
Self-Attractive Semiflexible Polymers under an External Force Field

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Method
3. Numerical Results
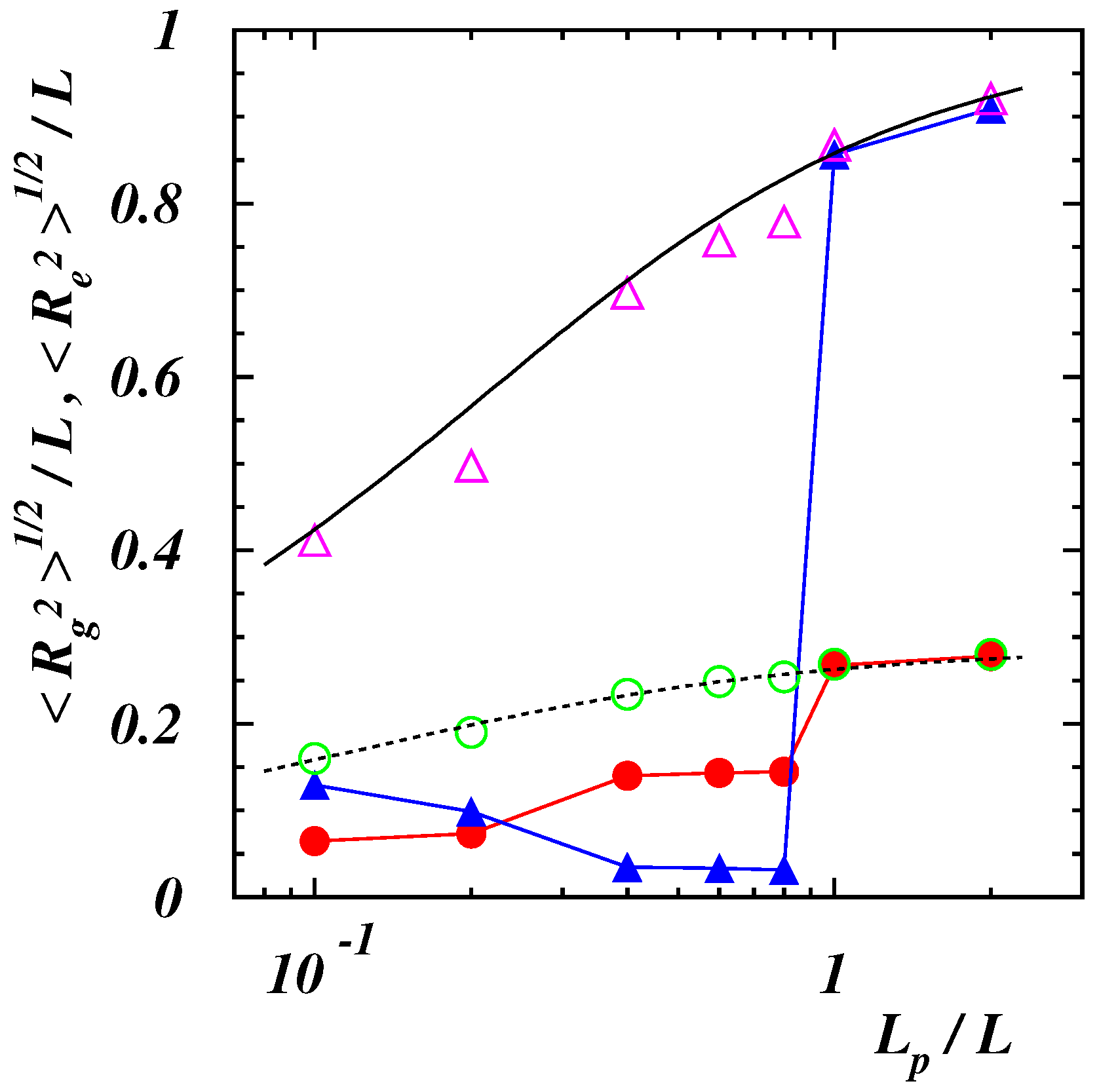
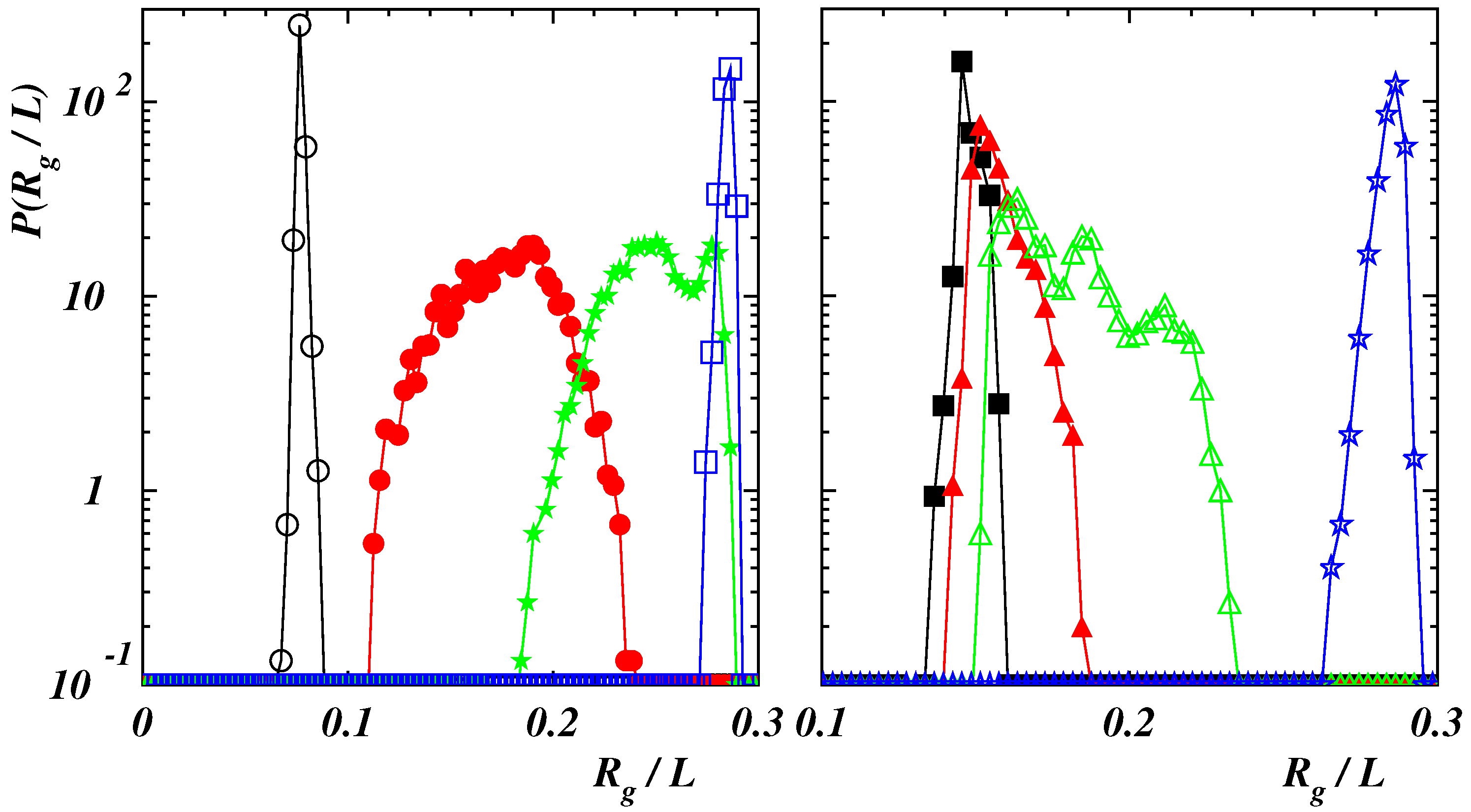
3.1. Equilibrium Polymer Conformations
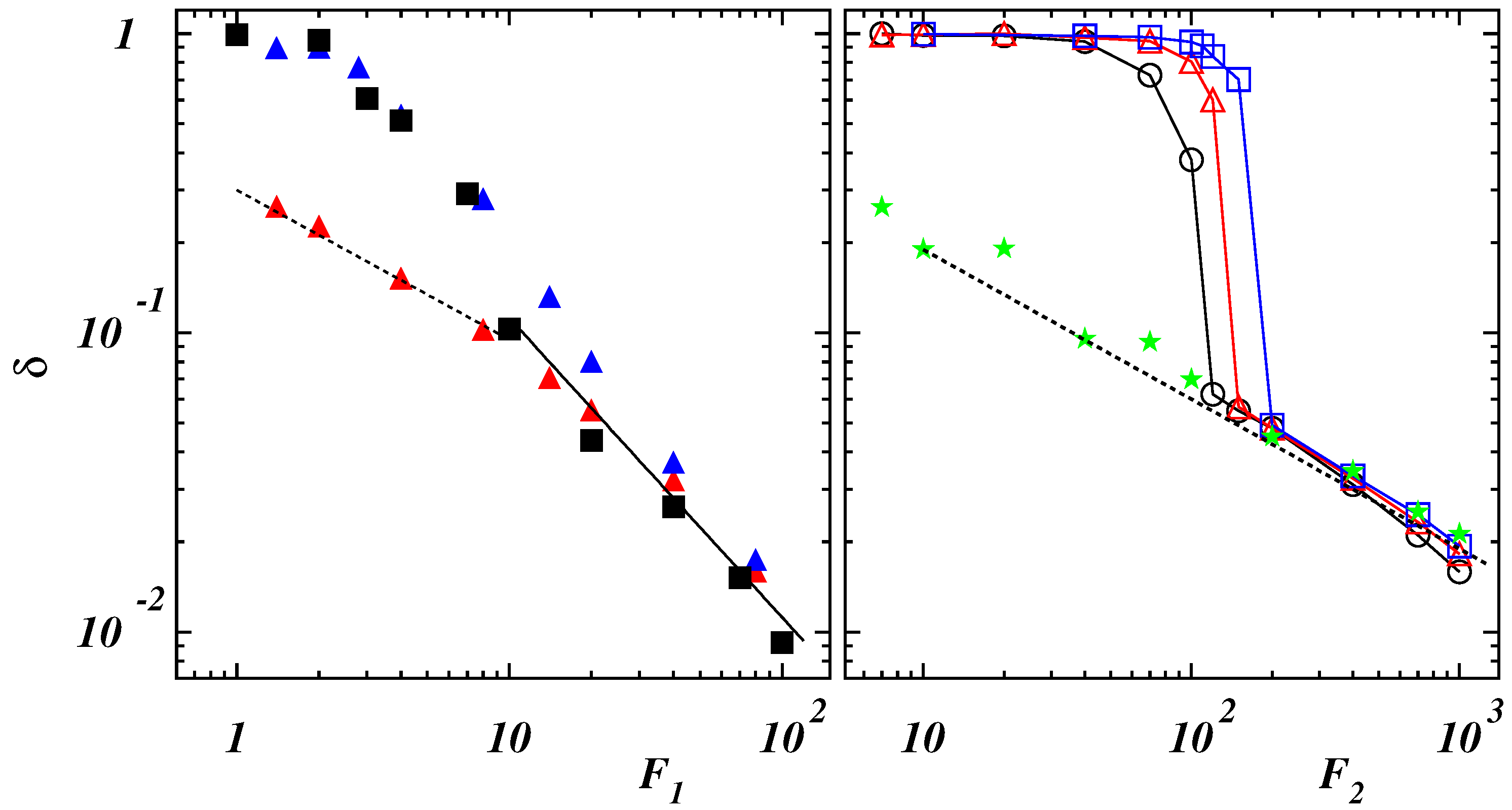
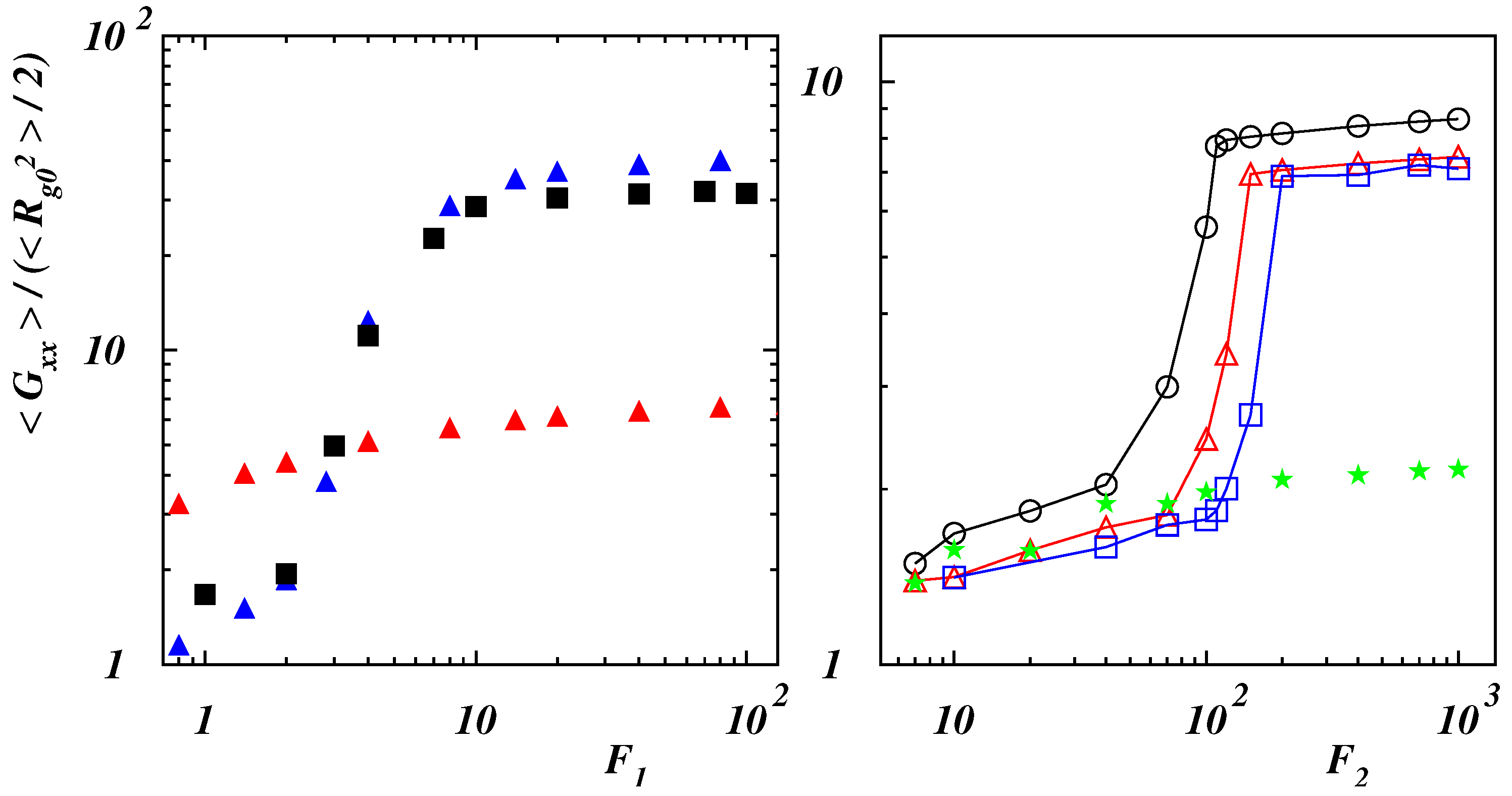
3.2. Polymer Stretching in Uniform Force Field
4. Discussion and Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaqfeh, E.S.G. The dynamics of single-molecule DNA in flow. J. Non-Newtonian Fluid Mech. 2005, 130, 1. [Google Scholar] [CrossRef]
- Perkins, T.T.; Smith, D.E.; Larson, R.G.; Chu, S. Stretching of a single tethered polymer in a uniform flow. Science 1995, 268, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, J.; Frey, E. Radial distribution function of semiflexible polymers. Phys. Rev. Lett. 1996, 77, 2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götter, R.; Kroy, K.; Frey, E.; Bärmann, M.; Sackmann, E. Dynamic Light Scattering from Semidilute Actin Solutions: A Study of Hydrodynamic Screening, Filament Bending Stiffness, and the Effect of Tropomyosin/Troponin-Binding. Macromolecules 1996, 29, 30. [Google Scholar] [CrossRef] [Green Version]
- Harnau, L.; Winkler, R.G.; Reineker, P. Dynamic Structure Factor of Semiflexible Macromolecules in Dilute Solution. J. Chem. Phys. 1996, 104, 6355. [Google Scholar] [CrossRef]
- Everaers, R.; Jülicher, F.; Ajdari, A.; Maggs, A.C. Dynamic Fluctuations of Semiflexible Filaments. Phys. Rev. Lett. 1999, 82, 3717. [Google Scholar] [CrossRef] [Green Version]
- Samuel, J.; Sinha, S. Elasticity of semiflexible polymers. Phys. Rev. E 2002, 66, 050801. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, L.; Hallatschek, O.; Frey, E.; Amblard, F. Tracer Studies on F-Actin Fluctuations. Phys. Rev. Lett. 2002, 89, 258101. [Google Scholar] [CrossRef]
- Winkler, R.G. Deformation of semiflexible chains. J. Chem. Phys. 2003, 118, 2919. [Google Scholar] [CrossRef] [Green Version]
- Petrov, E.P.; Ohrt, T.; Winkler, R.G.; Schwille, P. Diffusion and Segmental Dynamics of Double-Stranded DNA. Phys. Rev. Lett. 2006, 97, 258101. [Google Scholar] [CrossRef]
- Schroeder, C.M. Single polymer dynamics for molecular rheology. J. Rheol. 2018, 62, 371. [Google Scholar] [CrossRef] [Green Version]
- Yeou, S.; Lee, N.K. Single-Molecule Methods for Investigating the Double-Stranded DNA Bendability. Mol. Cells 2022, 45, 33. [Google Scholar] [CrossRef]
- Bird, R.B.; Curtiss, C.F.; Armstrong, R.C.; Hassager, O. Dynamics of Polymer Liquids; John Wiley & Sons: New York, NY, USA, 1987; Volume 2. [Google Scholar]
- Öttinger, H.C. Stochastic Processes in Polymeric Fluids; Springer: Berlin, Germany, 1996. [Google Scholar]
- Winkler, R.G. Semiflexible polymers in shear flow. Phys. Rev. Lett. 2006, 97, 128301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munk, T.; Hallatschek, O.; Wiggins, C.H.; Frey, E. Dynamics of semiflexible polymers in a flow field. Phys. Rev. E 2006, 74, 041911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, R.G. Conformational and rheological properties of semiflexible polymers in shear flow. J. Chem. Phys. 2010, 133, 164905. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.S.; Shaqfeh, E.S.G. Brownian dynamics simulations of single DNA molecules in shear flow. J. Rheol. 2000, 44, 713. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Larson, R.G. Modelling hydrodynamic interaction in Brownian dynamics: Simulation of extensional and shear flows of dilute solutions of high molecular weight polystyrene. J. Rheol. 2004, 48, 995. [Google Scholar] [CrossRef]
- Liu, S.; Ashok, B.; Muthukumar, M. Brownian dynamics simulations of bead-rod-chain in simple shear flow and elongational flow. Polymer 2004, 45, 1383. [Google Scholar] [CrossRef]
- Sendner, C.; Netz, R.R. Shear-induced repulsion of a semiflexible polymer from a wall. EPL 2008, 81, 54006. [Google Scholar] [CrossRef]
- He, G.L.; Messina, R.; Löwen, H.; Kiriy, A.; Bocharova, V.; Stamm, M. Shear-induced stretching of adsorbed polymer chains. Soft Matter 2009, 5, 3014. [Google Scholar] [CrossRef]
- Zhang, Y.; Donev, A.; Weisgraber, T.; Alder, B.J.; Graham, M.G.; de Pablo, J.J. Tethered DNA dynamics in shear flow. J. Chem. Phys. 2009, 130, 234902. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.S.; Obermayer, B.; Frey, E. Dynamics of a semiflexible polymer or polymer ring in shear flow. Phys. Rev. E 2014, 89, 022606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoubashman, A.; Howard, M.P. Equilibrium Dynamics and Shear Rheology of Semiflexible Polymers in Solution. Macromolecules 2017, 50, 8279. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G. Tethered semiflexible polymer under large amplitude oscillatory shear. Polymers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shee, A.; Gupta, N.; Chaudhuri, A.; Chaudhuri, D. A semiflexible polymer in a gliding assay: Reentrant transition, role of turnover and activity. Soft Matter 2021, 17, 2120. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G.; Gompper, G. Wall-anchored semiflexible polymer under large amplitude oscillatory shear flow. J. Chem. Phys. 2021, 154, 224901. [Google Scholar] [CrossRef]
- Andersen, N.T.; Teng, Y.; Chen, J.Z.Y. Stretching a Semiflexible Polymer of Finite Length. Macromolecules 2022, 55, 210. [Google Scholar] [CrossRef]
- Kratky, O.; Porod, G. Roentgenuntersuchung geloester Fadenmolekuele. Recl. Trav. Chim. Pays-Bas 1949, 68, 1106. [Google Scholar] [CrossRef]
- Wittmer, J.P.; Meyer, H.; Johner, A.; Kreer, T.; Baschnagel, J. Algebraic displacement correlation in two-dimensional polymer melts. Phys. Rev. Lett. 2010, 105, 037802. [Google Scholar] [CrossRef]
- Baumann, C.G.; Bloomfield, V.A.; Smith, S.B.; Bustamante, C.; Wang, M.D.; Block, S.M. Stretching of Single Collapsed DNA Molecules. Biophys. J. 2000, 78, 1965. [Google Scholar] [CrossRef]
- Gerland, U.; Bundschuh, R.; Hwa, T. Mechanically Probing the Folding Pathway of Single RNA Molecules. Biophys. J. 2003, 84, 2831. [Google Scholar] [CrossRef] [Green Version]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Austin, K.S.; Zierenberg, J.; Janke, W. Interplay of Adsorption and Semiflexibility: Structural Behavior of Grafted Polymers under Poor Solvent Conditions. Macromolecules 2017, 50, 4054. [Google Scholar] [CrossRef]
- Haupt, B.J.; Senden, T.J.; Sevick, E.M. AFM Evidence of Rayleigh Instability in Single Polymer Chains. Langmuir 2002, 18, 2174. [Google Scholar] [CrossRef]
- Gunari, N.; Balazs, A.C.; Walker, G.C. Force-Induced Globule-Coil Transition in Single Polystyrene Chains in Water. J. Am. Chem. Soc. 2007, 129, 10046. [Google Scholar] [CrossRef]
- Rosa, A.; Hoang, T.X.; Marenduzzo, D.; Maritan, A. Elasticity of Semiflexible Polymers with and without Self-Interactions. Macromolecules 2003, 36, 10095. [Google Scholar] [CrossRef] [Green Version]
- Tkachenko, A.V. Unfolding and unzipping of single-stranded DNA by stretching. Phys. Rev. E 2004, 70, 051901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Giri, D. Force-Induced Conformational Transition in a System of Interacting Stiff Polymers: Application to Unfolding. Phys. Rev. E 2005, 72, 052901. [Google Scholar] [CrossRef] [Green Version]
- Kneller, J.M.; Elvingson, C.; Arteca, G.A. Shape Transition Induced by Mechanical External Stretching of Grafted Self-Attractive Wormlike Chains. Chem. Phys. Lett. 2005, 407, 384. [Google Scholar] [CrossRef]
- Rosa, A.; Marenduzzo, D.; Kumar, S. Stretching a Self-Interacting Semiflexible Polymer. Europhys. Lett. 2006, 75, 818. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Jensen, I.; Jacobsen, J.L.; Guttmann, A.J. Role of Conformational Entropy in Force-Induced Biopolymer Unfolding. Phys. Rev. Lett. 2007, 98, 128101. [Google Scholar] [CrossRef]
- Cifra, P.; Bleha, T. Stretching of Self-Interacting Wormlike Macromolecules. Polymer 2007, 48, 2444. [Google Scholar] [CrossRef]
- Kapri, R. Can a double stranded DNA be unzipped by pulling a single strand?: Phases of adsorbed DNA. J. Chem. Phys. 2009, 130, 145105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttmann, A.J.; Jacobsen, J.L.; Jensen, I.; Kumar, S. Modeling Force-Induced Bio-polymer Unfolding. J. Math. Chem. 2009, 45, 223. [Google Scholar] [CrossRef] [Green Version]
- Maier, B.; Seifert, U.; Rädler, J.O. Elastic response of DNA to external electric fields in two dimensions. Europhys. Lett. 2002, 60, 622. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.S.; Kounovsky, K.L.; Chang, R.; Jung, G.Y.; DePablo, J.J.; Kyubong, J.; Schwartz, D.C. Nanochannel confinement: DNA Stretch Approaching Full Contour Length. Lab Chip 2011, 11, 1721. [Google Scholar] [CrossRef]
- Reisner, W.; Pedersen, J.N.; Austin, R.H. DNA Confinement in Nanochannels: Physics and Biological Applications. Rep. Prog. Phys. 2012, 75, 106601. [Google Scholar] [CrossRef]
- Lamura, A.; Burkhardt, T.W.; Gompper, G. Semiflexible polymer in a uniform force field in two dimensions. Phys. Rev. E 2001, 64, 061801. [Google Scholar] [CrossRef] [Green Version]
- Lamura, A.; Winkler, R.G. Semiflexible polymers under external fields confined to two dimensions. J. Chem. Phys. 2012, 137, 244909. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, M.; Winkler, R.G.; Gompper, G. Hydrodynamic screening of star polymers in shear flow. Eur. Phys. J. E 2007, 23, 349. [Google Scholar] [CrossRef]
- Kapral, R. Multiparticle Collision Dynamics: Simulations of complex systems on mesoscale. Adv. Chem. Phys. 2008, 140, 89. [Google Scholar]
- Gompper, G.; Ihle, T.; Kroll, D.M.; Winkler, R.G. Multi-Particle Collision Dynamics: A particle-based mesoscale simulation approach to the hydrodynamics of complex Fluids. Adv. Polym. Sci. 2009, 221, 1. [Google Scholar]
- Winkler, R.G.; Reineker, P.; Harnau, L. Models and equilibrium properties of stiff molecular chains. J. Chem. Phys. 1994, 101, 8119. [Google Scholar] [CrossRef]
- Hsu, H.P.; Paul, W.; Binder, K. Standard Definitions of Persistence Length Do Not Describe the Local “Intrinsic” Stiffness of Real Polymer Chains. Macromolecules 2010, 43, 3094. [Google Scholar] [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Kikuchi, N.; Pooley, C.M.; Ryder, J.F.; Yeomans, J.M. Transport coefficients of a mesoscopic fluid dynamics model. J. Chem. Phys. 2003, 119, 6388–6395. [Google Scholar] [CrossRef] [Green Version]
- Ihle, T.; Kroll, D.M. Stochastic rotation dynamics: A Galilean-invariant mesoscopic model for fluid flow. Phys. Rev. E 2001, 63, 020201. [Google Scholar] [CrossRef]
- Lamura, A.; Gompper, G.; Ihle, T.; Kroll, D.M. Multiparticle collision dynamics: Flow around a circular and a square cylinder. Europhys. Lett. 2001, 56, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Marko, J.F.; Siggia, E.D. Stretching DNA. Macromolecules 1995, 28, 8759. [Google Scholar] [CrossRef]
- Livadaru, L.; Netz, R.R.; Kreuzer, H.J. Stretching response of discrete semiflexible polymers. Macromolecules 2003, 36, 3732. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.C. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Dünweg, B. Polymer Solutions. In Handbook of Materials Modelling; Andreoni, W., Yip, S., Eds.; Springer: Berlin, Germany, 2018; pp. 1–19. [Google Scholar]
- Marenduzzo, D.; Maritan, A.; Rosa, A.; Seno, F. Stepwise Unfolding of Collapsed Polymers. Eur. Phys. J. E 2004, 15, 83. [Google Scholar] [CrossRef]
- Cieplak, M.; Hoang, T.X.; Robbins, M.O. Stretching of homopolymers and contact order. Phys. Rev. E 2004, 70, 011917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemak, A.S.; Lepock, J.R.; Chen, J.Z.Y. Molecular Dynamics Simulations of a Protein Model in Uniform and Elongational Flows. Proteins 2003, 51, 224. [Google Scholar] [CrossRef] [PubMed]
- Lubensky, D.K.; Nelson, D.R. Single molecule statistics and the polynucleotide unzipping transition. Phys. Rev. E 2002, 65, 031917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaton, D.T.; Schnabel, S.; Landau, D.P.; Bachmann, M. From flexible to stiff: Systematic analysis of structural phases for single semiflexible polymers. Phys. Rev. Lett. 2013, 110, 028103. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamura, A. Self-Attractive Semiflexible Polymers under an External Force Field. Polymers 2022, 14, 4762. https://doi.org/10.3390/polym14214762
Lamura A. Self-Attractive Semiflexible Polymers under an External Force Field. Polymers. 2022; 14(21):4762. https://doi.org/10.3390/polym14214762
Chicago/Turabian StyleLamura, Antonio. 2022. "Self-Attractive Semiflexible Polymers under an External Force Field" Polymers 14, no. 21: 4762. https://doi.org/10.3390/polym14214762
APA StyleLamura, A. (2022). Self-Attractive Semiflexible Polymers under an External Force Field. Polymers, 14(21), 4762. https://doi.org/10.3390/polym14214762