3.3. Thermal Properties
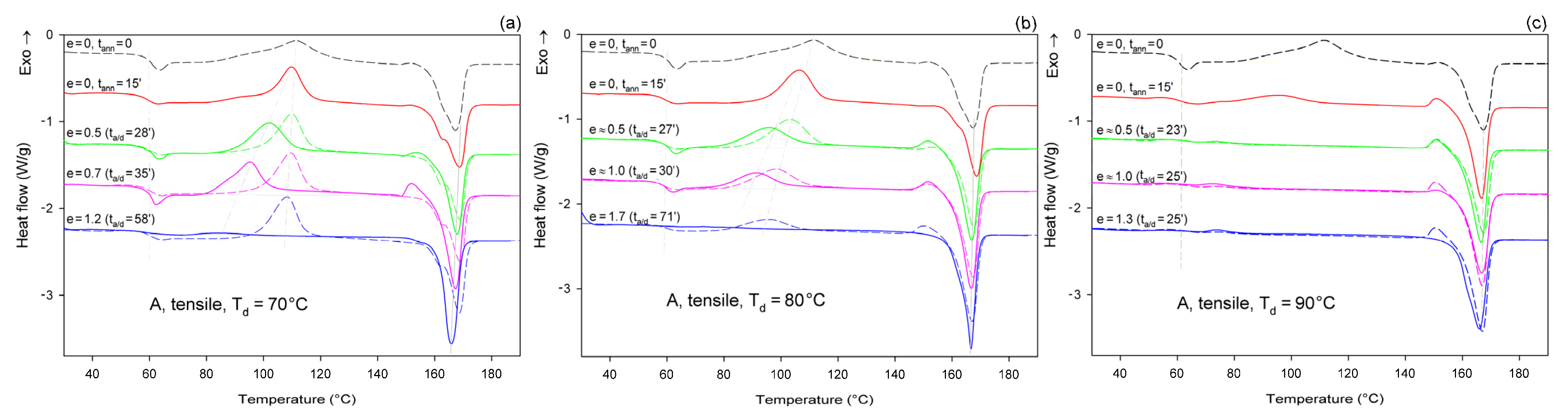
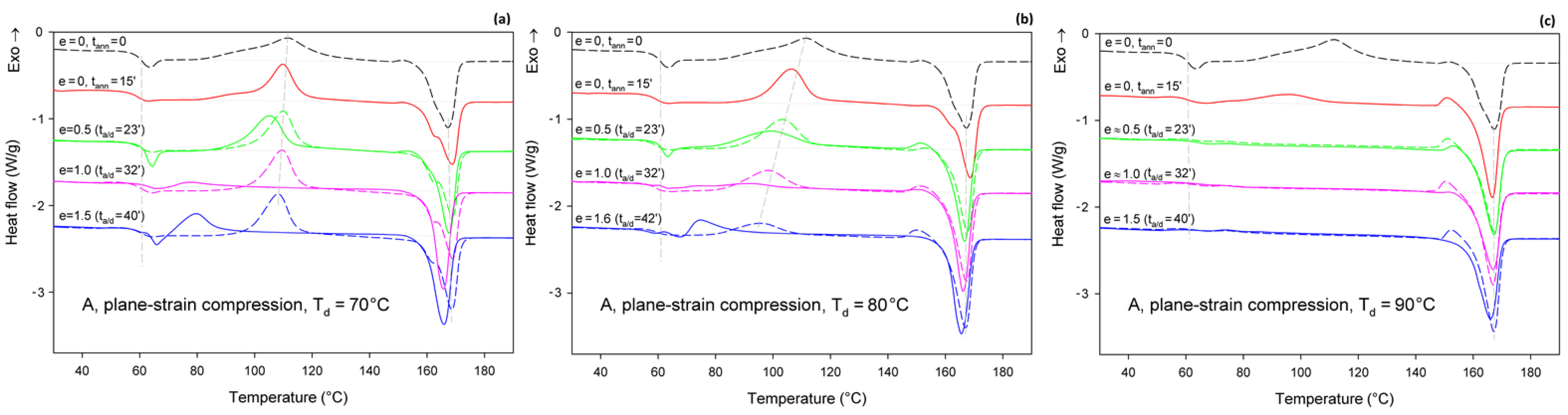
Figure 6 shows the DSC melting thermograms of initially amorphous A samples deformed in uniaxial tension at T
d = 70, 80, and 90 °C, respectively. For comparison, thermograms of the initial sample and samples that were only annealed at the same temperature and time as the deformed samples, but actually were not deformed, were plotted with dashed lines.
As can be seen in
Figure 6a, the samples that were annealed at T
a = 70 °C (dashed lines) show a cold crystallization peak at about 110 °C and a double melting peak with a major maximum around 168 °C, regardless of the annealing time. Such bimodal melting was frequently observed in PLLA when T
c was between 100 °C and 120 °C [
48,
55]. Both endothermic peaks were found to be related to the melting of the α phase: the lower temperature peak is related to the synchronous melting of the original α form crystals and the phase transition from α′ to α, while the higher temperature peak is connected to the melting of the crystals formed during the α′→α crystalline phase transition [
48]. The crystallinity of the samples after annealing at T
a = 70 °C, calculated from the enthalpy difference related to the melting and cold crystallization peaks, respectively, ranged from
Xc ≈ 0.4 wt.% up to approx. 2.3 wt.%. On the other hand, X-ray examination of these samples did not reveal any trace of crystalline phase (
Xc = 0%). All these very low values of crystallinity are close to this estimated for the initial A sample before further treatment:
Xc ≈ 2 wt.% (see
Table 1). This indicates that annealing at 70 °C, i.e., slightly above the T
g, does not affect the structure of the sample over the time scale of the experiment, and that the sample remains essentially amorphous. On the other hand, samples deformed at this temperature by uniaxial drawing demonstrate a shift of the cold crystallization peak to a lower temperature with an increasing strain and a significant decrease in size of this peak, which finally disappears at the true strain of e = 1.2 (which corresponds to the residence time at the deformation temperature t
a = 58 min). The crystallinity of the deformed samples estimated from the enthalpy difference was initially stable at a fairly low level—less than 2 wt.% at e = 0 (t
a = 15 min), 4.5 wt.% at e = 0.5, and 5.1 wt.% at e = 0.7—but it increased significantly to
Xc = 42.7 wt.% when the true strain reached e = 1.2. This result illustrates the development of the crystalline phase through the strain-induced crystallization process, which apparently takes place at T = 70 °C only in the high strain range. In addition, the post-Tg endothermic peak, similar to the enthalpy relaxation phenomenon observed for aged polymers, appeared at e = 0.5–0.7. According to Stoclet et al. [
9,
24], this endotherm in deformed samples can result from “melting” of the mesophase that was possibly formed upon drawing.
In samples annealed at T = 80 °C, the cold crystallization peak shifted to a lower temperature, below 100 °C, and decreased in size, while the double melting peak, previously observed in samples annealed at T
a = 70 °C, was replaced with a single peak with extended annealing time. Additionally, in samples annealed longer than 15 min, a small exothermic peak emerged just before the melting peak. Such a feature was usually observed in PLLA when T
c was below 100 °C and is probably related to the α′ to α phase transition, immediately followed by the melting of the newly formed α phase. This small exothermic peak just before melting—representing the α′→α transition—was found not to be a melt–recrystallization process, but rather a solid phase transition without the melting of the α′-phase [
48]. The crystallinity of the annealed samples increased moderately with the annealing time from less than 2 wt.% at t
a = 15 min to nearly 21 wt.% at t
a = 40 min (lower crystallinity,
Xc = 12.4 wt.%, was estimated from X-ray diffraction data). The crystals formed were in both α′ and α form. On the other hand, when the samples were deformed by drawing at T
d = 80 °C, the cold crystallization peak shifted to lower temperatures and decreased even faster than in the samples subjected only to annealing, while the small exothermic peak before the melting peak associated with the α′→α transition could already be seen at e ≥ 0.5. A post-Tg endothermic peak, similar to that observed at T
d = 70 °C and possibly related to the mesophase, can be recognized at e = 0.5 and 1.0. The crystallinity estimated for A samples deformed by drawing T
d = 80 °C increased from a low initial value of about 2 wt.% to
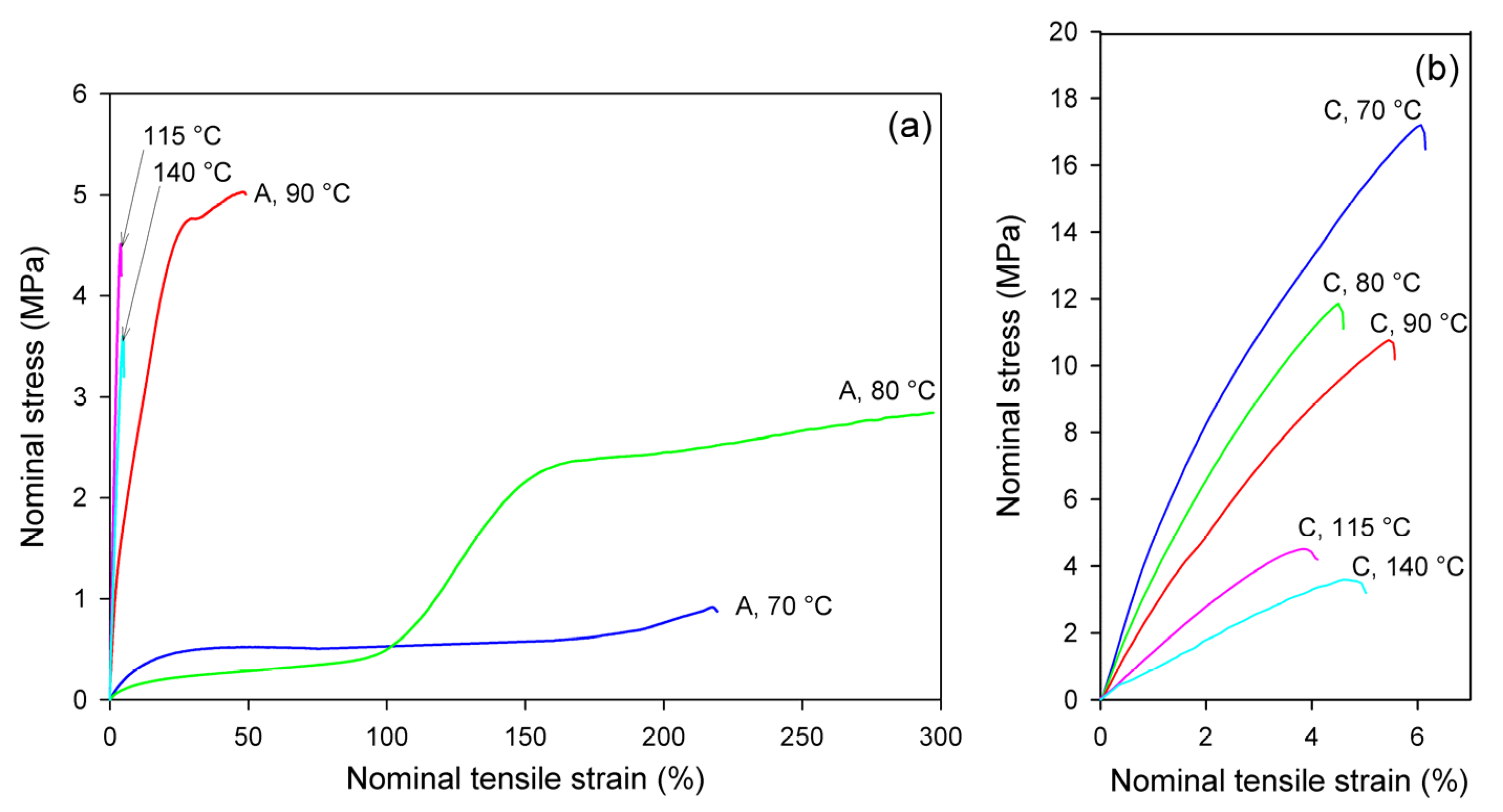
Xc = 12.1 wt.% at e = 0.5, next up to 25.8 wt.% at e = 1.0, and further up to 48.9 wt.% at e = 1.75. This crystallinity is much higher than that resulting from annealing alone, so that a significant fraction of crystals must have arisen due to strain-induced crystallization. Comparing the stress–time relationship, which is similar to the nominal stress–nominal strain curve (see
Figure 4a) and the change in crystallinity with time, it can be concluded that the upward stress jump in the stress–strain curve observed around the nominal strain of 100% is directly related to an increasing fraction of the well-oriented strain-induced crystalline phase.
Amorphous samples annealed at T
a = 90 °C for t
a > 15 min show no peak of cold crystallization after heating in DSC, which indicates that the crystallization was complete at this time. The overall crystallinity of the annealed samples increased from about 0% at t
a = 0 to approx. 27 wt.% at t
a = 15 min and, further, up to more than 40 wt.% at t
a ≥ 20 min (crystallinity estimated from X-ray diffraction data:
Xc = 0 at t
a = 0,
Xc = 19.3 wt.% at t
a = 15 min, and 37–40 wt.% at t
a = 24–40 min). An exothermic peak just before the melting peak associated with the α′→α phase transition and a single melting peak were present in the thermograms, regardless of the time of annealing, which could be expected for T
c = 90 °C [
48]. This confirms that the crystalline phase formed upon annealing at T
a = 90 °C consisted of a mixture of α′ and α form crystals, similar to the samples annealed at 80 °C. Deformation upon drawing of A samples at T
d = 90 °C was non-uniform, with the formation of a distinct neck. Therefore, it was possible to find parts deformed to different strains in the same sample. The cold crystallization peak was observed in the deformed material at a temperature only slightly lower than in the annealed only samples, whereas the small exothermic peak before the melting peak associated with the α′→α transition, seen in the annealed samples and in that deformed to e ≈ 0.5, decreased with strain and eventually disappeared at e = 1.3. At the same time, a low temperature shoulder could be recognized in the melting peak of the material deformed to e = 1.3, which suggests that some recrystallization phenomena might occur at high strain. The crystallinity of the stretched samples (T
d = 90 °C) increased from approx. 27 wt.% at e = 0 (t
a = 15 min) to
Xc = 52.3 wt.% at e = 1.3. This crystallinity is notably higher than that resulting from the annealing alone (up to 40 wt.%, depending on the annealing time), so that a significant fraction of crystals must have arisen due to strain-induced crystallization during drawing.
Figure 7 presents the DSC thermograms of initially amorphous A samples, deformed by plane-strain compression in a channel-die at the same temperatures as drawing: T
d = 70, 80, and 90 °C. Similar to tension, here thermograms of annealed-only samples are also presented for reference with dashed lines. Thermograms of samples compressed at T
d = 70 °C show the same features as those of samples drawn at this temperature (cf.
Figure 6a and
Figure 7a)—the cold crystallization peak shifts to a lower temperature and decreases in size with increasing strain; a single melting peak (α phase) and a post-Tg endothermic peak (mesophase) can be also seen in the deformed samples. The crystallinity of the compressed samples increases significantly between true strain of e = 0.5 and 1.0—from
Xc ≈ 1.4 wt.% to more than 40 wt.%. This indicates an intense strain-induced crystallization in this strain range, which is responsible for heavy strain hardening beginning below the true strain of e = 1 (see
Figure 5a).
Additionally, the samples compressed at T
d = 80 °C (
Figure 7b) and T
d = 90 °C (
Figure 7c) show thermal behavior similar to the respective tensile samples. The same features can be recognized in both tensile and compressed samples. These include a post-T
g endothermic peak, possibly related to the mesophase, which can be observed in samples deformed at T
d = 80 °C. This peak, however, does not occur in samples compressed at T
d = 90 °C. The crystallinity of the samples compressed at 80 °C, calculated from the difference between the enthalpy of the melting and cold crystallization peaks, increases clearly, from 12.8 wt.% to over 40 wt.% with an increase in the true strain from e = 0.5 to 1.0. This again indicates a significant contribution of the strain-induced crystallization. When compression is carried out at T
d = 90 °C, the material shows
Xc = 26.9 wt.% before the deformation (after 15 min of soak time) and quickly increases its crystallinity to above 40 wt.% already at e = 0.5 (t
a/d = 23 min). This means that deformation proceeded in the semi-crystalline material with a significant crystalline fraction from the very beginning, which explains why the stress observed during the deformation of A samples at low strains is higher at T
d = 90 °C than at T
d = 70 °C or 80 °C, where the nearly amorphous material is deformed in the initial deformation stages (cf.
Figure 4a or
Figure 5a).
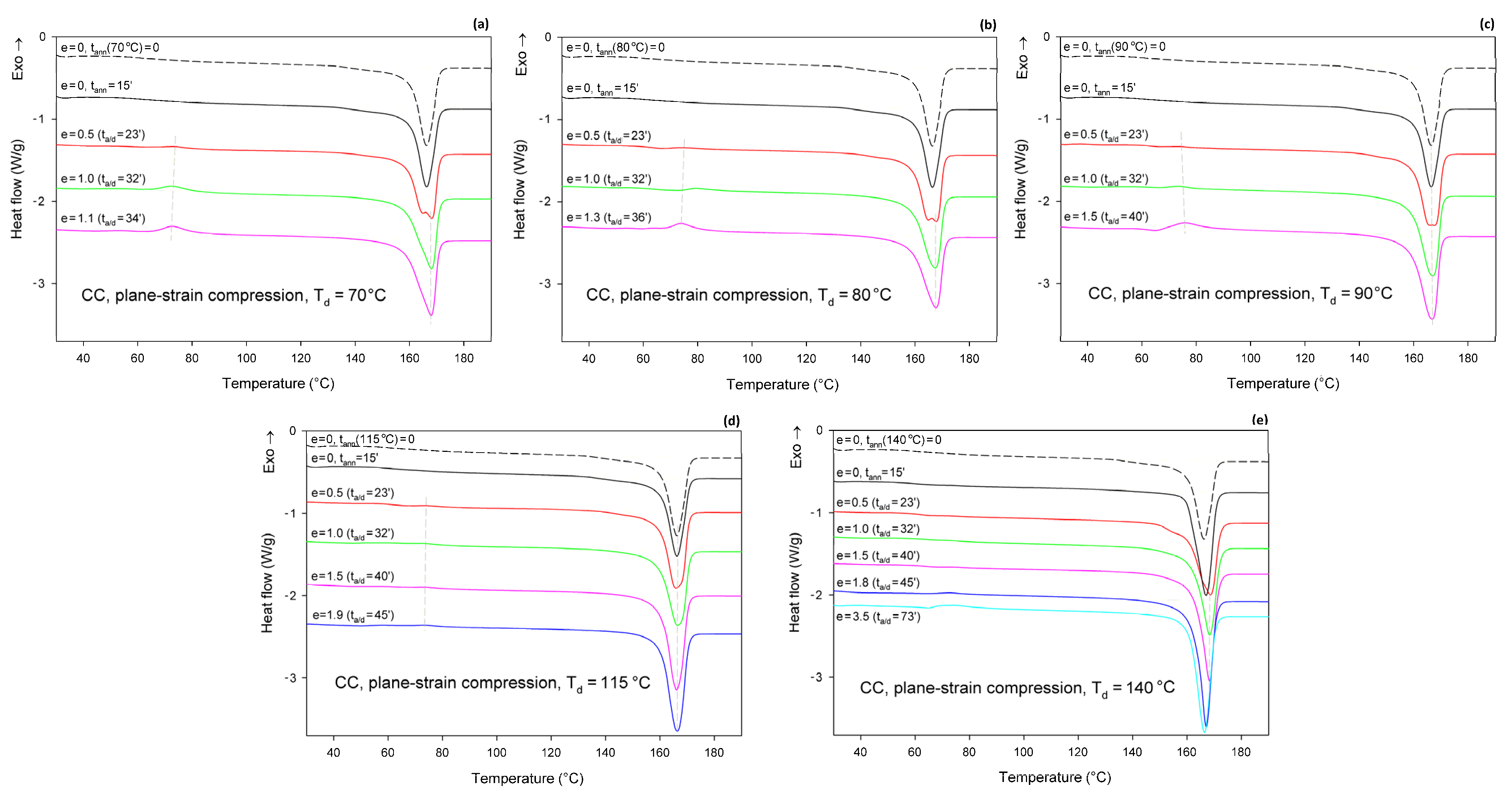
Figure 8 shows the DSC thermograms of CC samples (cold crystallized during 1 h annealing at T = 122 °C), compressed in a channel-die at T
d = 70, 80, 90, 115, and 140 °C. The thermogram of the initial CC sample is also provided for reference. The thermograms of the initial CC sample, as well as the CC samples subjected to 15 min annealing at any set temperature, show no peak of cold crystallization. A poorly developed glass transition and a single endothermic peak associated with the melting of α form crystals can be observed. These features indicate that annealing at T = 122°C resulted in the highly crystalline material (
Xc = 54 wt.%) of pure α form. Thermograms of CC samples that were subjected to plane-strain compression at T
d = 70–115 °C show an additional exothermic peak centered well below 80 °C, related to cold crystallization. This peak is generally small; it is barely visible at e = 0.5 and increases only slightly with increasing strain. It was bigger at T
d = 70–90 °C than at 115 °C. At T
d = 140 °C, this peak was not observed below e = 1.8 (corresponding to the compression ratio of λ = 6). At the same time, the crystallinity calculated from the enthalpy difference decreased with increasing strain from initial 54 wt.% to approx. 47–48 wt.% at the maximum strain when T
d = 70 or 80 °C and to about 51 wt.% at T
d = 90 °C, or remained roughly constant at T
d = 115 °C. In contrast, during deformation at T
d = 140 °C, which temperature was notably higher than the annealing temperature of the CC sample, the crystallinity tended to increase with strain (time), which was probably an annealing effect. The observations reported above for T
d = 70–115 °C suggest that a limited fraction of the crystalline structure, growing with strain, was destroyed during the deformation process. Interestingly, samples that were deformed to a relative low strain e = 0.5 demonstrated a double melting peak, suggesting that a fraction of the initial α crystals was converted into crystals of the α′ form of relatively high stability, similar to those α′ crystals formed at T
c = 100–120 °C together with the crystals of the α form [
48]. At higher strains, only a single melting peak can be observed, although this peak demonstrates a remarkable asymmetry, which may suggest a low-temperature shoulder due to the second melting peak, associated with the synchrotronous α′→α phase transition and melting of α crystals.
3.4. Structrure and Orientation of the Crystalline Phase (WAXS and SAXS)
As already reported in
Section 3.2, the initially amorphous samples deformed at temperature T
d ≤ 90 °C demonstrated ductile behavior and could be deformed to the true strain exceeding 1.2 (nominal strain ε ≈ 230%). The A samples, when deformed at higher temperatures of T
d = 115–140 °C, fractured before reaching the yield point, similarly to CC samples drawn at any temperature. The behavior of A in this temperature range is brittle, similar to CC samples, as the cold crystallization of PLLA is now very fast and almost complete during the soak period before deformation begins. Consequently, the structure of this A sample becomes very similar to that of CC sample, as must their deformation behavior.
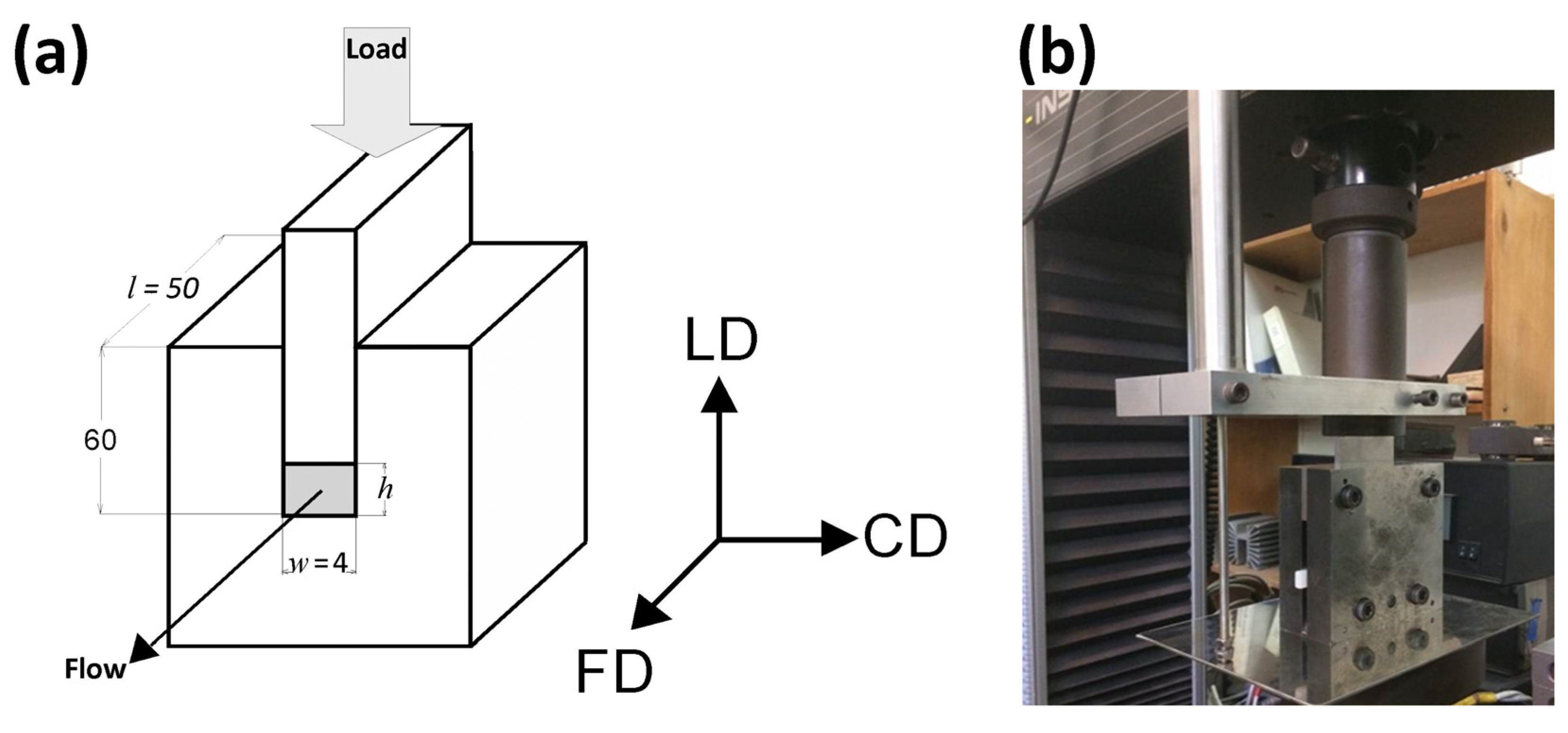
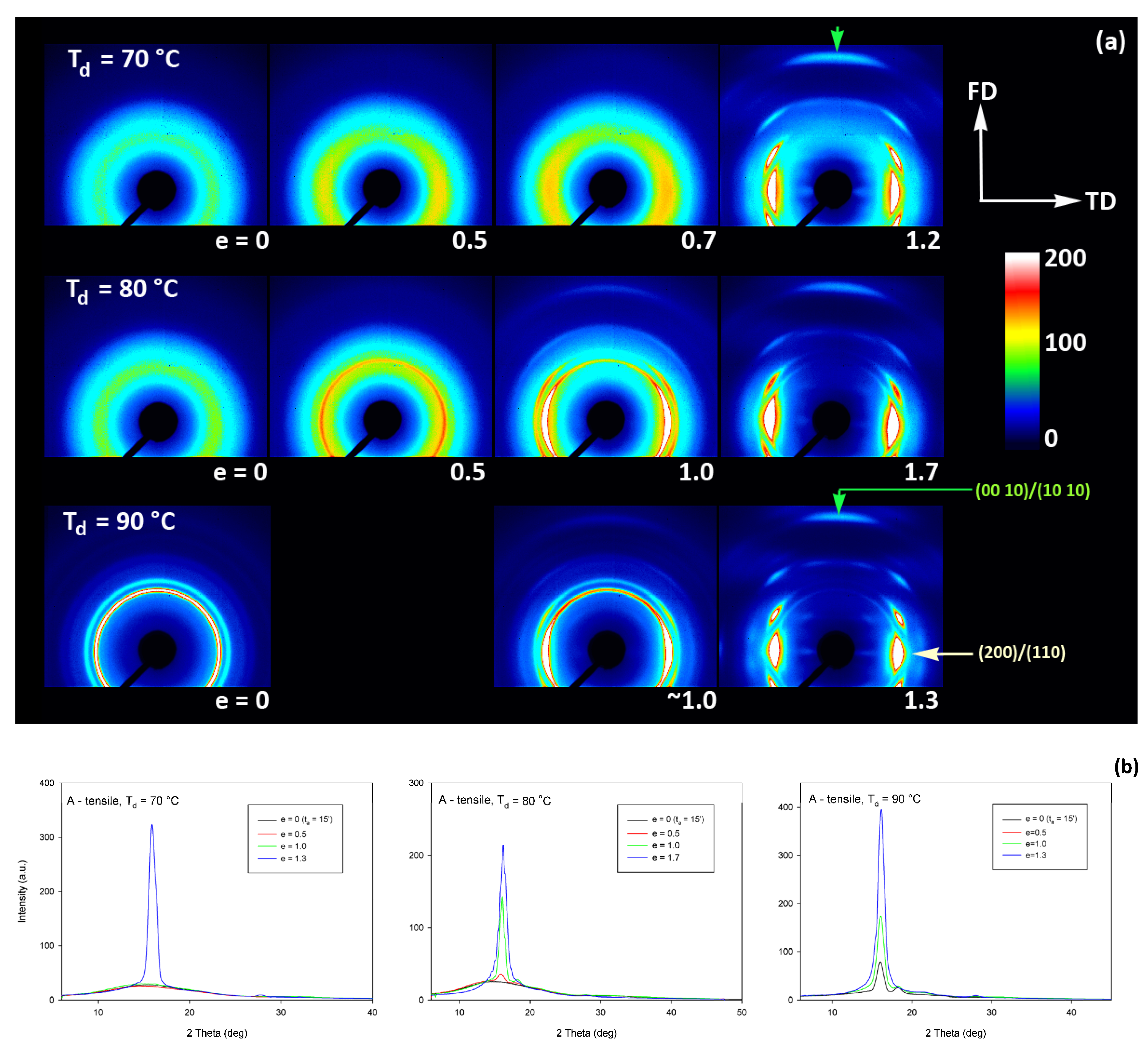
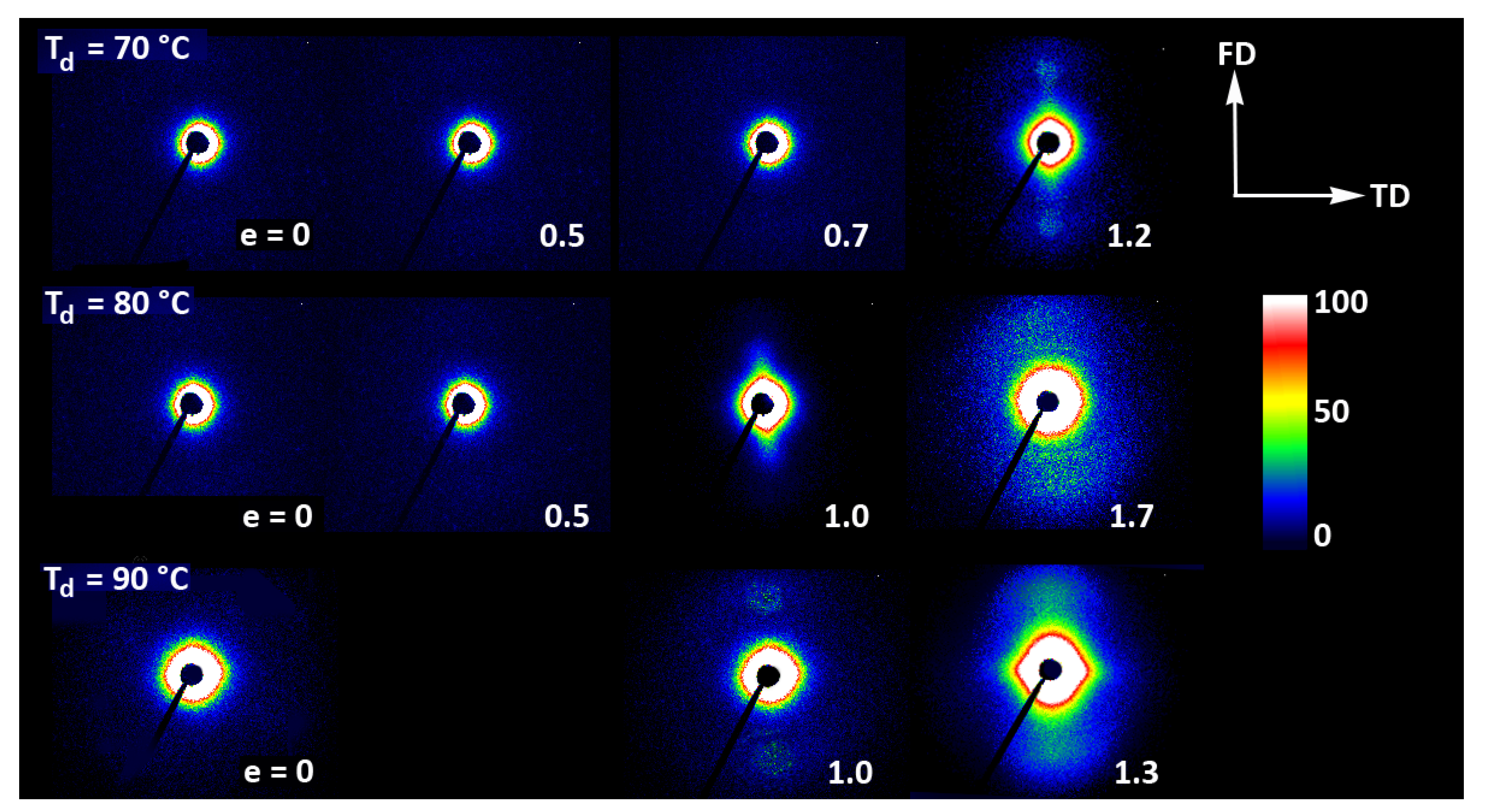
Figure 9 presents the evolution of the 2D diffraction image with strain observed in the drawn A samples, deformed to the indicated true strain, at temperatures T
d = 70, 80, and 90 °C, respectively. It can be noted that all drawn samples described here demonstrated axial symmetry along the drawing direction, FD, as the 2D-WAXS patterns recorded for a given specimen in the front-view (X-ray illumination perpendicular to the sample surface; the patterns shown in
Figure 9) and in the edge-view were very similar, almost identical. This symmetry along FD was actually expected as for uniaxial drawing.
The A samples deformed at 70 °C showed initially and at low strains a non-oriented amorphous structure, manifested in the form of a uniform diffuse ring observed in the 2D-WAXS pattern. As the strain increased to e = 0.5 and further to 0.7–1.0 the sample remained essentially amorphous (cf. one-dimensional scans presented
Figure 9b) but underwent some molecular orientation along FD, which resulted in a concentration of the scattering along the transverse direction, TD. Finally, at the true strain of e≈1.0, some crystals developed due to strain-induced crystallization. As suggested by the position of the diffraction spots in the 2D-WAXS pattern, these newly formed crystals were already well oriented with their c axis (the chain direction) along the drawing direction. In particular, at 2Θ ≈ 31–32°, in the polar region of the pattern, one can observe the reflections of the (0010) and (1010) planes (2Θ = 30.99° and 32.09°, respectively), which are apparently oriented roughly perpendicular to FD, suggesting a preferred orientation of crystals with the chain direction along FD. Billimoria et al. [
37] assigned the peak located above 32° to the (018)
α′ plane instead of to (1010). However, we observed the peak at approx. 32.1–32.3°, so assigned it to (1010) plane rather than to (018) (expected at 2Θ = 32.09 and 32.80°, respectively). The diffraction observed in the discussed range was assigned in the past to the development of crystals oriented along FD [
18] or to the contribution of the amorphous phase strongly oriented along FD—the third amorphous halo at 2Θ ≈ 31° arising from the characteristic interchain spacing of the methyl group in PLLA three-fold helices [
24]. However, in the collected data, one can distinguish easily and separate the contribution of amorphous halo from contributions of two crystalline peaks that were assigned here to (0010) and (1010) planes. These peaks can be observed only in these patterns which also show other crystalline reflections, i.e., belong to crystalline samples, while they were not seen in images of oriented amorphous samples, where only the almost uniform ‘intrachain’ amorphous halo was observed. As the normal to the (0010) plane is parallel to the chain direction in the orthorhombic α crystal while the normal to the (1010) plane is close to this direction, a presence and azimuth position of diffraction peaks of assigned to these planes can be used for tracking the orientation of the chain direction in crystals.
Similar orientation behavior was observed in samples drawn at Td = 80 °C and 90 °C, except that the crystalline phase appeared in these samples earlier than during drawing at 70 °C due to cold crystallization associated with annealing, which apparently started earlier than the strain-induced crystallization, occurring only at high strains, as confirmed by the DSC results reported earlier. Similar to the amorphous phase, the orientation of the crystals produced by cold crystallization remains relatively low at strains below e = 1. The preferred orientation of the crystals along FD becomes significantly stronger above e = 1, when the strain-induced crystallization also becomes active.
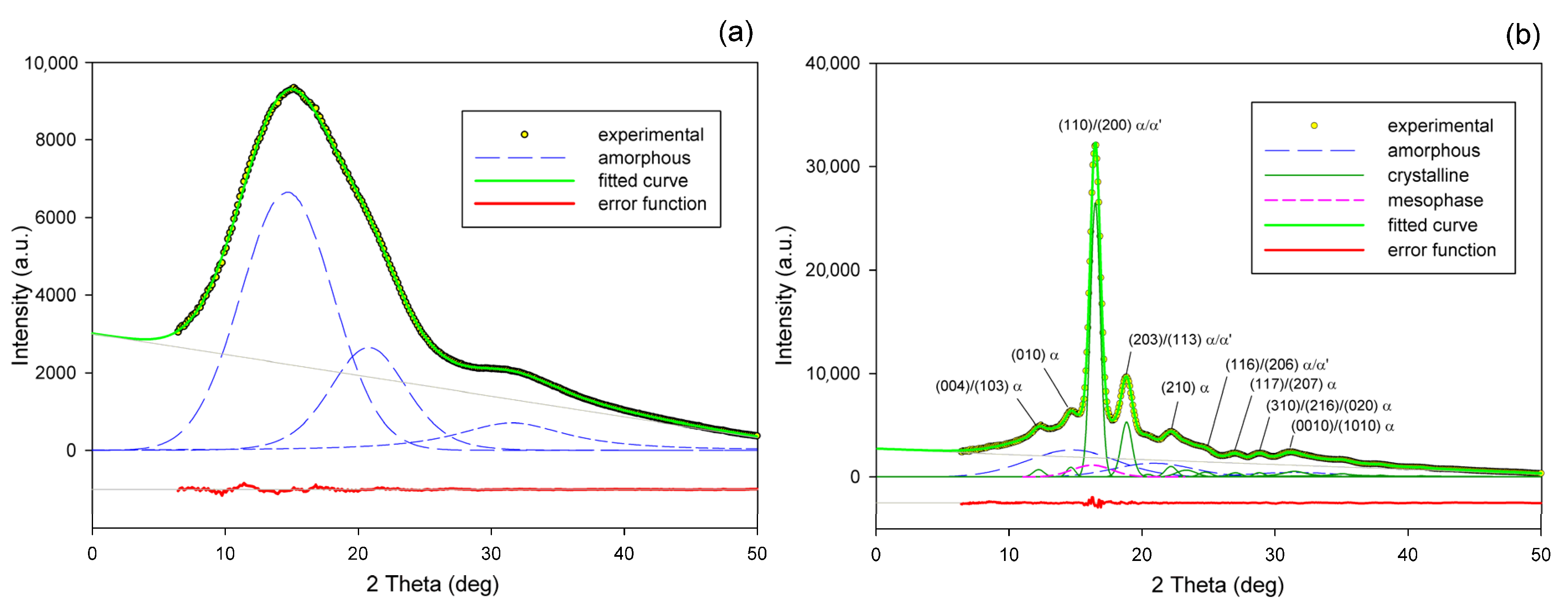
Detailed examination of the diffraction curves extracted from the 2D-WAXS patterns by azimuth integration allowed to determine the phase structure of the deformed samples and to estimate their crystallinity using curve-fitting procedures, as described in the Materials and Methods section. It turned out that, for T
d = 70 °C and 80 °C, the ‘best fit’ to the diffraction curves of deformed samples was obtained when the peak located at 2Θ = 16.2° and with a moderate FWHM ≈ 3.5° was taken into consideration, in addition to broad amorphous halos and narrow crystal reflections (cf.
Figure 2b). This peak corresponds to the mesophase [
9,
24,
25] developed from the strain-oriented amorphous chains at a strain well above e = 0.5. This result, together with the observation of the post-T
g endotherm in thermograms of the respective deformed samples (see
Figure 6), which, according to Stoclet et al. [
9], may be associated with the “melting” of the mesophase, strongly suggest the formation of the mesophase during deformation at T
d = 70 °C or 80 °C. For samples deformed at higher temperature (T
d = 90 °C), the fits of diffractograms that did not include the mesophase peak demonstrated comparable or perhaps slightly better quality than those assuming the presence of the mesophase (similar values of the weighted sum of square roots were obtained in both cases). Moreover, the characteristic post-T
g endotherm mentioned above was not present in thermograms of the samples deformed at T
d = 90 °C (see
Figure 6c). Therefore, the formation of the mesophase during drawing at T
d = 90 °C cannot be confirmed. This finding is in line with the conclusion of Stoclet et al. [
9,
24], who observed generation of mesophase at T
d = 70 °C and 80 °C and found the upper limit of mesophase stability in PLLA below T = 90 °C.
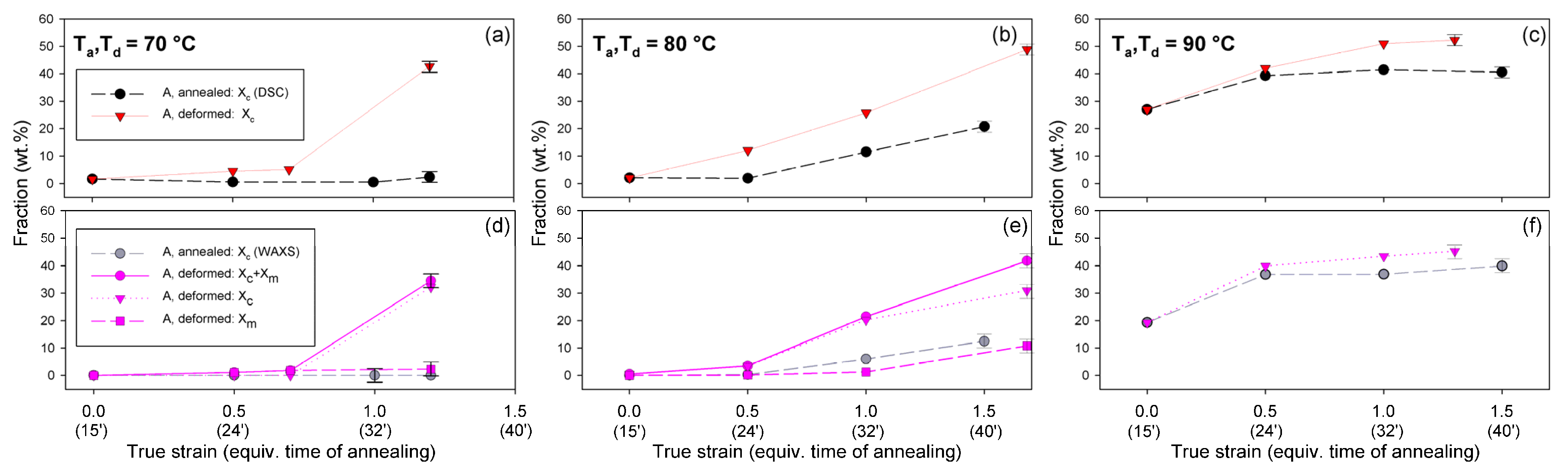
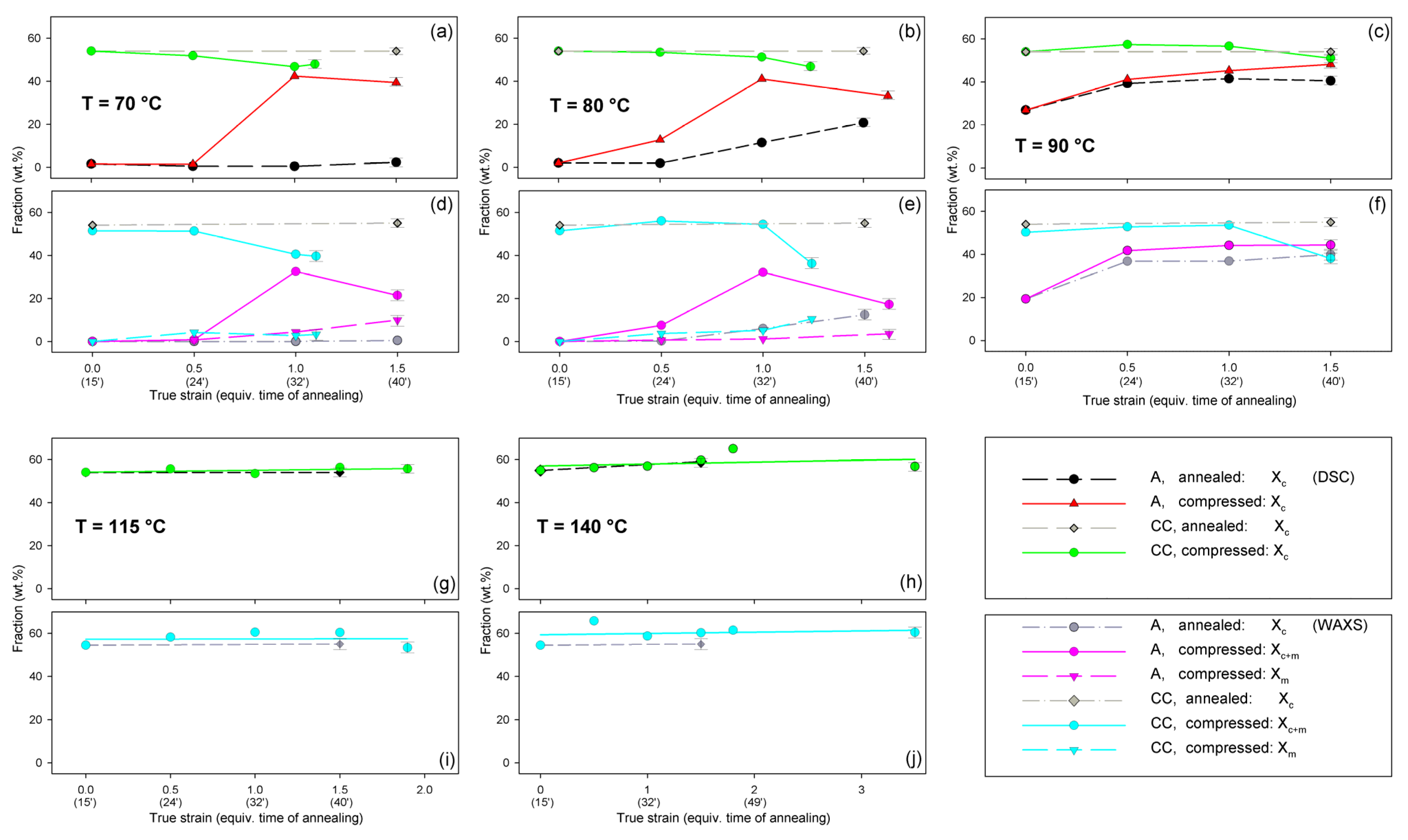
Figure 10 presents the phase composition of the drawn A samples (fraction of crystalline phase and mesophase) determined through the peak-fitting procedure applied to the X-ray diffraction data, compared with the crystallinity estimated from the DSC melting thermograms. A fairly good agreement of the sum of the crystalline and mesophase fraction determined from WAXS and the DSC-based crystallinity can be observed (note that the mesophase present in the sample after its deformation was converted into crystalline phase when heated in DSC). The content of both the crystalline and mesomorphic phases detected in samples deformed at T
d = 70–80 °C tends to increase with strain, especially in the range of high strains, above e = 1.0 (corresponding nominal strain, ε = 170%), which confirms that the mesophase as well as crystalline phase were formed from the amorphous phase, presumably well oriented due to plastic flow. Both the meso- and crystalline phase show a preferred orientation along the drawing direction, FD, as indicated by the major reflections concentrated in the equatorial plane of the WAXS pattern. In samples deformed at T
d = 70 °C, the mesophase fraction is relatively low, not exceeding 2.5 wt.% with e = 1.2, while at T
d = 80 °C this fraction is clearly higher, increasing to over 10 wt.% at e = 1.7. Most likely, the mesophase developed directly from the oriented amorphous phase at the same time as the crystalline component rather than through destruction of the previously formed crystals.
Figure 11 presents the 2D-SAXS images of the drawn A samples, complementing the 2D-WAXS patterns of
Figure 9. It can be seen here that the scattering of the deformed samples that remained essentially amorphous (e.g., e < 1 at T
d = 70 °C or 80 °C) is a more-or-less uniform diffuse scattering. As oriented crystals developed in the material due to strain-induced crystallization, two faint peaks emerged in the SAXS patterns along the drawing direction, FD. These maxima indicate some periodic stacking of the lamellar crystals developed along the FD direction. The signal is weak, suggesting that the periodicity of the lamellar structure that was formed during the deformation is rather low. This stacking is perhaps less regular than in cold crystallized CC samples, where, at similar level of crystallinity, stronger scattering due to the lamellar structure was observed.
Strong scattering developed in samples deformed at 80 °C and 90 °C in the final stages of their deformation, at e = 1.3–1.4, in the range of very small scattering angles, which is observed near the beam-stop, especially in the equatorial plane (along TD). This scattering is probably related to cavitation and voiding phenomena preceding fracture or right after sample unloading (samples were probed with SAXS in the unloaded state).
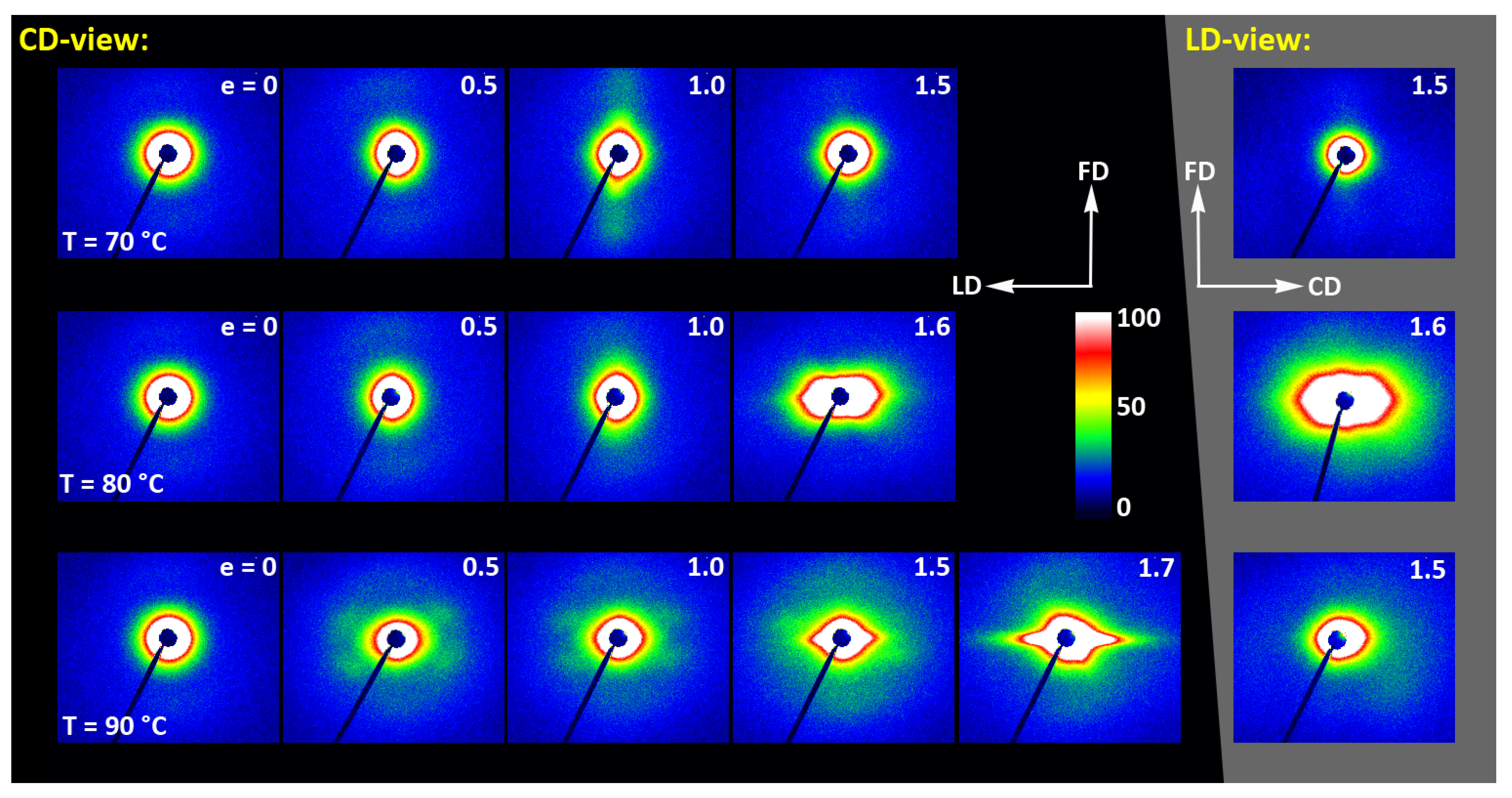
The 2D-WAXS and 2D-SAXS patterns of similar A samples, but now deformed by plane-strain compression, are presented in
Figure 12 and
Figure 13. Only the samples deformed in the temperature range of 70–90 °C are presented here as, as already discussed, at higher temperatures the cold crystallization of PLLA was so fast that the A sample quickly converted to CC, even before deformation began. Consequently, the deformation behavior of A had to be almost identical to that of CC sample, discussed later in this section.
The WAXS images of
Figure 12, recoded for compressed samples, are qualitatively similar to the respective images of the tensile samples shown in
Figure 9. Both sets show similar features. Interestingly, the images recorded with X-ray illumination along the constrained direction (CD-view) and those along the loading direction (LD-view) are very similar. This suggests an axial symmetry of the crystalline texture along the flow direction, FD, similar to that observed earlier in tensile samples. Such a uniaxial texture was surprising, as the strong constraints imposed on the sample by the walls of the channel lead to a triaxial stress state under load, therefore development of rather triaxial texture with preferred orientation direction and preferred plane could be expected, as, e.g., the (100)[001] texture (quasi-single crystal) observed in polyethylene [
57]. Apparently, when the sample was initially amorphous and isotropic, its deformation resulted primarily in an axial flow that led to molecular orientation along FD and consequently to strain-induced crystallization, fed by well oriented chains, resulting in uniaxial crystalline texture, which was not modified markedly by the stress distribution in transverse directions due to lateral constraints. The resultant crystal orientation is quite distinct. Examination of the strongest reflection, associated with (110) and (200) planes, along the azimuth demonstrated that normals to the (200) plane (2Θ = 16.46°) are oriented preferentially in the equatorial plane (LD-CD), while the normals of the (110) plane (2Θ = 16.32°), which is tilted approx. 60° from (200) in the unit cell, form a broad and much lower maximum centered along FD. Moreover, the reflection of the (0010) and (1010) planes, directly illustrating the orientation of chain direction [001], can be also seen in the FD direction. All these features suggest a relatively sharp uniaxial crystalline texture with the chain direction oriented preferentially along the drawing direction, FD. Such a sharp texture is due to strain-induced crystallization that converted highly oriented amorphous material into crystals oriented along FD, and probably not due to the activity of any crystallographic mechanism that would cause the crystallites to progressively rotate towards FD.
Figure 13 compares the results of determination of the phase structure and crystallinity of the compressed samples, based on DSC and WAXS data. As with the tensile samples, the curve-fitting procedure was applied to the diffractograms. As can be seen, there is a clear agreement between DSC- and WAXS-based estimates. The crystallinity of compressed A samples increases with strain up to e = 1.0, similarly to the respective tensile samples (cf.
Figure 10). The same mechanisms are responsible for such structural evolution: the strain-induced crystallization leading to highly oriented crystals, which prevails at T
d = 70–80 °C, and the cold crystallization (effect of annealing) only supported by the strain-induced crystallization at T
d = 90 °C, resulting in an overall less oriented material. This orientation, however, increases later with increasing strain due to activation of deformation mechanisms characteristic for semicrystalline polymers. At e ≥ 1.0, a decrease in crystallinity was detected by both DSC and WAXS. This is probably a result of a partial destruction of the crystalline ordering due to the fragmentation of the lamellae—see the SAXS results presented later in this section.
Similar to the drawn samples, the ‘best fit’ to the diffraction curves of samples deformed in a channel-die at T
d = 70 °C and 80 °C was obtained when mesophase formation during deformation was assumed, which resulted in the appearance of the peak at 2Θ = 16.2° and FWHM ≈ 3.5°. For higher deformation temperatures, the fits not taking into account the mesophase demonstrated comparable (T
d = 90 °C) or better quality (T
d = 115–140 °C) than those assuming the presence of the mesophase, hence the appearance of the mesophase in this temperature range was unlikely. This finding again confirms the upper limit of mesophase stability in PLLA below
T = 90 °C, suggested by Stoclet et al. [
9]. In A samples deformed at T
d = 70 °C or T
d = 80 °C the mesophase fraction increases with the increase in strain, although it remains at a low level of a few percent, similar to the tensile samples. Most probably, the mesophase developed during the deformation directly from strongly oriented amorphous chains, simultaneously with the crystalline phase (strain-induced crystallization), which is seen clearly up to the strain e = 1.0. At higher strains, a certain reduction in crystallinity was observed, therefore it is possible that apart from these strongly oriented amorphous chains, the remnants of damaged or destroyed crystals may also participate in the formation of a new mesophase fraction [
9,
24,
25].
Figure 14 presents a set of 2D-SAXS images of A samples compressed in a channel-die. It can be noticed that the patterns of samples compressed at T
d = 70 °C, when the developed crystallinity is relatively low, are similar to those observed for drawn samples (cf.
Figure 11)—for low strain, the pattern reveals mainly the features of amorphous material, although a very weak signal related to lamellar ordering can be distinguished, manifested by two extremely low maxima along the flow direction, FD (difficult to visualize in the image, but visible in its 1D cross-section along FD). The maxima are very low as the crystallinity of the sample is still low, below 2wt.%. At e = 1.0 these maxima become stronger and can already be observed directly in the pattern, while they seem to fade out again when the strain increases to e = 1.5, which is probably related to a partial loss of crystallinity, decreasing from approx. 42 wt.% at e=1.0 to about 39 wt.% at e = 1.5 (DSC estimates). As with the corresponding tensile samples, the low scattering intensity at the maxima discussed here, even at e = 1.0, suggests that, in addition to the low crystallinity, the periodicity of the lamellar structure that was created during deformation due to strain-induced crystallization, must be also rather poor. Comparing the patterns recorded in the CD- and LD-view, one can notice their similarity, which indicates the axial symmetry of the structure, when probed at the level of the lamellar structure. This symmetry is in fact analogous to that described earlier for the lower level of the crystalline texture.
Similar evolution of the lamellar structure was observed in samples compressed at T
d = 80 °C. In this case, however, in addition to the features discussed above, the characteristic hexagon-like shaped scattering can be seen in the strongly deformed sample (e = 1.6) at the lowest angles in SAXS images captured in both CD- and LD-view. The strength and angular position of this scattering component suggest that it is related to cavitation and voiding phenomena preceding fracture or following unloading. In such a case, these hypothetical voids should be elongated and oriented at an acute angle with respect to the flow direction in order to produce scattering of the observed shape. These might be voids and fissures located in the interlamellar layers, while lamellae had been tilted due to their shearing or sliding, i.e., the mechanisms known as active in deformation of semicrystalline polymers [
57].
At T
d = 90 °C, the amorphous material started to crystallize already during the 15 min soaking period prior to deformation, and demonstrated a crystallinity about 27 wt.% at the start of deformation, increasing further up to approx. 40 wt.% so that the deformation took place in a semicrystalline material from the very beginning. At e = 0, a scattering in the form of a weak yet uniform ring indicated that the initial orientation of lamellae was random. When the strain approached e = 0.5, the SAXS image transformed into a weak four-point type pattern, which suggests the development of a preferred orientation of lamellae with their normals oriented preferentially around 60° from the flow direction. A similar four-point pattern was also observed at higher strains, e = 1.0–1.5, however the angle of the preferred orientation with respect to FD gradually increased to over 75°—i.e., the lamellae tilted progressively towards FD and eventually oriented almost along FD. The development of such a preferred orientation was probably due to inter- and intralamellar shear that is commonly observed in other semicrystalline polymers subjected to plastic deformation [
57]. Another feature was observed above e = 1.0—two weak scattering spots appearing in the direction of FD. Such a signal is usually a signature of lamellae fragmentation and their restructuring into a new arrangement with the long period along FD [
52,
58,
59]. Finally, the strong scattering component developed at the lowest angles near beam stop, preferably along LD, at e ≥ 1.5. This scattering is probably related to cavitation and voiding preceding fracture or to voids and fissures parallel to LD formed upon sample unloading.
The long period was determined from the one-dimensional cross-sections of the 2D-SAXS images (CD-view) along the flow direction and the load direction, respectively. It turned out that, for all deformation temperatures, the long period along FD remains approximately constant with increasing strain, at the level of 13–14 nm, while the long period along LD tends to decrease slowly with strain from less than 13 nm to approx. 11 nm. This behavior probably reflects a squeezing of amorphous material from the interlamellar layers when oriented roughly normal to LD, due to the compressive loading. This can happen when the crystallinity is low, as in the case of T
d = 70 °C or 80 °C, and the separated lamellar stacks are embedded in a continuous amorphous matrix. When the crystallinity was higher and the crystalline lamellae formed a semi-continuous structure, as in the case of T
d = 90 °C, such squeezing is unlikely and the observed reduction in LP may be then the result of lamellae thinning that accompanies crystallographic (interlamellar) slip [
57].
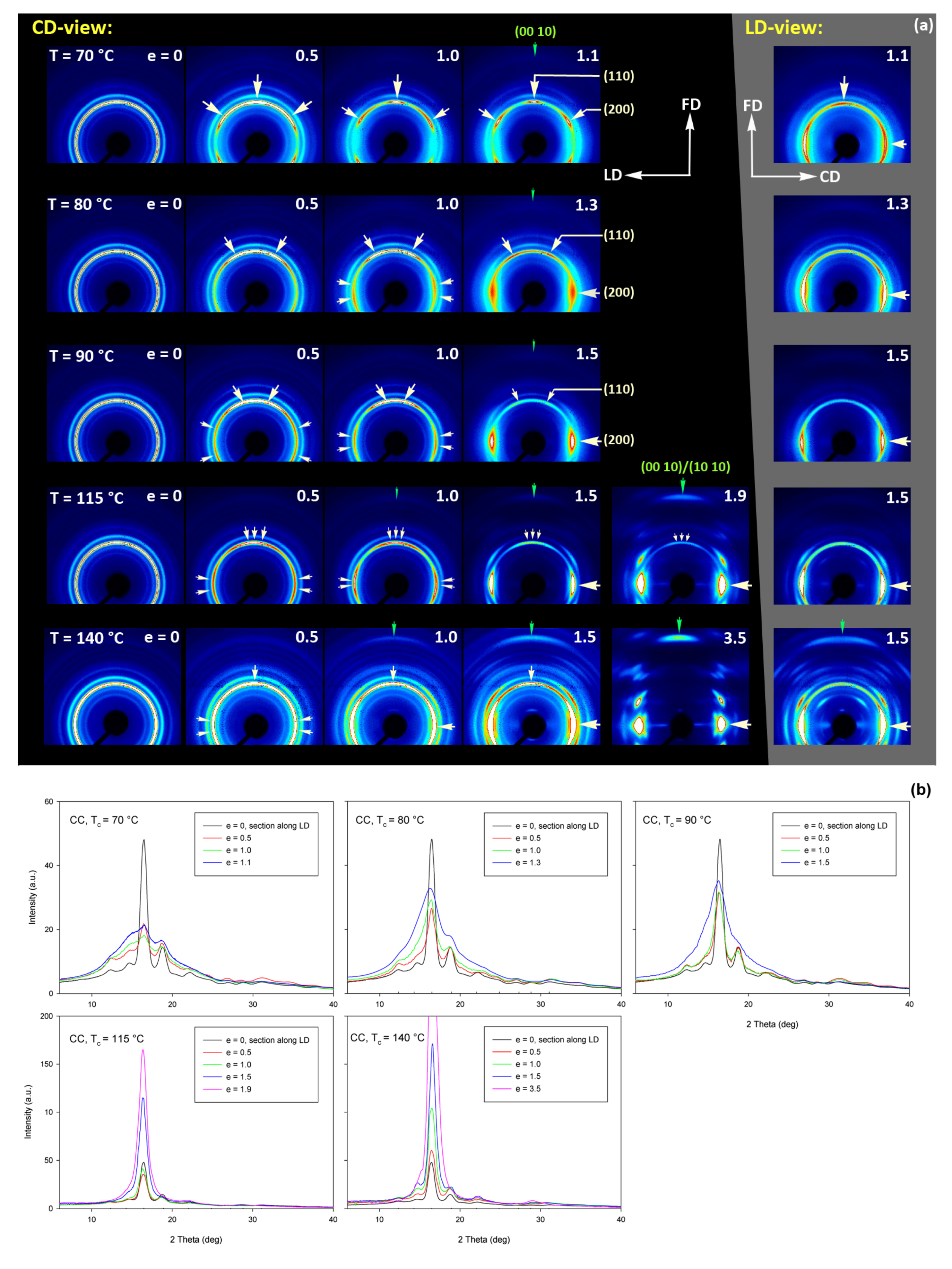
The deformation behavior of semicrystalline CC samples, obtained by annealing amorphous samples, deformed in the plane-strain compression mode, was studied in a wider temperature range, T
d = 70–140 °C, than amorphous A samples, which, at temperatures 115–140 °C, crystallized so fast they converted to CC material even before beginning deformation. In contrast to uniaxial tension, in which all CC samples easily fractured in a rather brittle fashion, regardless of the deformation temperature (cf.
Figure 4b), they were able to deform plastically to high true strains ranging from e = 1.1 at T
d = 70 °C to above e = 3.5 at T
d = 140 °C in the plane-strain compression (
Figure 5b).
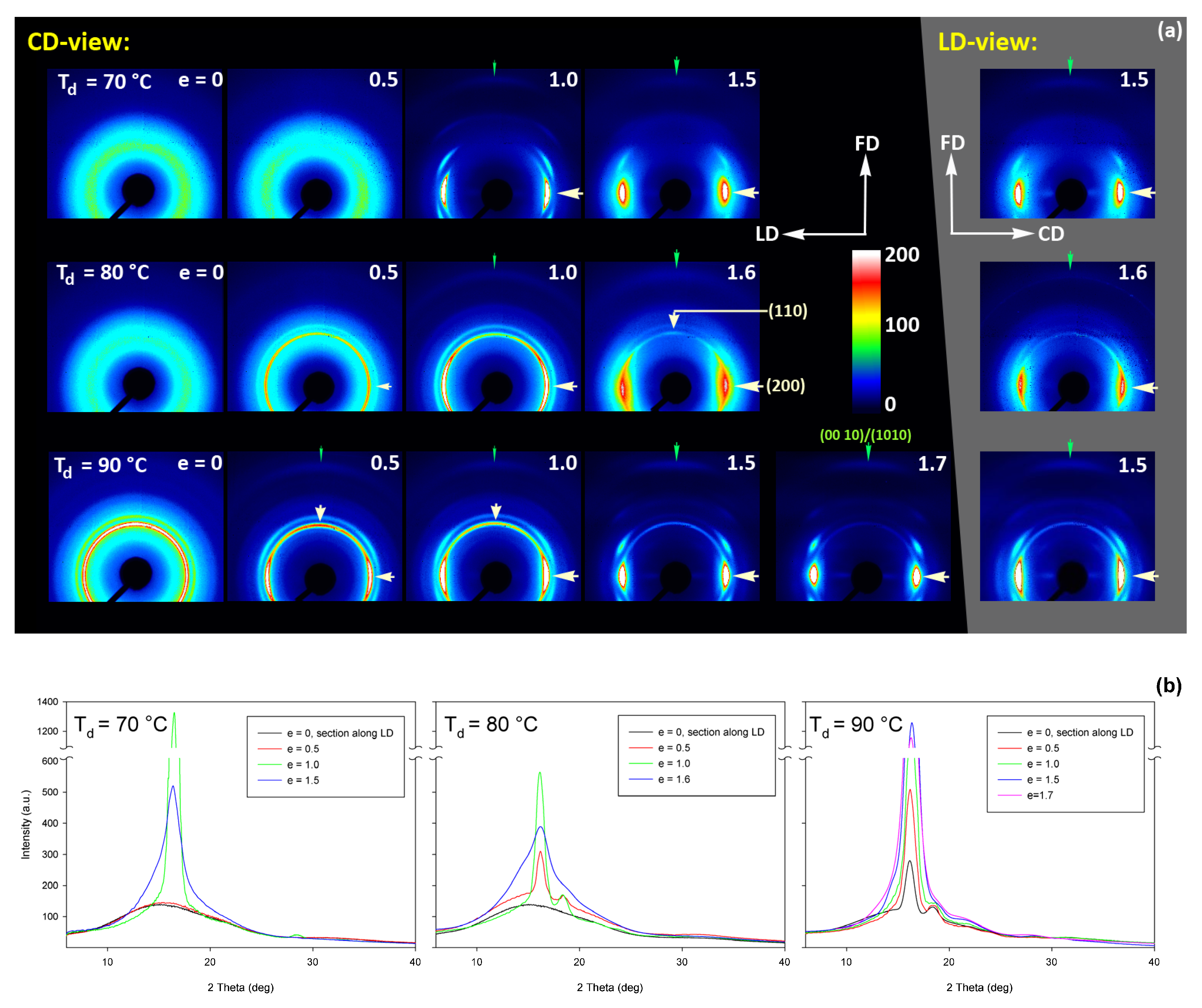
Figure 15 shows the results of WAXS examination of the compressed samples. As the initial crystallinity of the CC samples was high (54 wt.%, as estimated on the basis of DSC results), one can observe development of the preferred orientation of the crystalline phase due to deformation already at the low strains. The evolution of the crystal orientation can be easily observed by following the strain-related changes in the intensity distribution in (200)/(110) reflection along the azimuth in 2D-WAXS images. The (200) and (110) planes in α or α′ modifications of PLLA demonstrate similar interplanar distance, resulting in very close diffraction angles (e.g., 2Θ = 16.32° and 16.46°, for (200)
α and (110)
α, respectively) and consequently in merging their reflections into a single peak. In the oriented material, the maximum of this peaks can slightly shift towards lower or higher angles for a particular orientation of the sample with respect to the X-ray beam, depending on which plane contributes more to the peak. This allows the contribution of the (200) and (110) planes to 2D pattern to be distinguished.
At a low deformation temperature of 70 °C, the (200)/(110) diffraction ring, initially uniform in undeformed sample, developed a maximum along the flow direction in both the LD- and CD-view pattern, already at e = 0.5. Two clearly lower and very broad culminations, centered at approx. 45° from FD, can be also seen in the CD-view pattern. The maximum along FD was identified as related to the (110) plane, while the two tilted contributions were probably associated with the (200) plane. As the strain increased to e = 1.0–1.1, the (110) reflection was still observed along FD, while the (200) maxima turned to the position approx. 58° from FD in the CD-view pattern, sharpened, and increased in intensity to the level of the maximum associated with the (110) plane. The observed tilt angle of 58 °C corresponds well to the angle between normals of (200) and (100) planes in the unit cell (60.3°), thus the maxima in (200)/(110) diffraction observed in the CD-view pattern may originate from the same component of crystal orientation with the (110) plane normal to the FD and a chain direction along CD, i.e., perpendicular to FD. In the LD-view pattern, still relatively broad (200) maxima developed near CD, while a tilted orientation was not observed. Additionally, at e = 1.1, which exhibited the ultimate strain for this deformation temperature, a weak diffraction arc appeared at 2Θ ≈ 32° at the position near FD. This spot, related to reflections of the (0010) and (1010) planes, can be considered as the signature of an additional new component of the crystalline texture, with the chain direction preferentially oriented along FD. The described distribution of crystal orientation developed in CC samples deformed at T
d = 70 °C with the (110) planes oriented preferentially perpendicular to FD as the main component of texture, may probably be a result of interlamellar slip (shear), perhaps with some, rather limited, contribution of crystallographic (interlamellar) slip. Stoclet et al. [
25] suggested that the twinning mechanism, occurring in (200) or (110) plane in crystals that are oriented with these planes roughly normal to FD, may also be active at the initial stages of deformation at such a relatively low temperature.
At T
d = 80 °C, two very broad maxima in (200)/(110) ring, about 25–30° from FD, developed in the CD-view pattern at e = 0.5. Similar, although much weaker maxima can be observed in the LD-view. At e = 1.0, these maxima seen in the CD-view developed further at approx. 25° from FD and could be identified as corresponding to the (110) plane. Meanwhile, the maxima related to the (200) plane emerged near LD, about 80° from FD, and turned further towards LD and increased in intensity as the true strain increased to e = 1.3. It was observed that similar maxima due to diffraction on the (200) planes developed in the LD-view pattern along the CD direction, perpendicular to FD, while the maxima of (110) gradually faded out. As in the case of samples deformed at 70 °C, weak diffraction arcs (albeit notably stronger than those seen at T
d = 70 °C) associated with (0010) and (1010) planes appeared in the direction of FD, which may indicate an increasing fraction of crystals oriented preferentially with the chain direction along FD, probably due to growing contribution of the crystallographic slip mechanism to the deformation process. It seems justified, as the critical shear stress for slip in the polymer crystal (plastic resistance) decreases strongly with temperature [
56].
A similar evolution of 2D-WAXS patterns was observed at T
d = 90 °C. The first maxima in (200)(110) ring appeared now, at e = 0.5, closer to FD and CD, respectively, than at low temperature: stronger maximum, related to the (110) plane, approximately 10° from FD, and weaker, associated with the (200) plane, close to CD—approx. 75–80° away from FD. As the strain increased, these maxima rotated to approx. 68° from FD and to 90° (i.e., towards CD), respectively. While the intensity of the maxima near LD increased, it became significantly weaker near FD, especially at e = 1.5. Meanwhile, in the LD-view patterns, the (110) maxima near FD also decreased notably in intensity and only strong maxima near CD could be eventually observed at e ≥ 1.0. The strong (200) diffraction perpendicular to FD, seen in both CD- and LD-view images, together with the (0010)/(1010) reflection observed along FD, indicates the preferred orientation of crystals with the chain direction along the flow direction, which was probably produced with a noticeable contribution of crystallographic slip mechanisms [
57] in addition to interlamellar shear.
At Td = 115 °C and 140 °C, almost the same features could be observed in 2D-WAXS images, although the maxima related to diffraction in the (110) plane seemed weaker, while those associated with the (200) plane became stronger than at lower deformation temperatures, and developed gradually closer into FD and CD, respectively. Moreover, the intensity of the (0010)/(1010) diffraction, observed at e ≥ 1.0, became notably higher, and this diffraction spot could be recognized easily in the patterns as early as at e = 1.0, especially in samples deformed at a high temperature of 140 °C. This indicates an increasing preferred orientation of the crystals with the chain direction along the flow direction, hence the probably increasing contribution of crystallographic slip mechanisms to the deformation process with the increase in the deformation temperature.
Figure 13, presented earlier, reported the changes of phase composition with temperature and strain. As with the samples of series A, in the CC samples, a small amount of mesophase, formed during deformation, was detected when the deformation took place at T
d = 70–80 °C, i.e., below the upper limit of mesophase stability. A low fraction of mesophase was detected already at e = 0.5. For the true strain e = 1.0, it was still below 4–5 wt.%, but increased even up to 10 wt.% at higher strains. At the same time, in the deformed samples, a clear reduction in the content of the crystalline phase was observed, which may indicate that, in addition to the mesophase, which was probably formed from oriented amorphous chains, the other fraction, appearing at higher strains, was probably formed from still oriented small residues survived from the crystalline lamellae that were damaged or destroyed during deformation due to deformation instabilities [
52,
59]. Indeed, Stoclet et al. [
9,
24,
25] suggested a partial destruction of initial α or α′ crystals and subsequent reorganization into mesophase and/or α′crystals, depending on the deformation temperature, i.e., the mechanism of the strain-induced “melting” followed by partial recrystallization. Interestingly, when the deformation temperature was high (T
d = 115–140 °C), not only was the mesophase not detected, but no reduction in crystallinity with strain was observed—for a given deformation temperature, the crystallinity estimated on the basis of either WAXS or DSC data remained almost constant over the entire strain range. This may indicate that the crystalline phase did not deteriorate during deformation at these temperatures, or that partial destruction (strain-induced ‘melting’) did indeed occur in this temperature range as well, but that immediate recrystallization took place, keeping the sample crystallinity constant. The SAXS results discussed later in this section suggest that the second scenario, in line with the Stoclet’s hypothesis, is true.
Another crystal transformation, α→β, was postulated to occur during deformation of PLLA, [
5,
6,
8]. Lotz suggested that the β-phase is possibly an intermediate phase that could convert into more stable α′ modification [
46]. On the other hand, Wang et al. [
3] explained formation of the β-modification as the strain-induced structural transition from the α to the β form occurring via the transient disordering of the α form to the α′ form (α→α′→β), by assuming the cooperative displacements of the upward and downward helical chains, as well as the conformational change. The crystals of the β-phase were first obtained by drawing fibers at high temperatures and high draw ratios [
45]. The crystal transformation from α to β was observed to occur upon tensile drawing [
8] and solid-state extrusion [
5,
6], more efficiently at higher temperatures, larger draw ratios and/or higher draw rates [
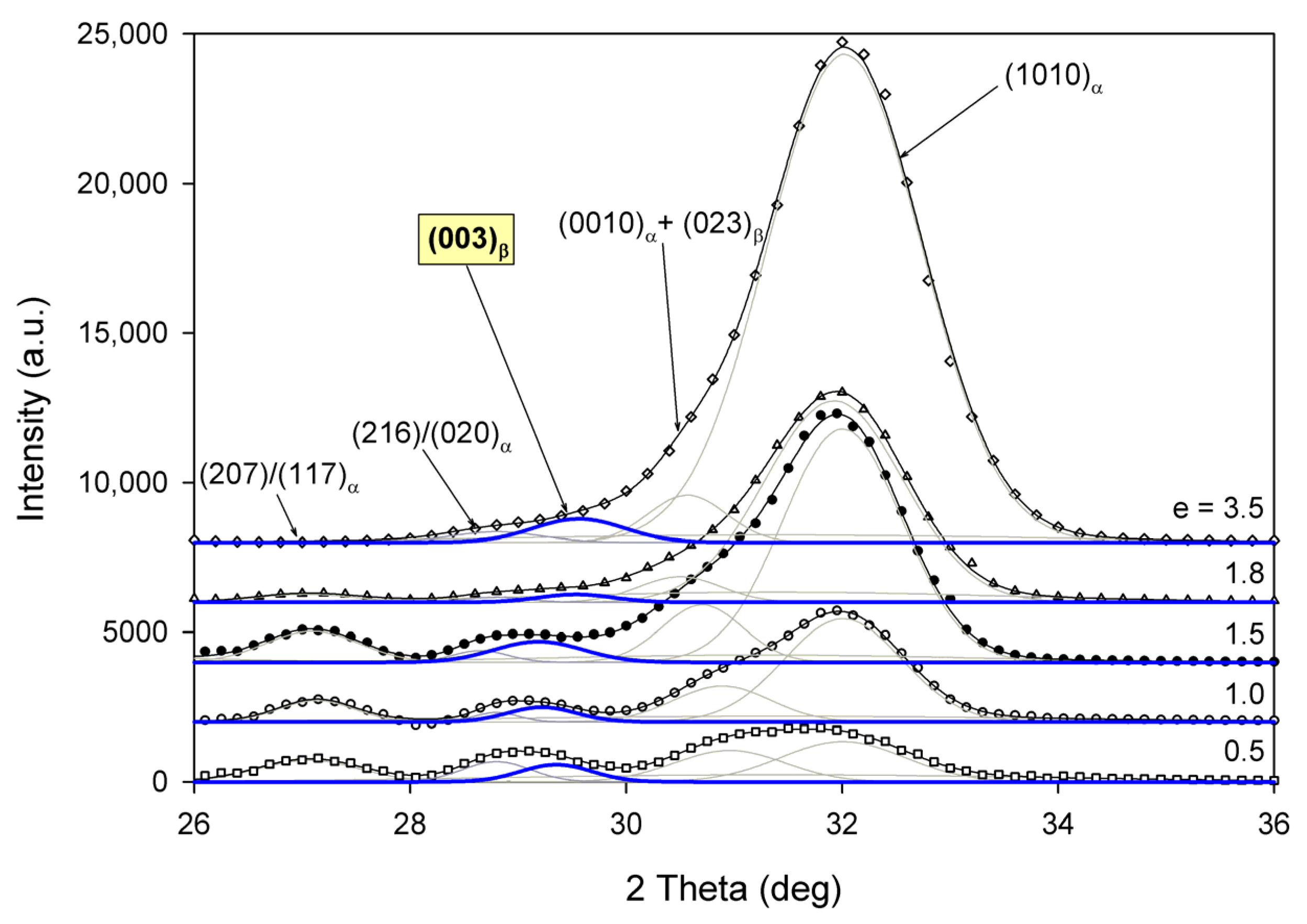
4]. We examined our data for the α→β transformation and presence of β-phase crystals in deformed samples. As the main (200)
β X-ray reflection strongly overlaps with (200)/(110) peak of α modification, the activity of the α→β crystal transformation can be alternatively confirmed by checking the presence of the (003)
β reflection from β crystals at 2Θ ≈ 29.8° and, if found, by following the relative intensity of this reflection and the (0010)
α or (1010)
α reflections from α crystals in the WAXS meridional scan [
6,
8] (one-dimensional section of the CD-view image along FD), as shown in
Figure 16. Careful examination of the experimental data, again employing the peak-fitting procedure, indicated the presence of a very small (003)
β peak in the CC samples deformed at T
d = 115 °C and 140 °C, which can confirm the small quantities of β crystals formed during deformation at these relatively high deformation temperatures, perhaps by transformation from crystals of the α–phase. At the lower deformation temperature, the β peak was hardly detected, which indicates that α→β transformation was most probably not active then.
Unfortunately, the relative amount of β crystals could not be directly correlated with the ratio of the intensities of (003)
β and (0010)
α reflections [
8]. Nevertheless, the low intensity of (003)
β compared to (0010)
β or (1010)
α suggests a very small amount of β-phase produced, so it can be concluded that the α→β transformation plays only a secondary role in the plastic deformation process.
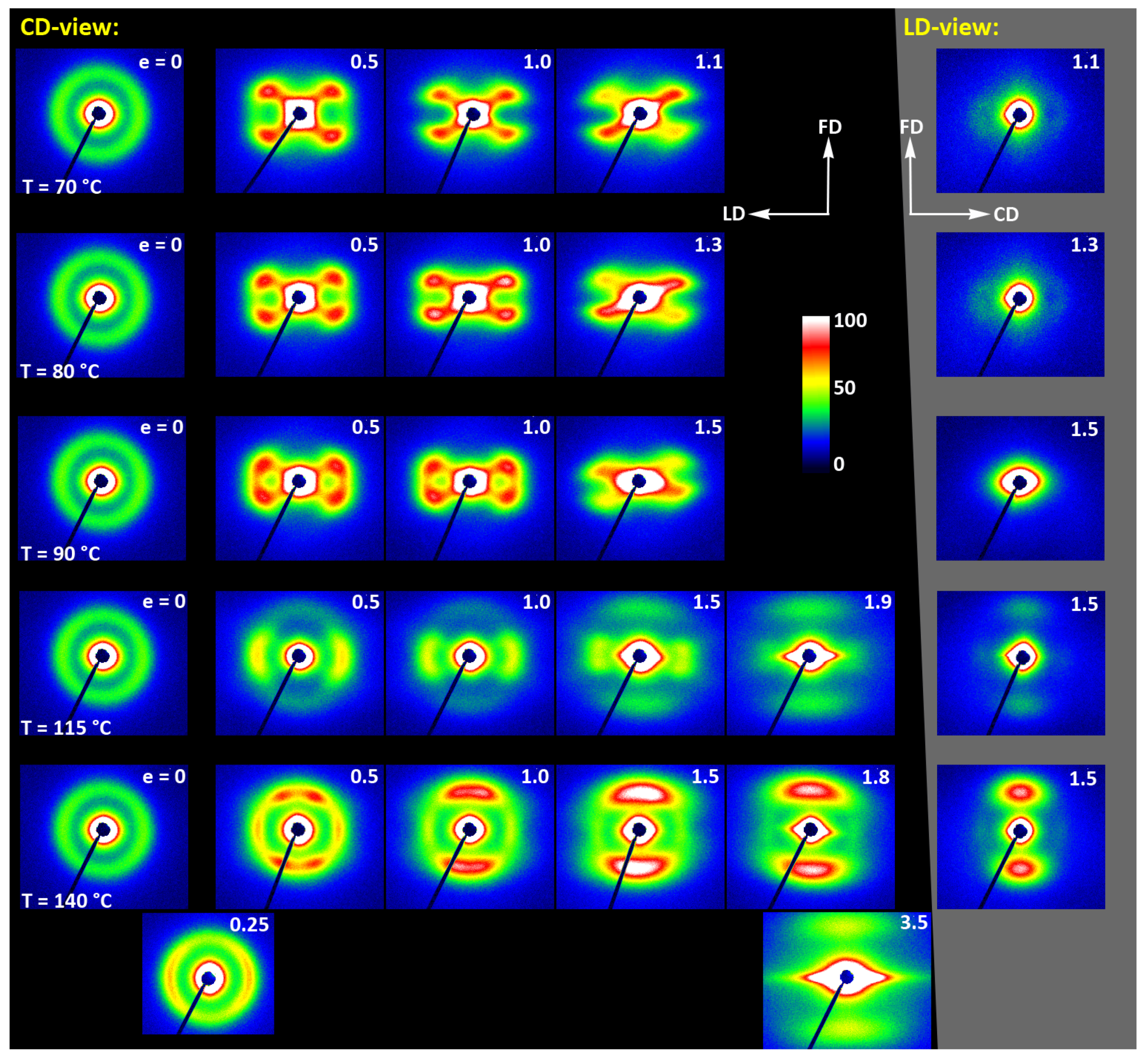
Figure 17 shows 2D-SAXS images of deformed CC samples. As the crystallinity of these samples is high, they all give a clear SAXS image. In general, the uniform ring observed for the initial non-oriented sample is transformed into a four-point pattern already at the true strain e=0.5, regardless of the deformation temperature. This temperature, however, affects the azimuthal position of the maxima in that pattern—the angle between azimuth at which the maximum is observed and the flow direction changes from ψ ≈ 54° at T
d = 70 °C to ψ ≈ 77° at T
d = 115 °C. At T
d = 140 °C, two broad maxima oriented along LD, i.e., perpendicular to FD, are visible (additionally, another, stronger four-point feature is also observed in this image near FD. This component of the pattern will be discussed later). Most likely, each of these wide maxima is the result of merging of two maxima belonging to the four-point pattern, similar to that observed at lower temperatures, yet now oriented very close to LD (ψ→90°). The formation of this component of the pattern was observed near LD already at e = 0.25, when the second four-point component near FD had not yet appeared. The maxima in the four-point pattern rotated slowly away from FD towards LD when the true strain increased to about 1.5, whereas the intensity at these maxima decreased, especially noticeably faster when e ≥ 1.0. The long period determined along the direction defined by the position of maxima near LD practically did not change with strain when T
d = 70–90 °C and it decreased slowly with increasing strain when T
d = 115–140 °C. The rotation of the maxima in SAXS pattern away from FD suggests the progressive tilting of lamellae towards the plane containing FD as the strain increased. The long period remaining constant at low deformation temperatures may suggest that such lamellar rotation was primarily due to interlamellar slip, i.e., the shear taking place in amorphous layers between lamellae [
57,
60], which proved to be the dominant mechanism at low temperatures. Changes in crystal orientation deduced from 2D-WAXS images (
Figure 15) support this view, although a limited contribution of crystallographic slip and twinning was also considered. At higher deformation temperature, the long period in the tilted lamellae considered here tends to slightly decrease with strain. Thinning of interlamellar layers due to interlamellar shear is unlikely, therefore for the observed shortening of the long period, the lamellae has to become thinner. A probable deformation mechanism that could lead to the described orientation of the lamellae and their thinning is the crystallographic chain slip [
60], which increasingly supported the main deformation mechanism through interlamellar slip. The intensity of the chain slip increases with temperature as the plastic resistance of polymer crystals decrease strongly with increasing temperature [
56]. The emerging preferred orientation of the chain direction in the crystals along the FD direction, as shown by the WAXS results, fully supports this hypothesis. As the easy slip planes are generally the closest packing planes, i.e., of large interplanar distance [
60], a suggested active crystallographic slip may be (010)[001], (100)[001], or (110)[001] slip system (i.e., a slip proceeding in the (010), (100), or (110) plane along the [100] direction), as those close-packed planes exhibit the largest interplanar distance of 0.62 nm and around 0.54 nm, respectively.
At the deformation temperature of 140 °C, SAXS images for e = 0.5 and 1.0 seem to consist of two superimposed four-point patterns, each perpendicular to the other. That which formed first, oriented similarly to those observed at lower temperatures, was described above. Its intensity decreased with increasing strain and gradually faded away until it finally disappeared at approx. e = 1.5. The second four-point component with strong maxima oriented near FD increased the intensity to e = 1.5. These maxima turned toward FD with increasing strain and eventually merged at about e = 1.5 creating a strong two-point pattern oriented along FD. A similar, even sharper two-point feature could be observed in the image captured in the LD-view. The origin of this second component is not clear. It can be guessed that the lamellae contributing to such a pattern, strongly deviating from FD, acquired this preferred orientation owing the joint action of crystallographic and interlamellar slip, perhaps with a stronger contribution of the crystallographic mechanism, but it occurred in another population of lamellae of different initial orientation than those contributing to the first component of the SAXS pattern.
At strains above 1.5, the maxima observed in the image along FD become significantly weaker in intensity and, moreover, above e = 1.8 they jump to a new position at a larger angle, which indicates the formation of a new long period along FD, yet shorter than the original. Such a transition is usually a signature of heavy fragmentation of lamellae already severely deformed by slip processes and restructuring the small crystalline residues that have left such a fragmentation to a new ordering giving new long period along FD [
52,
58,
59]. These recreated crystals are smaller and possibly stacked less regularly than the original lamellae, thereby reducing the intensity of scattering. Usually, this new structure can be improved by annealing. The fragmentation process requires that lamellae deformation by chain slip is active and well advanced, as fragmentation usually results from deformation instabilities in the lamellae that have already been significantly thinned by an advanced slip action [
52,
59]. Similar lamella fragmentation probably also occurred at T
d = 115 °C and possibly at 90 °C, above e = 1.5, as suggested by the relatively weak scattering streak appearing along FD in both CD- and LD-view images at e ≥ 1.5 (clearly visible at T
d = 115 °C).






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}