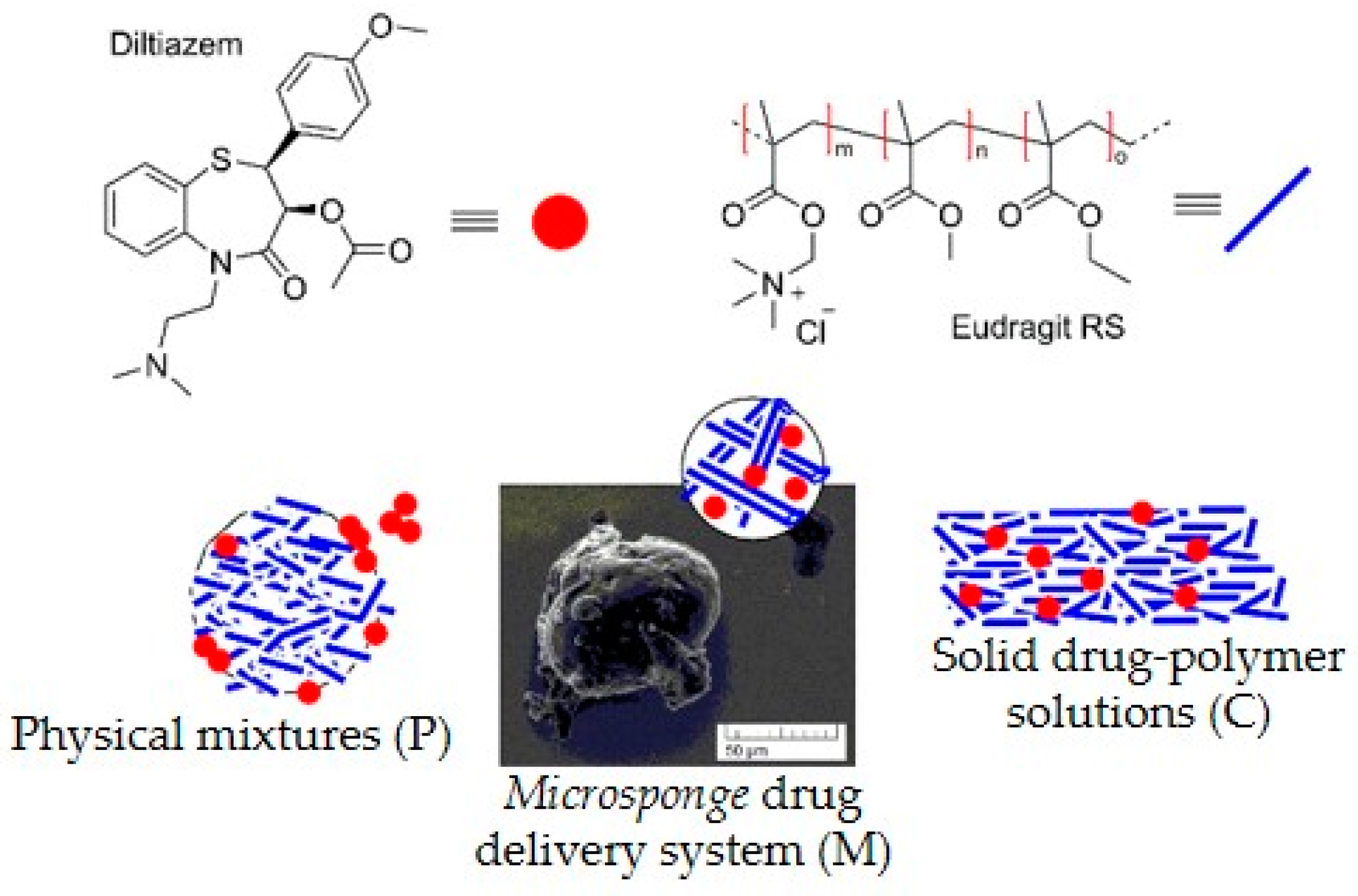
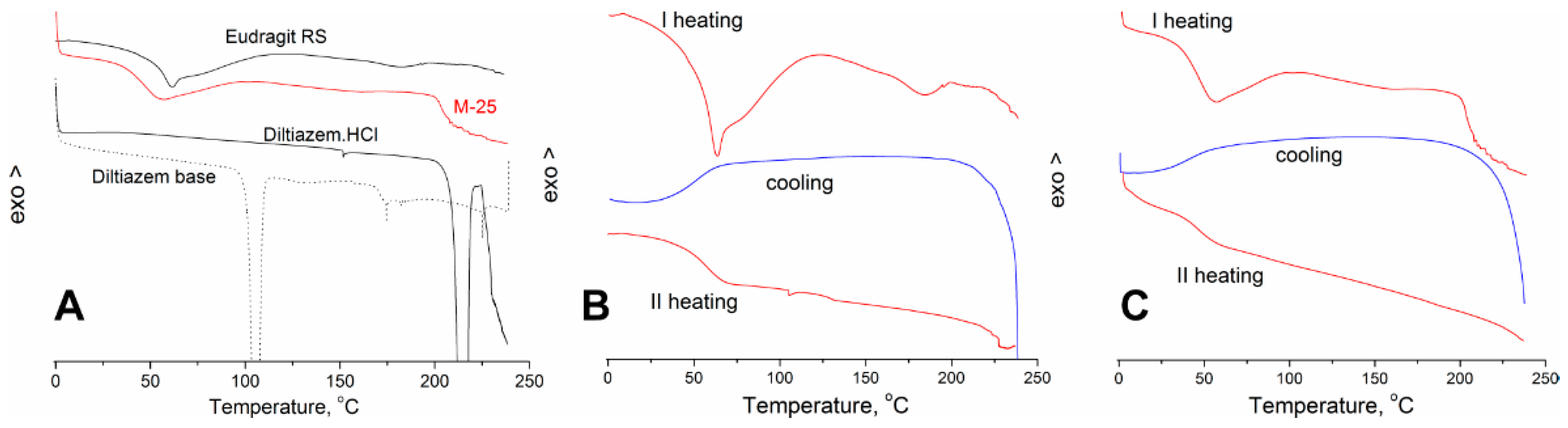
Carbonyl Group Vibrations
In the progress of our observations, we found out that the proposed ATR-FTIR method acquires satisfactory performance (with the required high analytical sensitivity and specificity) in a relatively narrow wavenumber range of about 50 cm
−1 (from 1700 to 1650 cm
−1) (
Figure 3). Similarly, in many other circumstances, it has been assumed relevant to track out the changes in the carbonyl frequency (νCO) region when carbonyl-bearing drug molecules are examined [
19,
20,
21].
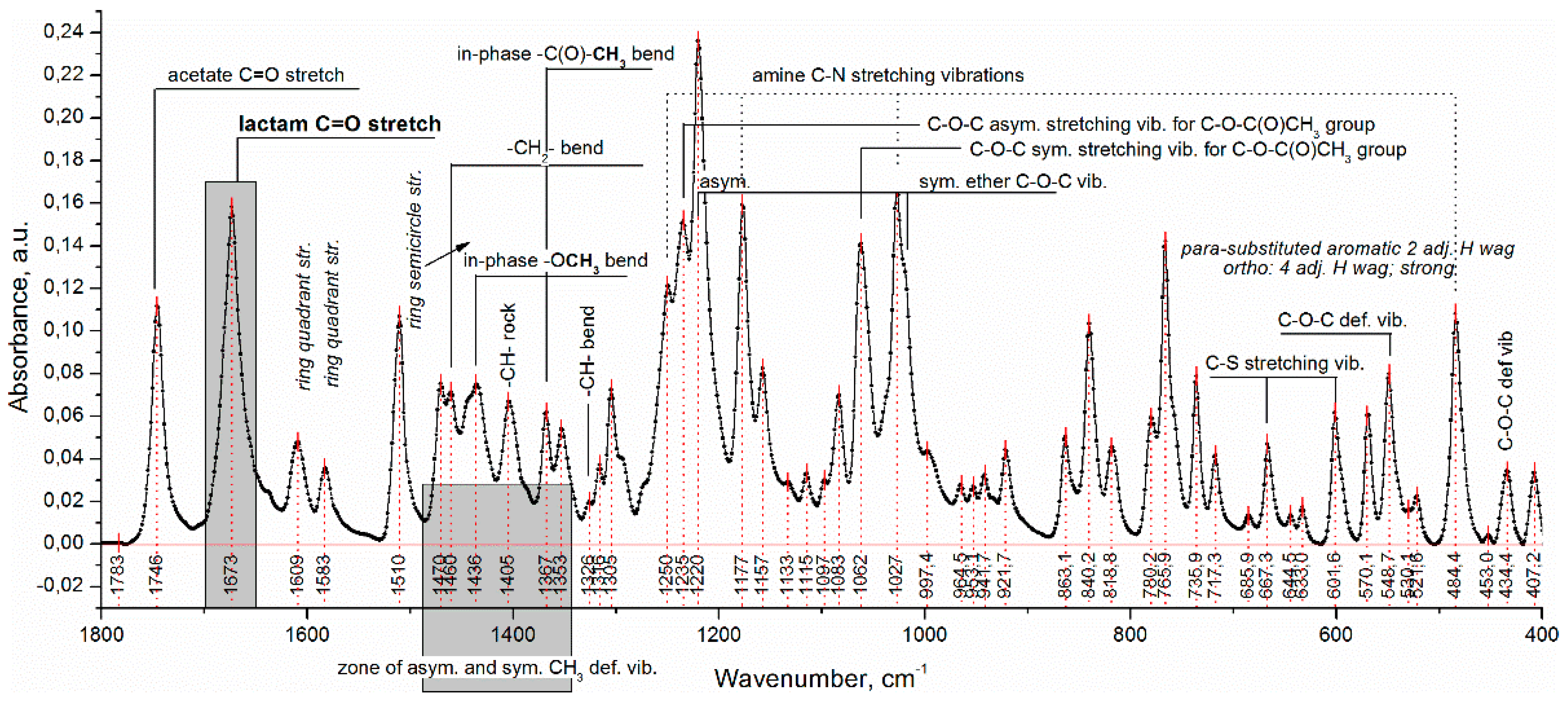
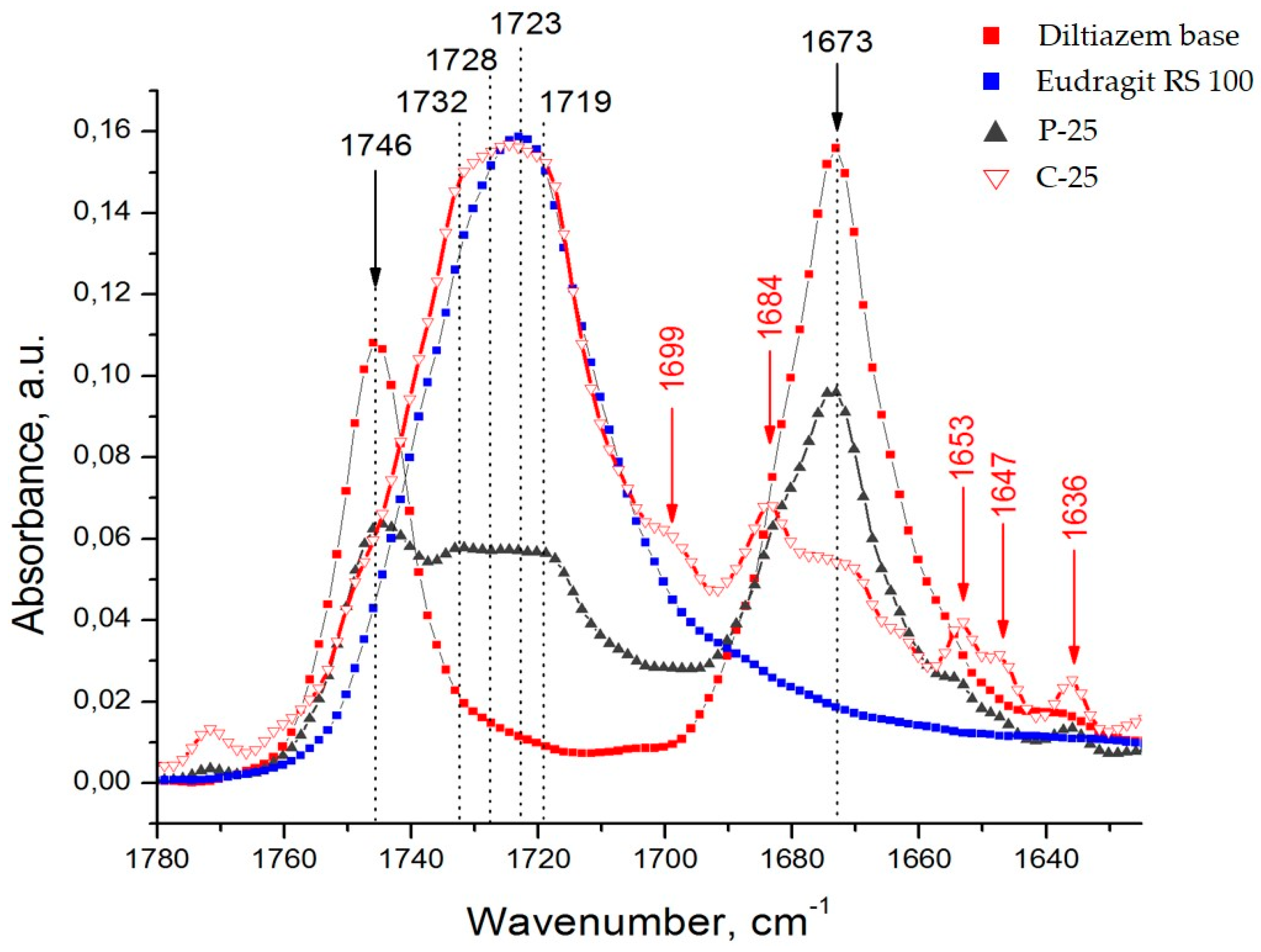
Diltiazem shows two highly characteristic, well-defined, and readily recognizable C=O stretching bands with relatively high intensity in the range of 1760–1660 cm
−1 (
Figure 2 and
Figure 3). The band observed at 1746 cm
−1 is assigned to the CO group of the acetate functional, and that positioned at lower frequencies (with a maximum at 1673 cm
–1) is assigned to the CO functional group of the seven-membered lactam ring [
22]. In the same spectral interval, however, the presence of a very intense and broad IR band (with a max. at 1723 cm
−1) in the spectrum of the polymer Eudragit RS 100 was also registered (
Figure 3). Certainly, this last-mentioned absorption band should be considered as consisting of at least three overlapping bands corresponding to the three different kinds of grafted carbonyl (ester) groups on the polymeric backbone (
Figure 1). According to the generally accepted theory of infrared spectroscopy, however, when solvent–solute mixtures of two or more constituents are examined (as in the case here studied), considerable frequency shifts of all carbonyl groups due to mutual interference effects are expected to appear. Usually, the used solvent (a liquid of low-molecular-weight organic substance) or a high-molecular organic matrix (polymer) affects both the position and the intensity of each absorption band of the examined solute (diltiazem) via the so-called solvent effect. Indeed, when the spectra of diltiazem–Eudragit RS physical mixtures (P) and solid solutions (C) were analyzed, considerable differences in the CO frequency pattern of each participant from its native spectrum were registered (
Figure 3).
Concerning the lower-frequency subzone, from 1690 to 1660 cm
−1 (
Figure 3), the appearance of a new absorption band at 1684 cm
−1 in the spectra of both types of mixtures (P and C models) was recorded. In the case of the physical mixtures, the presence of this band (normally detected as a shoulder on the high-energy tail of the primary band at 1673 cm
−1) was, as a whole, less pronounced. In the spectra of the solid solutions (C), however, this band occurs as spectrally predominant and thus indicative of the successful dispersing of drug molecules in the polymer matrix. It is reasonable to assume that the emerging new band increases its intensity as a function of the extent of mixing (dispersing) the drug molecules into the polymeric bulk, whereas the lower one, at 1673 cm
−1, accordingly decreases its integral transparency. The shift in the observed CO frequency of about 11 cm
−1 should, in principle, be attributed to the effect of strong dipole–dipole interaction between molecules of the used solvent, Eudragit RS 100, and the dispersed solute, diltiazem.
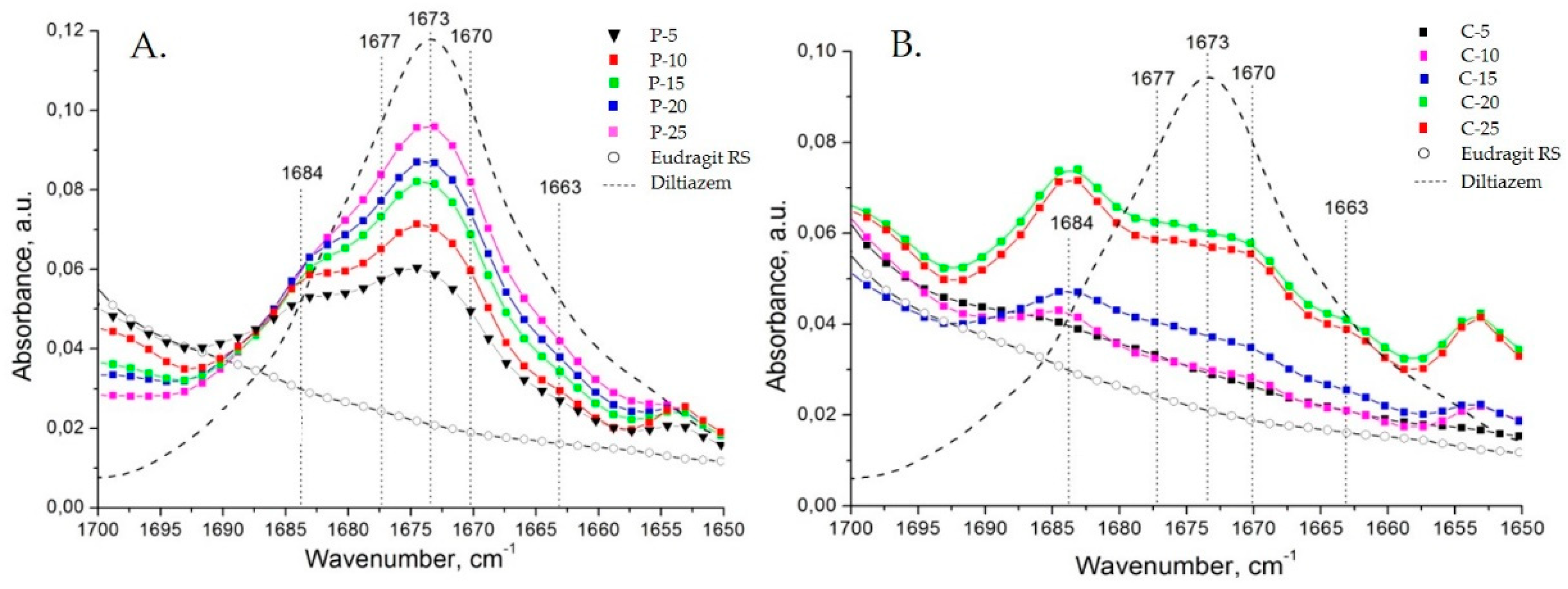
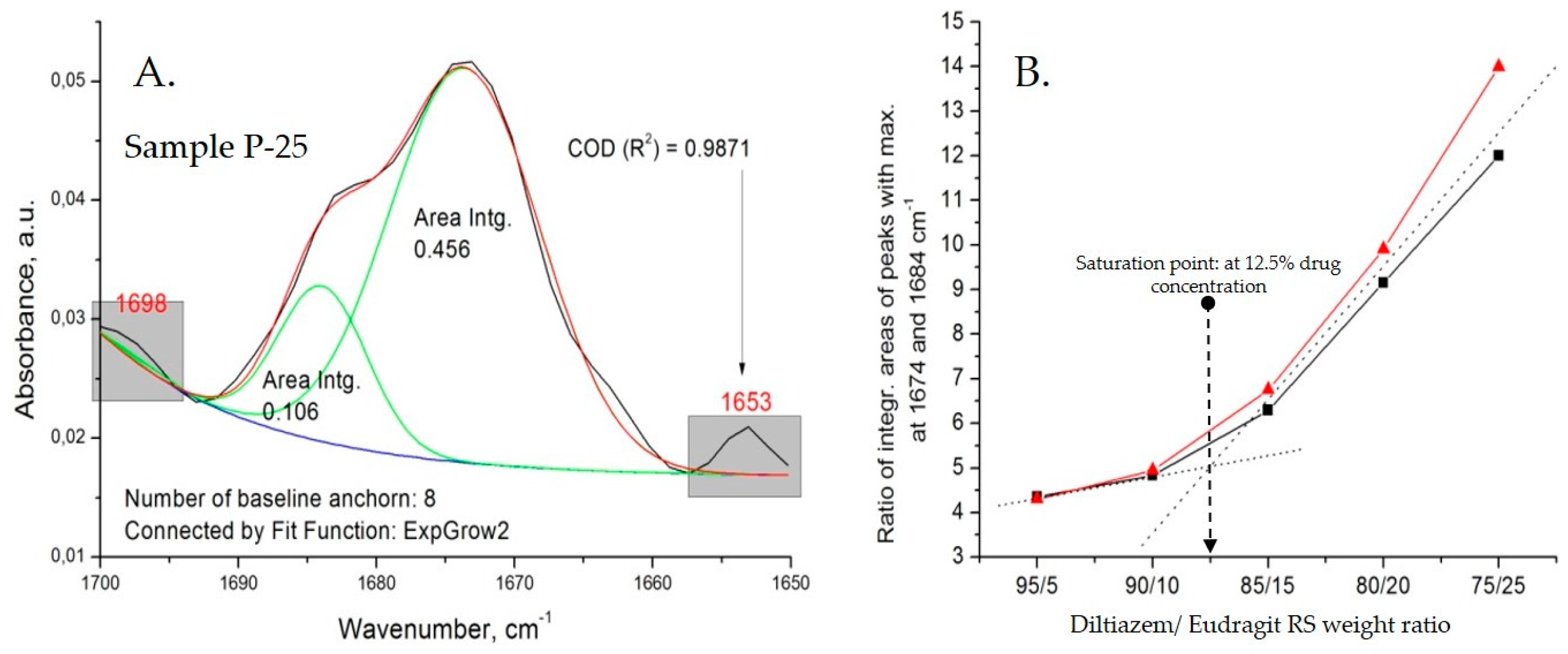
All types of diltiazem–Eudragit RS mixtures were studied at varying concentrations in order to determine the point of saturation for each of the techniques applied and follow other concentration-dependent changes within the polymer matrix (
Figure 4).
The appearance of a C=O stretching vibration band at 1684 cm
−1 (as a “shoulder” to the intrinsic 1673 cm
−1 band) in the spectra of all diltiazem–Eudragit RS 100 physical mixtures (P) should be directly ascribed to the particularities of the preparation technique. Upon mixing of the two constituents, due to significant variations in their hardness and the relatively high coefficient of applied friction, an effect of partial mutual diffusion (penetration) between both ground constituents is expected to occur at the points of contact. Thus, the “distinct” neighboring molecules diffuse, creating a heterogeneous composite mixture of microparticles consisting of a polymeric core and an outer “functionalized” shell of irregularly organized diltiazem molecules. As the drug content increases, however, the population of diltiazem molecules included in the polymeric volume reaches its limit (the point of saturation), while the molecules in excess form an intact diltiazem phase; the latter displays the characteristic for the drug IR absorption behavior (i.e., abs. maximum at 1673 cm
−1). Approaching the drug-saturation point of the polymeric surface, no further increase in the intensity of the 1684 cm
−1 band is observed, whereas the 1673 cm
−1 band continues to “grow”. A deconvolution analysis in the C=O (lactamic) stretching vibrations range (from 1700 to 1650 cm
−1), performed with the aid of specialized peak-fitting software, Origin 8.1 (
Table A1,
Table A2,
Table A3,
Table A4,
Table A5,
Table A6,
Table A7,
Table A8,
Table A9 and
Table A10,
Appendix A), allowed us to define the exact point of polymer saturation with diltiazem when the drug is introduced via mechanical grinding and mixing (P models) (
Figure 5).
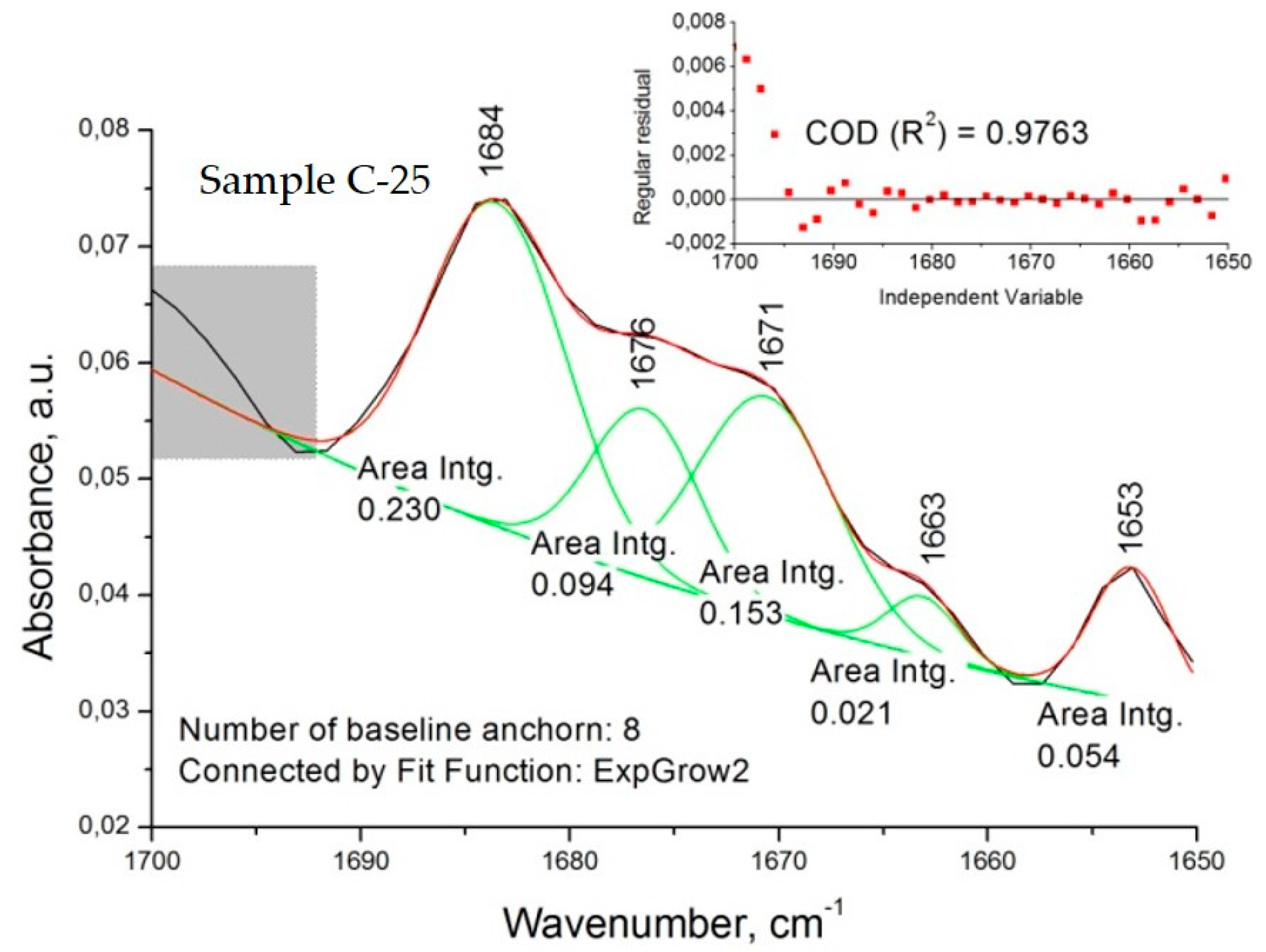
Concerning the spectra of diltiazem–Eudragit RS solid solutions (C models), significant changes in the considered wavenumber range were registered. The appearance of several new bands was observed for all concentrations examined (
Figure 4B and
Figure 6). Each of the newly appearing bands may be assigned to a complex intermolecular interaction between the two constituents, diltiazem and Eudragit RS. The good mutual solubility between them was illustrated and confirmed by the appearance of three new bands in the C=O stretching region (at 1663, 1670/1671, and 1684 cm
−1) and by the reduction in the intensity of the absorption band at 1673 cm
−1. Nonetheless, the spectral patterns of the highly dispersed mixtures (C probes) were considered too complex for a clear identification to be made. Still, it is reasonable to assume that during the process of homogeneous dispersion, the drug molecules occupy regions of the polymeric matrix with different degree of crystallinity, and to a certain extent dielectricity, which produce detectable frequency variations in νCO of the drug solute.
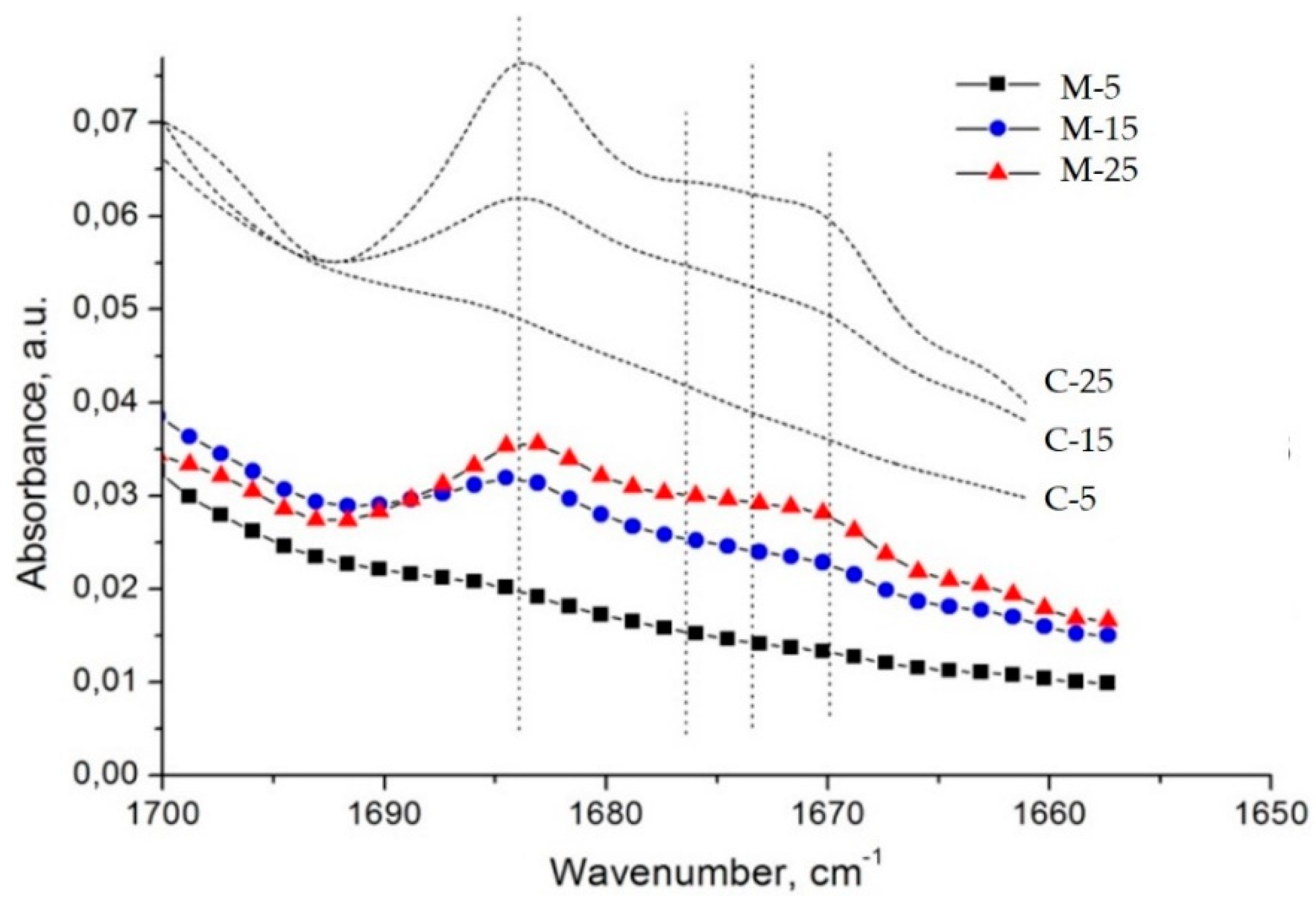
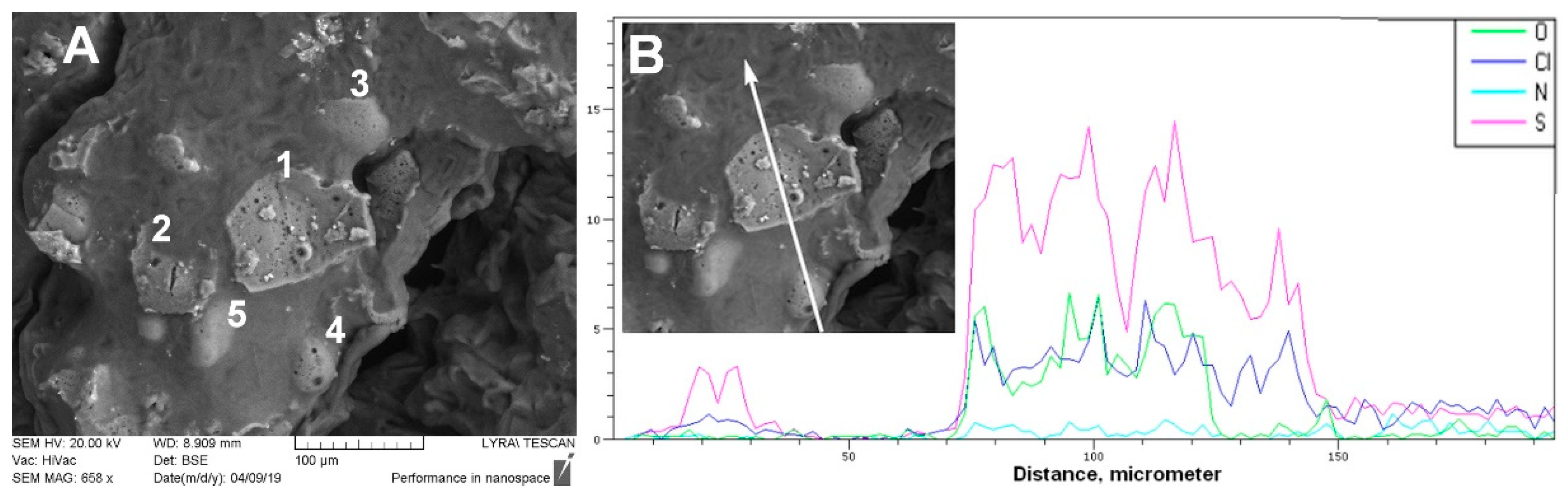
Strangely enough, the FTIR spectra of the
microsponge samples (M samples) imitate the spectral profile of the solid drug–polymer solutions (C samples) (
Figure 7). This observation does not correspond to the fact that—as shown in
Figure 7—the population of dispersed diltiazem molecules (absorbing at 1684 cm
−1) in the bulk of the two kinds of materials is different. Comparing the respective peak heights of the relevant bands in the spectrum of the M-25 sample, one could see that the proportion of occluded to easily diffusing (absorbing at 1673 cm
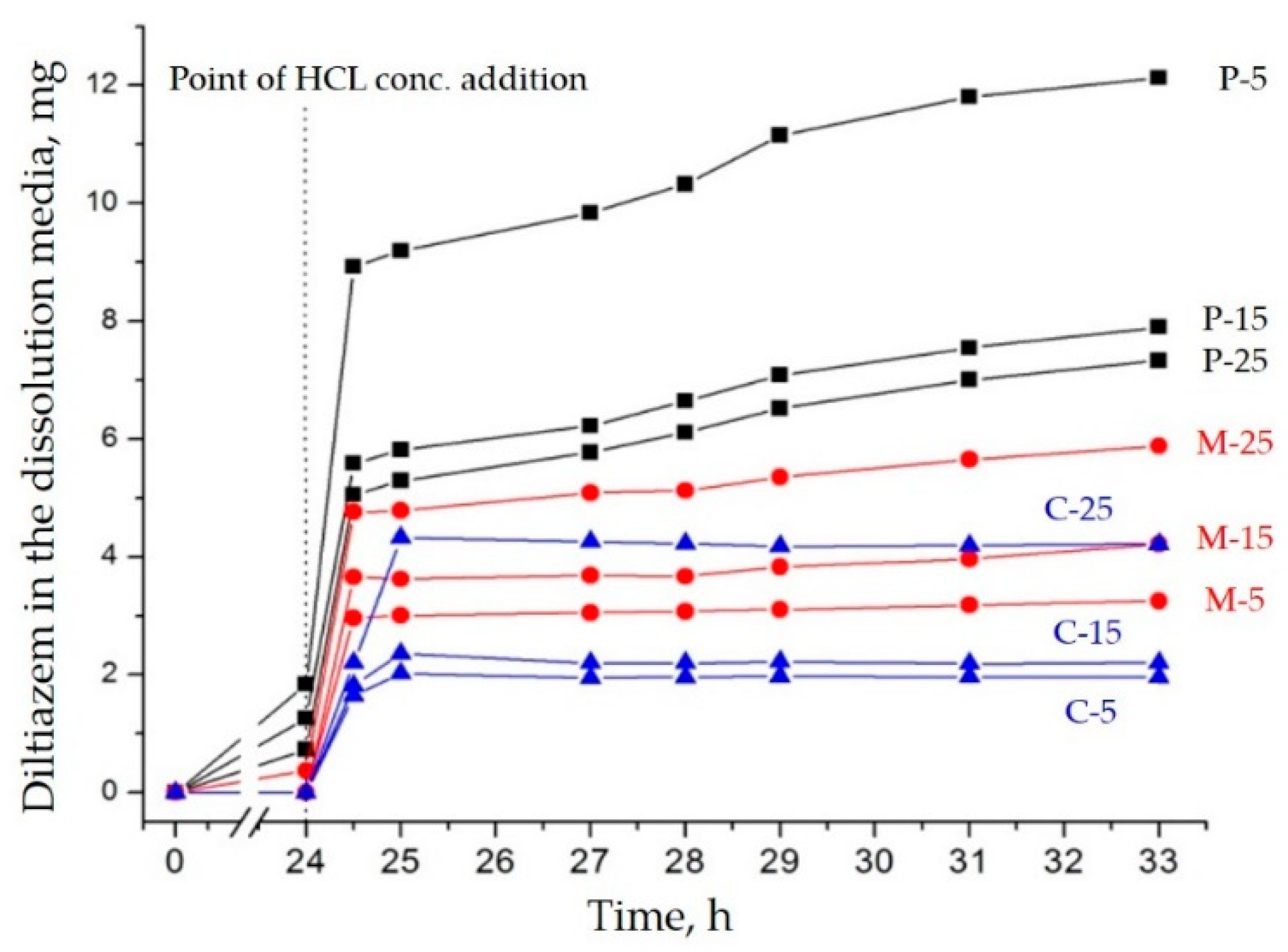
−1) diltiazem molecules appears to be significantly lower than that of its C-25 equivalent. As it will be shown below, these observations have led us to an adequate explanation of the unusual desorption behavior of the sample in question at neutral pH. However, such a result testifies for good homogeneity and amorphization of diltiazem within the
microsponge-type polymeric matrix.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















