.; Höfer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A. pH-Controlled two-step uncoating of influenza virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Otto, H.H.; Schirmeister, T. Cysteine proteases and their inhibitors. Chem. Rev. 1997, 97, 133–172. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Tang, J.; Wong, R.N. Evolution in the structure and function of aspartic proteases. J. Cell. Biochem. 1987, 33, 53–63. [Google Scholar] [CrossRef]
- Szecsi, P.B. The aspartic proteases. Scand. J. Clin. Lab. Investig. 1992, 52, 5–22. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2021, 39, 3409–3418. [Google Scholar] [CrossRef] [Green Version]
- Kanhed, A.M.; Patel, D.V.; Teli, D.M.; Patel, N.R.; Chhabria, M.T.; Yadav, M.R. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol. Divers. 2021, 25, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, J.; Dheeman, S.; Sharma, V.; Katiyar, P.; Karn, S.K.; Sarangi, M.K.; Chauhan, A.K.; Verma, G.; Baliyan, N. Insights of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) pandemic: A current review. Biol. Proced. Online 2021, 23, 1–22. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL M pro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Römer, R.A.; Römer, N.S.; Wallis, A.K. Flexibility and mobility of SARS-CoV-2-related protein structures. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, S.P. Fundamentals of viruses and their proteases. In Viral Proteases and Their Inhibitors; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–24. [Google Scholar]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Verschueren, K.H.; Anand, K.; Shen, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: Molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, B.; Goyal, D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Talley, K.; Alexov, E. On the pH-optimum of activity and stability of proteins. Proteins Struct. Funct. Bioinform. 2010, 78, 2699–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.P.; Chou, C.Y.; Chang, G.G. Reversible unfolding of the severe acute respiratory syndrome coronavirus main protease in guanidinium chloride. Biophys. J. 2007, 92, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Yang, Z.; Song, H.; Wang, K.; Yang, Y.; Xie, L.; Huang, S.; Liu, J.; Ran, L.; Song, Z. Three Main Inducers of Alphacoronavirus Infection of Enterocytes: Sialic Acid, Proteases, and Low pH. Intervirology 2018, 61, 53–63. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamic Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef] [PubMed]
- Díaz, N.; Suárez, D. Influence of charge configuration on substrate binding to SARS-CoV-2 main protease. Chem. Commun. 2021, 57, 5314–5317. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Deep, S. pH Effect on the Dynamics of SARS-CoV-2 Main Protease (Mpro). bioRxiv 2020. [Google Scholar] [CrossRef]
- White, J.M.; Whittaker, G.R. Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Chen, X.; Geiger, J.D. Role of endolysosomes in severe acute respiratory syndrome coronavirus-2 infection and coronavirus disease 2019 pathogenesis: Implications for potential treatments. Front. Pharmacol. 2020, 11, 1739. [Google Scholar] [CrossRef]
- Jimenez, L.; Campos Codo, A.; Sampaio, V.d.S.; Oliveira, A.E.; Ferreira, L.K.K.; Davanzo, G.G.; Brito Monteiro, L.d.; Victor Virgilio-da Silva, J.; Borba, M.G.S.; Fabiano de Souza, G.; et al. Acid pH Increases SARS-CoV-2 Infection and the Risk of Death by COVID-19. Front. Med. 2021, 1358. [Google Scholar] [CrossRef]
- Nechipurenko, Y.D.; Semyonov, D.A.; Lavrinenko, I.A.; Lagutkin, D.A.; Generalov, E.A.; Zaitceva, A.Y.; Matveeva, O.V.; Yegorov, Y.E. The Role of Acidosis in the Pathogenesis of Severe Forms of COVID-19. Biology 2021, 10, 852. [Google Scholar] [CrossRef]
- Henry, B.M.; Aggarwal, G.; Wong, J.; Benoit, S.; Vikse, J.; Plebani, M.; Lippi, G. Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: A pooled analysis. Am. J. Emerg. Med. 2020, 38, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Reyburn, H.T.; Rizzuti, B.; Velazquez-Campoy, A. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Rabeh, W.M. Biochemical and biophysical characterization of the main protease, 3-chymotrypsin-like protease (3CLpro) from the novel coronavirus SARS-CoV 2. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hamdullah, K.S.; Tanzila, A.; ZainaSher, M.; Iqra, A.; Mohtasheemul, H. pH Dependent Differential Binding Behavior of Prtotease Inhibitor Molecular Drugs for SARS-CoV-2. 2020. Available online: https://pesquisa.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/resource/pt/ppcovidwho-148 (accessed on 10 January 2021).
- Vatansever, E.C.; Yang, K.S.; Drelich, A.K.; Kratch, K.C.; Cho, C.C.; Kempaiah, K.R.; Hsu, J.C.; Mellott, D.M.; Xu, S.; Tseng, C.T.K.; et al. Bepridil is potent against SARS-CoV-2 in vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2012201118. [Google Scholar] [CrossRef]
- Nguyen, T.T.H.; Jung, J.H.; Kim, M.K.; Lim, S.; Choi, J.M.; Chung, B.; Kim, D.W.; Kim, D. The Inhibitory Effects of Plant Derivate Polyphenols on the Main Protease of SARS Coronavirus 2 and Their Structure–Activity Relationship. Molecules 2021, 26, 1924. [Google Scholar] [CrossRef]
- Hamdi, M.; Abdel-Bar, H.M.; Elmowafy, E.; El-Khouly, A.; Mansour, M.; Awad, G.A. Investigating the Internalization and COVID-19 Antiviral Computational Analysis of Optimized Nanoscale Zinc Oxide. ACS Omega 2021, 6, 6848–6860. [Google Scholar] [CrossRef]
- Dražić, T.; Kühl, N.; Leuthold, M.M.; Behnam, M.A.; Klein, C.D. Efficiency Improvements and Discovery of New Substrates for a SARS-CoV-2 Main Protease FRET Assay. Slas Discov. Adv. Sci. Drug Discov. 2021, 24725552211020681. [Google Scholar] [CrossRef]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziano, V.; McGrath, W.J.; DeGruccio, A.M.; Dunn, J.J.; Mangel, W.F. Enzymatic activity of the SARS coronavirus main proteinase dimer. FEBS Lett. 2006, 580, 2577–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, L.L.; Luo, H.B.; Sun, T.; Chen, J.; Ye, F.; Cai, J.H.; Shen, J.K.; Shen, X.; Jiang, H.L. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique 1. Acta Pharmacol. Sin. 2005, 26, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Verma, N.; Henderson, J.A.; Shen, J. Proton-Coupled Conformational Activation of SARS Coronavirus Main Proteases and Opportunity for Designing Small-Molecule Broad-Spectrum Targeted Covalent Inhibitors. J. Am. Chem. Soc. 2020, 142, 21883–21890. [Google Scholar] [CrossRef]
- Pavlova, A.; Lynch, D.L.; Smith, M.D.; Smith, J.D.; Gumbart, J.C. Inhibitor Binding Influences the Protonation State of Histidines in SARS-CoV-2 Main Protease. Biophys. J. 2021, 120, 204a–205a. [Google Scholar] [CrossRef]
- Fan, S.; Xiao, D.; Wang, Y.; Liu, L.; Zhou, X.; Zhong, W. Research progress on repositioning drugs and specific therapeutic drugs for SARS-CoV-2. Future Med. Chem. 2020, 12, 1565–1578. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Coelho, C.; Gallo, G.; Campos, C.B.; Hardy, L.; Würtele, M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS ONE 2020, 15, e0240079. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; Hatamipour, M.; Banach, M.; Pirro, M.; Al-Rasadi, K.; Jamialahmadi, T.; Radenkovic, D.; Montecucco, F.; Sahebkar, A. Statins and the COVID-19 main protease: In silico evidence on direct interaction. Arch. Med. Sci. AMS 2020, 16, 490. [Google Scholar] [CrossRef]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Kumar, N.; Awasthi, A.; Kumari, A.; Sood, D.; Jain, P.; Singh, T.; Sharma, N.; Grover, A.; Chandra, R. Antitussive noscapine and antiviral drug conjugates as arsenal against COVID-19: A comprehensive chemoinformatics analysis. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moneriz, C.; Castro-Salguedo, C. Fármacos prometedores y potenciales para el tratamiento de COVID-19. Rev. Chil. Infectología 2020, 37, 205–215. [Google Scholar] [CrossRef]
- Yang, H.; Yang, J. A review of latest research on Mpro targeted SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. [Google Scholar] [CrossRef]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin Inhibits SARS-CoV-2 Viral Replication by Targeting Mpro and Ameliorates Pulmonary Inflammation. Front. Pharmacol. 2021, 12, 1012. [Google Scholar] [CrossRef]
- Mulu, A.; Gajaa, M.; Woldekidan, H.B. The impact of curcumin derived polyphenols on the structure and flexibility COVID-19 main protease binding pocket: A molecular dynamics simulation study. PeerJ 2021, 9, e11590. [Google Scholar] [CrossRef]
- Silvestrini, L.; Belhaj, N.; Comez, L.; Gerelli, Y.; Lauria, A.; Libera, V.; Mariani, P.; Marzullo, P.; Ortore, M.G.; Piccionello, A.P.; et al. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Roviello, V.; Roviello, G.N. In Silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficus-indica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041. [Google Scholar] [CrossRef]
- Roviello, V.; Musumeci, D.; Mokhir, A.; Roviello, G.N. Evidence of protein binding by a nucleopeptide based on a thymine-decorated L-diaminopropanoic acid through CD and in silico studies. Curr. Med. Chem. 2021, 28, 5004–5015. [Google Scholar] [CrossRef]
- Goyzueta-Mamani, L.D.; Barazorda-Ccahuana, H.L.; Mena-Ulecia, K.; Chávez-Fumagalli, M.A. Antiviral Activity of Metabolites from Peruvian Plants against SARS-CoV-2: An In Silico Approach. Molecules 2021, 26, 3882. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin is a low micromolar inhibitor of SARS-CoV-2 main protease 3CLpro: Implications for drug design of quercetin analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef] [PubMed]
- Madurga, S.; Garcés, J.L.; Companys, E.; Rey-Castro, C.; Salvador, J.; Galceran, J.; Vilaseca, E.; Puy, J.; Mas, F. Ion binding to polyelectrolytes: Monte Carlo simulations versus classical mean field theories. Theor. Chem. Accounts 2009, 123, 127–135. [Google Scholar] [CrossRef]
- Madurga, S.; Rey-Castro, C.; Pastor, I.; Vilaseca, E.; David, C.; Garcés, J.L.; Puy, J.; Mas, F. A semi-grand canonical Monte Carlo simulation model for ion binding to ionizable surfaces: Proton binding of carboxylated latex particles as a case study. J. Chem. Phys. 2011, 135, 184103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, P.M.; Madurga, S.; Mas, F.; Garcés, J.L. Coupling of charge regulation and conformational equilibria in linear weak polyelectrolytes: Treatment of long-range interactions via effective short-ranged and pH-dependent interaction parameters. Polymers 2018, 10, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, P.M.; Madurga, S.; Narambuena, C.F.; Mas, F.; Garcés, J.L. Role of charge regulation and fluctuations in the conformational and mechanical properties of weak flexible polyelectrolytes. Polymers 2019, 11, 1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barazorda-Ccahuana, H.L.; Gómez, B.; Mas, F.; Madurga, S. Effect of pH on the Supramolecular Structure of Helicobacter pylori Urease by Molecular Dynamics Simulations. Polymers 2020, 12, 2713. [Google Scholar] [CrossRef] [PubMed]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a novel inhibitor of coronavirus 3CL protease as a clinical candidate for the potential treatment of COVID-19. BioRxiv 2021, 2020-09. [Google Scholar] [CrossRef]
- de Vries, M.; Mohamed, A.; Prescott, R.A.; Valero-Jimenez, A.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A comparative analysis of SARS-CoV-2 antivirals in human airway models characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. bioRxiv 2021, 2020-08. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332. J. Enzym. Inhib. Med. Chem. 2021, 36, 1646–1650. [Google Scholar] [CrossRef]
- Anirudhan, V.; Lee, H.; Cheng, H.; Cooper, L.; Rong, L. Targeting SARS-CoV-2 viral proteases as a therapeutic strategy to treat COVID-19. J. Med. Virol. 2021, 93, 2722–2734. [Google Scholar] [CrossRef]
- Baig, M.H.; Sharma, T.; Ahmad, I.; Abohashrh, M.; Alam, M.M.; Dong, J.J. Is PF-00835231 a Pan-SARS-CoV-2 Mpro Inhibitor? A Comparative Study. Molecules 2021, 26, 1678. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Makov, G.; Payne, M. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, M.W.; Jorgensen, W.L. Diffusion constant of the TIP5P model of liquid water. J. Chem. Phys. 2001, 114, 363–366. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Racine, J. gnuplot 4.0: A portable interactive plotting utility. J. Appl. Econom. 2006, 21, 133–141. [Google Scholar] [CrossRef]
- Scheller, C.; Krebs, F.; Minkner, R.; Astner, I.; Gil-Moles, M.; Wätzig, H. Physicochemical properties of SARS-CoV-2 for drug targeting, virus inactivation and attenuation, vaccine formulation and quality control. Electrophoresis 2020, 41, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Buck, M. Molecular simulations of a dynamic protein complex: Role of salt-bridges and polar interactions in configurational transitions. Biophys. J. 2013, 105, 2412–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Świderek, K.; Moliner, V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M pro by QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef] [PubMed]












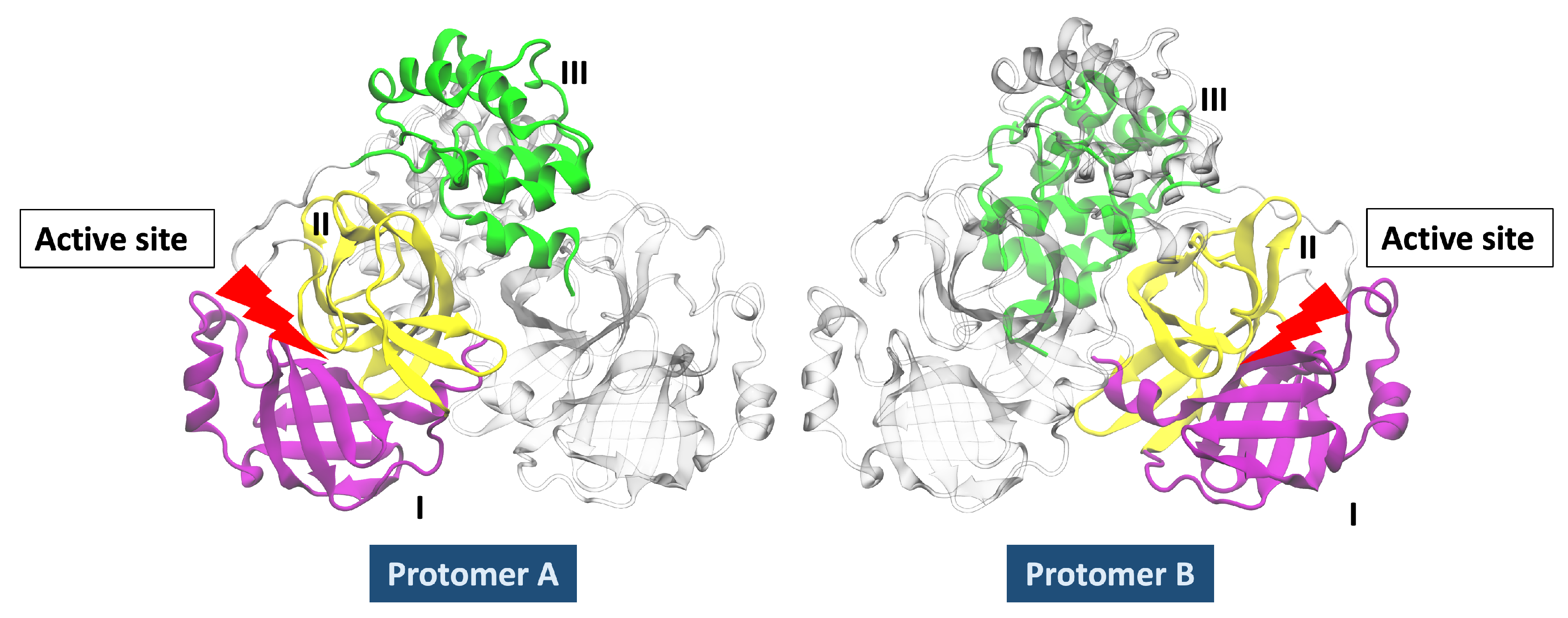
{kind=link}
{kind=link}
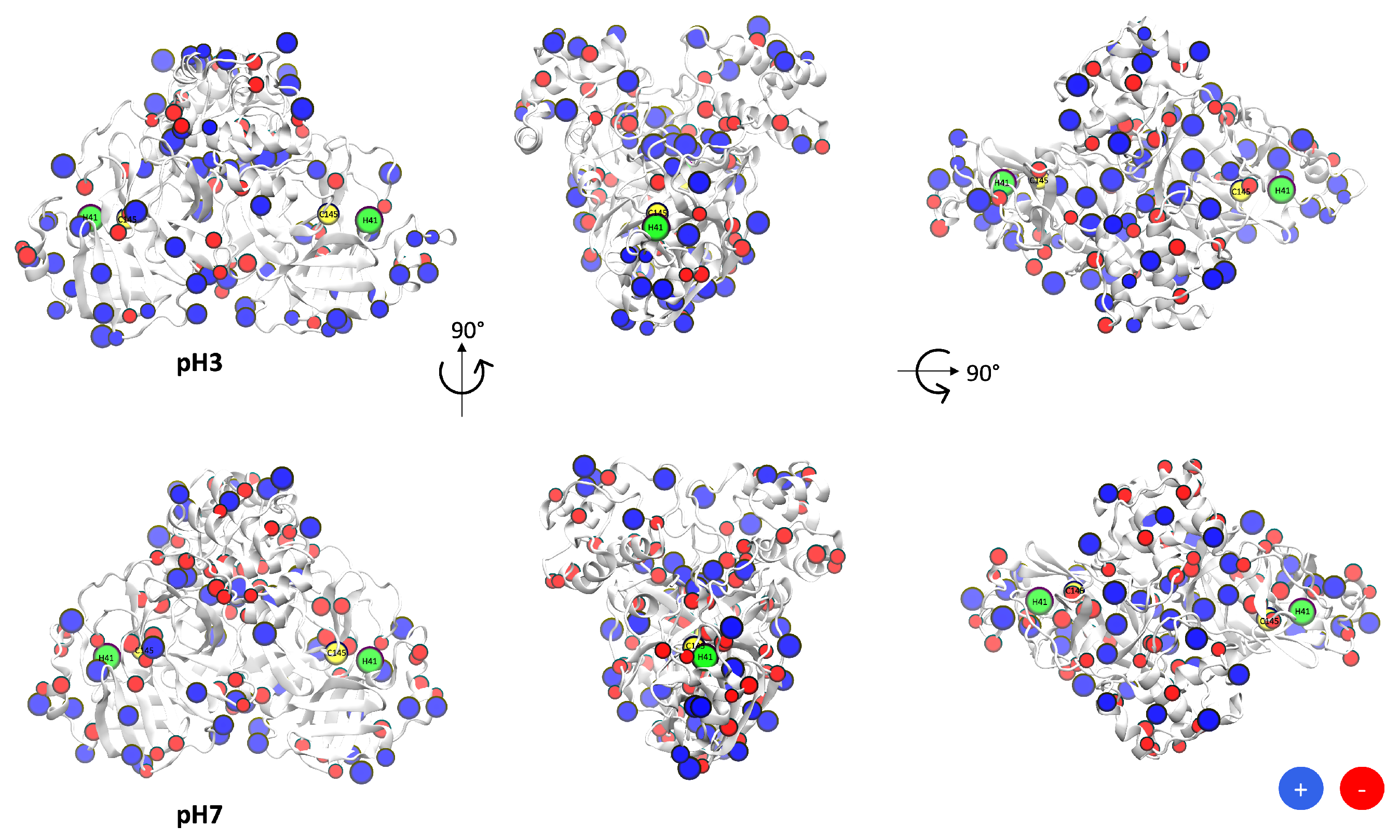{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroS-1 | MicroS-2 | MicroS-3 | MicroS-4 | MicroS-5 | MicroS-6 | MicroS-7 | MicroS-8 | MicroS-9 | MicroS-10 | Average Value | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| pH3 | 38 | 40 | 41 | 42 | 43 | 43 | 42 | 41 | 39 | 41 | 41 ± 2 |
| pH4 | 24 | 27 | 20 | 22 | 23 | 20 | 26 | 22 | 22 | 22 | 23 ± 2 |
| pH5 | 7 | 4 | 8 | 6 | 2 | 5 | 1 | 5 | 2 | 7 | 5 ± 2 |
| pH6 | −6 | −5 | −4 | −4 | −5 | −6 | −8 | −7 | −4 | −6 | −(5.5 ± 1.4) |
| pH7 | −8 | −9 | −9 | −8 | −8 | −7 | −8 | −9 | −9 | −9 | −(8.4 ± 0.7) |
| pH | RMSD | RG | SASA | Hbond | Hbond |
|---|---|---|---|---|---|
| (nm) | (nm) | (nm) | (Protein–Protein) | (Protein–Waters) | |
| pH3 | 0.26 ± 0.06 | 2.553 ± 0.010 | 268 ± 2 | 401 ± 1 | (107 ± 4) × 10 |
| pH4 | 0.20 ± 0.02 | 2.538 ± 0.002 | 261 ± 2 | 437 ± 10 | (110 ± 2) × 10 |
| pH5 | 0.21 ± 0.04 | 2.537 ± 0.005 | 259 ± 3 | 448 ± 3 | (114 ± 8) × 10 |
| pH6 | 0.20 ± 0.02 | 2.532 ± 0.005 | 260 ± 2 | 456 ± 6 | (114 ± 12) × 10 |
| pH7 | 0.19 ± 0.02 | 2.526 ± 0.007 | 259 ± 1 | 461 ± 1 | (114 ± 12) × 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barazorda-Ccahuana, H.L.; Nedyalkova, M.; Mas, F.; Madurga, S. Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations. Polymers 2021, 13, 3823. https://doi.org/10.3390/polym13213823
Barazorda-Ccahuana HL, Nedyalkova M, Mas F, Madurga S. Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations. Polymers. 2021; 13(21):3823. https://doi.org/10.3390/polym13213823
Chicago/Turabian StyleBarazorda-Ccahuana, Haruna Luz, Miroslava Nedyalkova, Francesc Mas, and Sergio Madurga. 2021. "Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations" Polymers 13, no. 21: 3823. https://doi.org/10.3390/polym13213823
APA StyleBarazorda-Ccahuana, H. L., Nedyalkova, M., Mas, F., & Madurga, S. (2021). Unveiling the Effect of Low pH on the SARS-CoV-2 Main Protease by Molecular Dynamics Simulations. Polymers, 13(21), 3823. https://doi.org/10.3390/polym13213823