1. Introduction
Iron is considered to be one of the essential trace elements necessary for all living organisms and it is involved in many indispensable functions in many life forms, such as electron transfer, oxygen transport, DNA synthesis, and detoxication reaction [
1]. Although it is necessary and involved in these functions, iron is potentially toxic in the body when present in excessive amounts owing to its low solubility in the stable oxidation state (i.e., Fe
3+) and its tendency to potentiate the production of high levels of oxidative stress [
2,
3,
4]. Accordingly, it is crucially important to maintain a dynamic balance between the benefits and toxic effects of iron by tightly regulating iron homeostasis, which is mostly accomplished by iron-binding proteins such as ferritin and transferrin [
3,
5].
Ferritins are well known as an ancient superfamily of globular proteins that are associated with the storing and detoxification of iron. In addition, they exhibit a capability to oxidize Fe
2+ to generate Fe
3+; and ferritins facilitate iron nucleation as their mineral core for storage inside the protein cavity, allowing the accommodation of up to 4500 iron atoms [
5]. In general, ferritin’s high capacity for iron storage is mainly due to antioxidant protection, which is achieved by preventing Fenton reactions and thus stopping the formation of the radical element hydroxyl (OH⸱), and in the reversible storage of iron [
3,
4]. Notably, the superfamily of these proteins is usually classified into three different types, i.e., classical ferritins, heme-binding bacterioferritins (Bfr), and DNA-binding proteins from starved cells (Dps) [
6]. The typical form of most common ferritins presents as a spherical protein composed of 24 identical or similar subunits with a molecular weight (MW) of ~450−500 kDa [
7]. In eukaryotes, ferritin is generally formed by self-assembly with two or three types of highly homologous subunits, namely heavy (H, 21 kDa), middle (M, 20 kDa), and light (L, 19 kDa) chains, in which the M subunit contains both the ferroxidase center and the ferrihydrite nucleation center, in stark contrast to the H and L subunits [
4,
6]. Specifically, M-ferritins are found in some lower vertebrate (e.g., amphibians and fish) and invertebrate (e.g., shellfish) species and closely resemble vertebrate H-ferritins (sequence identity of ~85%) [
6,
8]. Previous research has indicated that the marine invertebrate
Apostichopus japonicus ferritin (AjFER) shows very high sequence similarity with other invertebrates and comprises known conserved functional domains of both the di-iron ferroxidase sites of the H subunit and the iron nucleation site of the L subunit [
8,
9,
10], suggesting that AjFER is functionally similar to M-ferritin. However, knowledge regarding the iron uptake and oxidation mechanism of M-ferritins from marine invertebrates remains limited.
Structurally, each subunit of almost all ferritins is composed of a characteristic anti-parallel
α-helix bundle (helices A–D) and a fifth short E-helix (with the exception of Dps proteins) pointing inside the protein cavity [
11]. These subunits assemble into a spherical structure with octahedral (4-3-2) symmetry, resulting in 12 two-fold, 8 three-fold, and 6 four-fold channels. Specifically, the hydrophobic three-fold channel connecting the inner cavity to the outside in ferritins is considered a potential gateway for the entry and exit of iron ions and other small molecules [
5,
11,
12]. Numerous studies have demonstrated that the carboxylate side chains of Asp and Glu residues along the three-fold pores are essential for iron uptake, and these residues are involved in the translocation of Fe
2+ ions toward the ferroxidase center for rapid catalytic activity in human H-ferritin (HuHF) and frog M-ferritin [
6,
12]. Moreover, a previous X-ray crystal structure also indicated that the Asp129 and Glu132 residues of the three-fold channel in AjFER, corresponding to that of highly conserved residues in other marine invertebrates [
13,
14], indeed play a key role in iron uptake [
9]. Of course, after uptake, the Fe
2+ ions are transported by a series of metal-binding residues toward a specific catalytic site, which is known as the di-iron ferroxidase center and is situated in the subunit four-helix bundle [
5,
9,
13,
14,
15]. However, although the amino acid residues located at the ferroxidase center seem to be highly conserved in most eukaryotic ferritins [
7,
9], the ferroxidase reaction rate may vary. For instance, De Meulenaere et al. found that the ferroxidase velocity of the marine invertebrate
Chaetopterus ferritin (ChF) was up to eight times faster than that of HuHF, which is known as a conventionally accepted representative ferritin [
15]. Furthermore, recent studies have suggested that the Glu168 residue at the four-fold channel can act as a potential binding site of metal ions in some marine invertebrate ferritins [
9,
14,
15], but there is still a lack of sufficient evidence to determine whether this metal-binding site is related to the catalytic function of ferritin. Accordingly, some unique functions of marine invertebrate ferritins have not been observed.
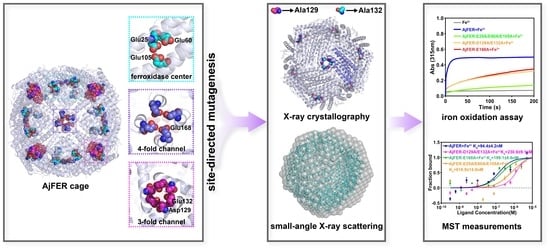
In this study, on the basis of the previously identified potential metal-binding sites of AjFER, we created the AjFER-E25A/E60A/E105A mutant with three amino acid substitutions at the ferroxidase center, the AjFER-D129A/E132A mutant with two amino acid substitutions at the three-fold channel, and the AjFER-E168A mutant with a single amino acid substitution at the four-fold channel using site-directed mutagenesis. We then determined the crystal structure of the AjFER-D129A/E132A mutant using X-ray crystallography. We further employed small-angle X-ray scattering (SAXS), which is one of the most powerful techniques, to obtain the structures of these proteins in solution. In addition, we performed biochemical characterizations related to the Fe2+ oxidation kinetics and metal ion-binding thermodynamics in comparison with AjFER. The present investigation aimed to provide new insight into the roles of potential metal-binding sites in AjFER.
2. Materials and Methods
2.1. Protein Expression and Purification
The expression construct for AjFER (pET-28a-AjFER) was produced in a previous work [
9]. Based on the crystal structure of AjFER, the pET-28a plasmid carrying the AjFER gene was used to introduce the AjFER-E25A/E60A/E105A mutant (MF), AjFER-D129A/E132A mutant (M3), and AjFER-E168A mutant (M4) using gene synthesis. All constructs were verified by gene sequencing. For overproduction of the AjFER variants, the constructs were transformed into
Escherichia coli BL21(DE3) cells (Thermo Fisher Scientific, Waltham, MA, USA).
The cells were grown at 37 °C in Luria–Bertani medium supplemented with 30 μg/mL kanamycin and 34 μg/mL chloromycetin until the OD600 reached 0.6–0.8. Protein expression was initiated with the addition of isopropyl-β-D-thiogalactopyranoside (IPTG, 0.5 mM final concentration) and the cells were then incubated at 18 °C for 20 h. The cells were harvested by centrifugation at 6500× g for 20 min at 4 °C and resuspended in 25 mM Tris, pH 8.0, containing 150 mM NaCl and 0.5% (v/v) Triton X-100. The cells were disrupted by sonication, and debris was removed by centrifugation at 12,000× g for 20 min at 4 °C. Contaminating proteins were removed using a Ni–NTA affinity column (GE Healthcare, Fairfield, CT, USA), and the His-SUMO tag was cleaved by incubation overnight at 4 °C with SUMO protease. The sample purity and oligomerization state were assessed using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The purified proteins were then concentrated using a 30 kDa molecular weight cutoff Amicon Ultra filter unit (Merck Millipore, Billerica, MA, USA). The protein concentrations were determined using a BCA assay kit (Beyotime, Shanghai, China) with bovine serum albumin as the standard.
2.2. Circular Dichroism (CD) Spectroscopy
The protein concentrations were adjusted to 0.1 mg/mL with binding buffer (25 mM Tris–HCl, pH 8.0, containing 150 mM NaCl). The CD spectra of AjFER and its variants were examined using a Jasco J-1500 CD spectrometer (Jasco, Tokyo, Japan) with a 0.1 cm path length at room temperature. The far-UV region was scanned from 190 to 260 nm in triplicate with a bandwidth of 1 nm. The CD spectral data are presented as the molar ellipticity (deg∙cm2∙dmol−1) versus wavelength [θ]. The content of secondary structure elements, including α-helixes, β-sheets, β-turns, and random coils, was analyzed using Yang’s method by Jasco-Corp.
2.3. Dynamic Light Scattering (DLS) Experiments
The DLS data were collected on a Zetasizer Nano Zs instrument (Malvern Instruments Ltd., Malvern, UK) using disposable polystyrene microcuvettes (VWR). The protein samples (1 mg/mL) in 25 mM Tris–HCl, pH 8.0, containing 150 mM NaCl were centrifuged at 12,000× g for 10 min at room temperature and then loaded into a quartz cuvette prior to measurement. The hydrodynamic diameter (DH) distribution of the prepared samples was calculated using Omni SIZE 2.0 software. Three measurements were performed with the instrument optimizing the number of runs for each measurement.
2.4. Transmission Electron Microscopy (TEM)
The purified recombinant AjFER and its variants were placed on carbon-coated copper grids. After removing the excess solution by blotting with filter paper, the samples were negatively stained with 1% uranyl acetate for 1 min. The TEM images were captured at 80 kV using a Model H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
2.5. Crystallization, Data Collection and Structure Determination
The preliminary screening for the crystallization conditions was optimized using Crystal Screen I and II (Hampton Research, Riverside, CA, USA) with the sitting-drop vapor-diffusion method. A volume of 1 μL protein solution (approx. 15 mg/mL in 25 mM Tris-HCl, pH 8.0, containing 150 mM NaCl) was mixed with 1 μL reservoir solution in the well of a 48-well plate and incubated at 18 °C. The M3 protein crystals were obtained in a solution containing 0.05 M cadmium sulfate hydrate, 0.1 M HEPES, pH 7.5, and 1.0 M sodium acetate trihydrate. These crystals were harvested using a CryoLoop (Hampton Research). After soaking in a cryoprotection solution supplemented with 20% (
v/v) glycerol, the crystals were immediately flash-cooled and stored in liquid nitrogen. X-ray data were collected on a BL19U1 beamline with a Pilatus3 6M detector at SSRF [
16]. The X-ray wavelength was set to 0.97892 Å. The diffracting data were indexed, integrated, and scaled using the HKL-3000 program suite [
17]. A model of the structure was automatically generated by molecular replacement using the BABLES server [
18]. The manual model building was performed using COOT software [
19], and the automated model building and refinement were conducted using the REFMAC5 program in the CCP4 suite [
20]. The superimposed structure of the ferritins was determined using the superpose online server (
http://superpose.wishartlab.com/, accessed on 27 June 2022) [
21]. All crystallographic figures were generated using the PyMOL molecular graphics system [
22]. The collected data and final refinement statistics are summarized in
Table 1.
2.6. Small-Angle X-ray Scattering (SAXS) Analysis
To study the conformation of ferritins in solution, SAXS experiments were performed at the BL19U2 beamline of the National Center for Protein Science Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility (SSRF). The SAXS data were collected as 20 × 1 s exposures, and the scattering profiles for the 20 passes were compared at 10 °C using 60 μL sample in 25 mM Tris-HCl, pH 8.0, containing 150 mM NaCl. A total of 1500 successive frames were recorded using a Pilatus 1 M detector (Dectris Ltd., Baden, Switzerland) with an exposure time of 1.0 s for each frame. The wavelength of the incoming monochromatic X-ray radiation was 1.24 Å with a
q-range from 0.0084 to 0.4764 Å
−1 (
q = 4π⸳sinθ/λ, where 2θ is the scattering angle). All measurements were performed at 293 K. Protein samples with different concentrations (1, 2, 2.5, and 4 mg/mL) in buffer (25 mM Tris–HCl, pH 8.0, containing 150 mM NaCl) were prepared for the X-ray scattering analysis. SAXS data of the protein samples were collected between each buffer. The 2D scattering images were converted to 1D SAXS curves using the BioXTAS RAW software package [
23]. The scattering data were processed for background subtraction, concentration scaling, and curve merging using the PRIMUS program in the ATSAS software package [
24]. The scattering intensity extrapolated to zero angle
I(0), pair distance distribution function
p(r), radius of gyration
Rg, and maximum dimension
Dmax were determined by Guinier analysis and primus distance distribution analysis using GNOM in ATSAS [
25,
26]. The data were converted into Kratky plots [
I⁎
q2 vs.
q] to evaluate the shape and fold of the proteins, providing information regarding the oligomeric state of the biomolecule [
27]. The molecular weights were calculated using the SAXSMow2 program [
28]. Ten low-resolution ab initio shape models were calculated using the DAMMIF program [
29]. The bead models were generated using the DAMMIN program [
30] and the MultiFoXS server [
27]. The docking of the sample crystal structures into the SAXS envelopes was superimposed using the SUPCOMB program and visualized using PyMOL [
22]. The structural parameters and processed SAXS data are summarized in
Table S3.
2.7. Iron Oxidation Assay
The iron oxidation assay for AjFER and its variants was conducted by measuring an increase in ultraviolet (UV) absorbance at 315 nm with a UV–visible (UV–vis) spectrophotometer (Shanghai Mapada Instruments Co., Ltd., Shanghai, China) [
31]. Oxygen-free aliquots of Fe
2+ ion (7.2 mM) samples were prepared by dissolving FeSO
4·7H
2O in 0.1% (
v/v) HCl under anaerobic conditions. The protein samples were diluted to a final concentration of 0.5 μM in 20 mM HEPES, pH 7.0, containing 150 mM NaCl. The ferritins and Fe
2+ ions were added to a 1 mL quartz cuvette (Hellma) at final concentrations of 0.357 and 205.7 μM, respectively. The Fe/protein molar ratio was 576. All experiments were repeated in triplicate.
2.8. Inductively Coupled Plasma–Mass Spectrometry (ICP–MS) and CD Analyses
The metal ion content of the protein samples was then quantified with three replicates using a Thermo X Series II ICP–MS instrument (Thermo Fisher Scientific Inc., MA, USA). The standard curves for the Fe atoms were obtained using multielement standard solutions. The atoms per ferritin cage were calculated as previously described [
32]. Then, the samples were subjected to CD spectroscopy for secondary structure analysis, as described above.
2.9. Microscale Thermophoresis (MST) Measurements
MST, a powerful technique for characterizing biomolecular interactions, depends on the thermophoresis principle of detecting optical fluorescence properties to quantify the binding affinity of different molecules [
33]. To determine the binding affinity of Fe
2+ ions toward AjFER and its variants, MST experiments were performed using a Monolith NT.115 (NanoTemper Technologies, Munich, Germany) and standard capillaries. Each protein sample was labeled using a Protein Labeling Kit RED-NHS 2nd Generation (NanoTemper Technologies, Munich, Germany; catalog no. MO-L011) according to the manufacturer’s instructions. Briefly, 90 µL of protein solution (10 µM) in 20 mM HEPES, pH 7.0, was mixed with 10 µL of 300 µM RED-NHS fluorophore in labeling buffer and incubated for 30 min in the dark at room temperature. Then, the labeled protein was centrifuged at 12,000×
g at 4 °C for 10 min to remove the protein aggregates. Five microliters of the labeled protein (a concentration of approx. 200 nM) and increasing concentrations of non-labeled ferrous solution (from 3.05 nM to 10 µM) were loaded into NT.115 standard treated capillaries (NanoTemper Technologies, Munich, Germany; catalog no. MO-K022). Subsequently, the capillaries were inserted into the chip tray of the MST instrument, followed by thermophoresis analysis and the appraisal of the binding affinity (K
d) values. The MST measurements were performed in triplicate at 25 °C, with medium MST power and the red channel using 20% excitation power. Normalization of the fluorescence signal and curve fitting were carried out using MO Affinity Analysis v2.3 software (NanoTemper Technologies, Munich, Germany).
2.10. Thermal Stability Analysis
The thermostability of the AjFER, MF, M3, and M4 proteins was determined using a label-free thermal shift assay with the Tycho NT.6 (NanoTemper Technologies, Munich, Germany) via the intrinsic tryptophan and tyrosine fluorescence. The 10 µL solutions of protein samples (approx. 5 mg/mL) were prepared and loaded into Tycho NT.6 capillaries (NanoTemper Technologies, Munich, Germany; catalog no. TYC001). The thermal profiles of the proteins were recorded during a quick thermal ramp from 35 to 95 °C, with a heating rate of 30 °C/min. The inflection unfolding temperatures (Ti) were assessed based on the changes in the 350/330 nm fluorescence emission ratio values. Thereafter, AjFER and its variants were heated at 90 °C for 10 min, and their structural integrity was analyzed using TEM and DLS methods, as described above.
2.11. Statistical Analysis
All data were plotted using GraphPad 8.3 (San Diego, CA, USA) and OriginPro 9.0 (Northampton, MA, USA) software. Significant differences were determined by one-way ANOVA. All data were collected in triplicate and are presented as the mean ± standard deviation (SD). A p-value of less than 0.05, 0.01, and 0.001 was considered statistically significant.
4. Discussion
Since its discovery, ferritin has been observed to be directly related to the transport and storage of iron in a nontoxic and biologically available form, and it maintains redox balance during iron metabolism for most species of both prokaryotes and eukaryotes [
5]. Ferritin has been widely reported in vertebrates and gradually studied in invertebrates. The 3D structures of ferritins for most species exhibit many similarities; however, in contrast to mammalian ferritins, there are some exceptions for ferritins in invertebrate species [
7]. For instance, although both Fer147 and PeFer stemmed from the marine invertebrate
Phascolosoma esculenta, the interiors of their three- and four-fold channels were significantly distinct in terms of amino acid composition and electrostatic potential distribution [
13]. In turn, the biochemical characteristics of these channels directly affect their transport properties and determine the preferred transfer pathways of ferrous ions from the exterior of the cage to the ferroxidase center [
39]. In previous research, we established a method to express and purify AjFER and solved its crystal structure at a resolution of 2.75 Å [
9]. Several critical metal-binding sites were found in AjFER, including six highly conserved residues (Glu25, Tyr32, Glu60, His63, Glu105, and Gln139) at the ferroxidase center, two residues (Asp129 and Glu132) at the three-fold channel, and a Glu168 residue at the four-fold channel. Nonetheless, whether AjFER has a different transport property or iron oxidation function from known ferritins remains to be determined. In this study, we prepared a triple AjFER-E25A/E60A/E105A mutant (MF), a double AjFER-D129A/E132A mutant (M3), and a single AjFER-E168A mutant (M4) by site-directed mutagenesis; heterologously expressed the three mutants; and further performed detailed structural and biochemical characterizations using a multitechnical approach.
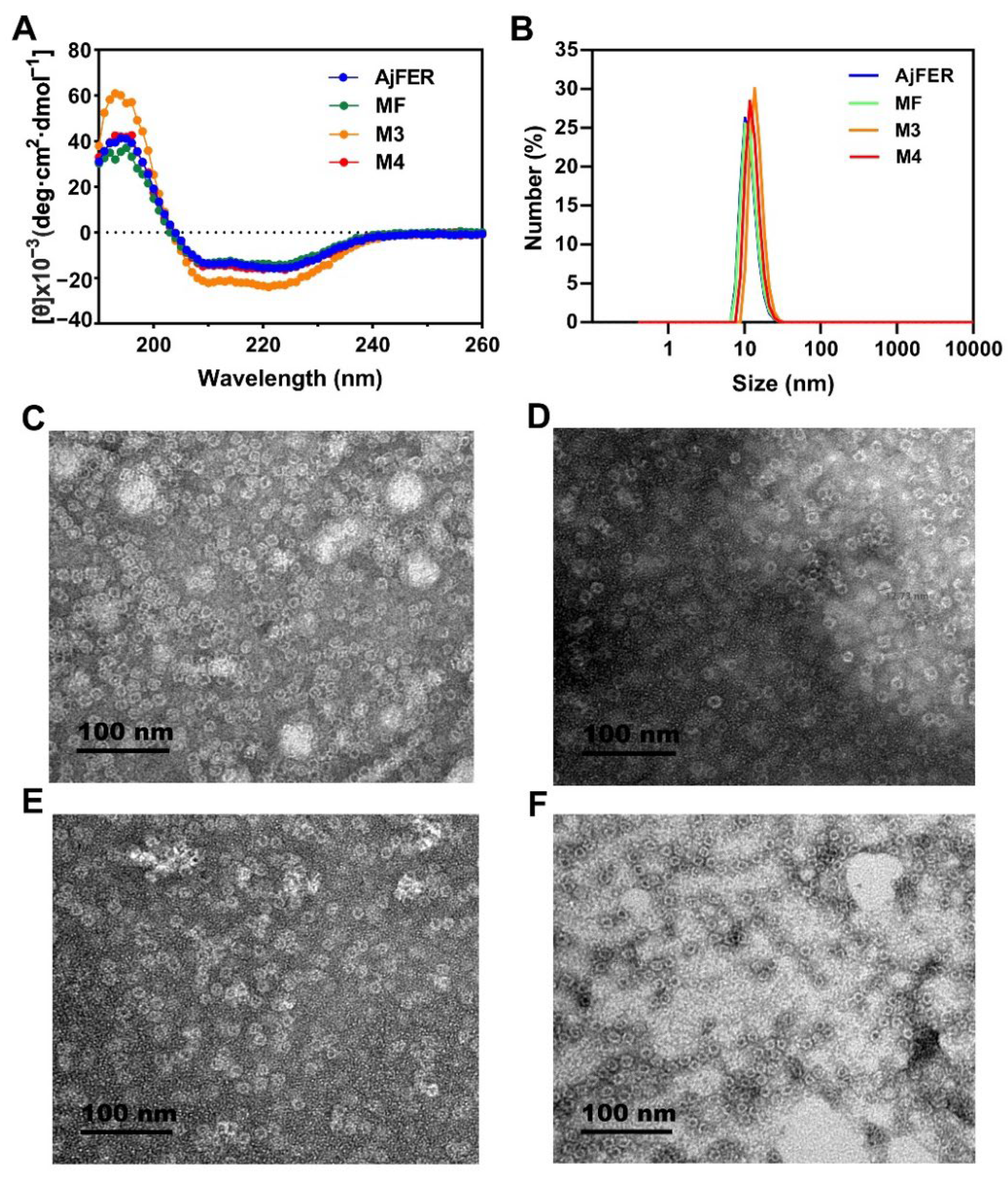
To identify the structure of AjFER before and after mutation, several supporting experimental methods were performed, including CD spectrum, DLS, and TEM analyses. The CD spectra revealed that the AjFER, MF, M3, and M4 proteins all had two very similar negative absorption peaks in the far-UV spectrum (
Figure 1A), which was in good agreement with the previously reported secondary structures of plant, animal, and bacterial ferritins [
13,
40,
41]. The CD results suggested that such mutations hardly affected the stability and integrity of AjFER, which was fully consistent with the results of the DLS and TEM analyses (
Figure 1B–F). To obtain more insight into the structural information, two powerful X-ray techniques (macromolecular X-ray crystallography and SAXS) were used to investigate the 3D structures of the mutant ferritins in crystal and solution states, respectively. Generally, numerous experiments have implied that eight hydrophilic three-fold channels in most ferritins are considered potential gates for the entry of metal ions into the cage with the assistance of highly conserved residues, such as Glu and Asp [
12,
42]. As previously described [
9], the negatively charged residues Asp129 and Glu132 are located at the three-fold pores of AjFER and play a pivotal role in Fe
2+ translocation to the internal cavity of the protein, at which Fe
2+ ions reach the ferroxidase center. This is similar to the reported crystal structure of bullfrog M-ferritin (BfMF) [
43]. Accordingly, we prepared the M3 protein by site-directed mutagenesis and solved the crystallographic structure of the mutant at a resolution of 1.98 Å (
Figure 2). It is well accepted that the internal entrance to the three-fold channel is generally surrounded by regions of the very lowest negative potential at any location in most ferritins, allowing for the attraction and insertion of many divalent ions, such as Fe, Mg, and Mn [
12,
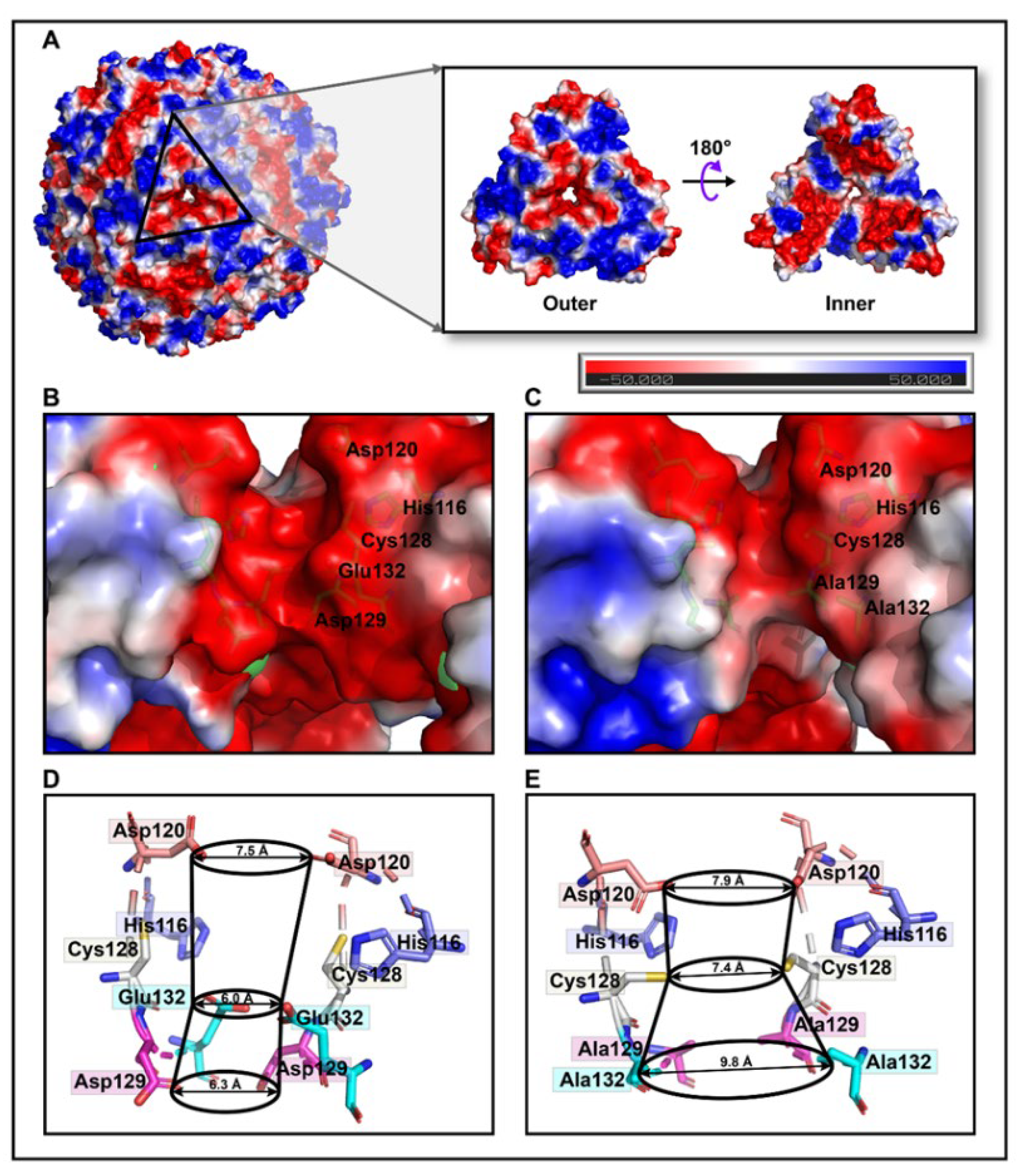
35]. However, due to replacement with relatively weaker charged alanine residues in the M3 protein, a small group of negative patches located inside the hourglass channel are replaced by neutral electrostatic surface potentials compared to that of AjFER (
Figure 3A–C), which most likely affected the entry of metal ions into the cage [
39].
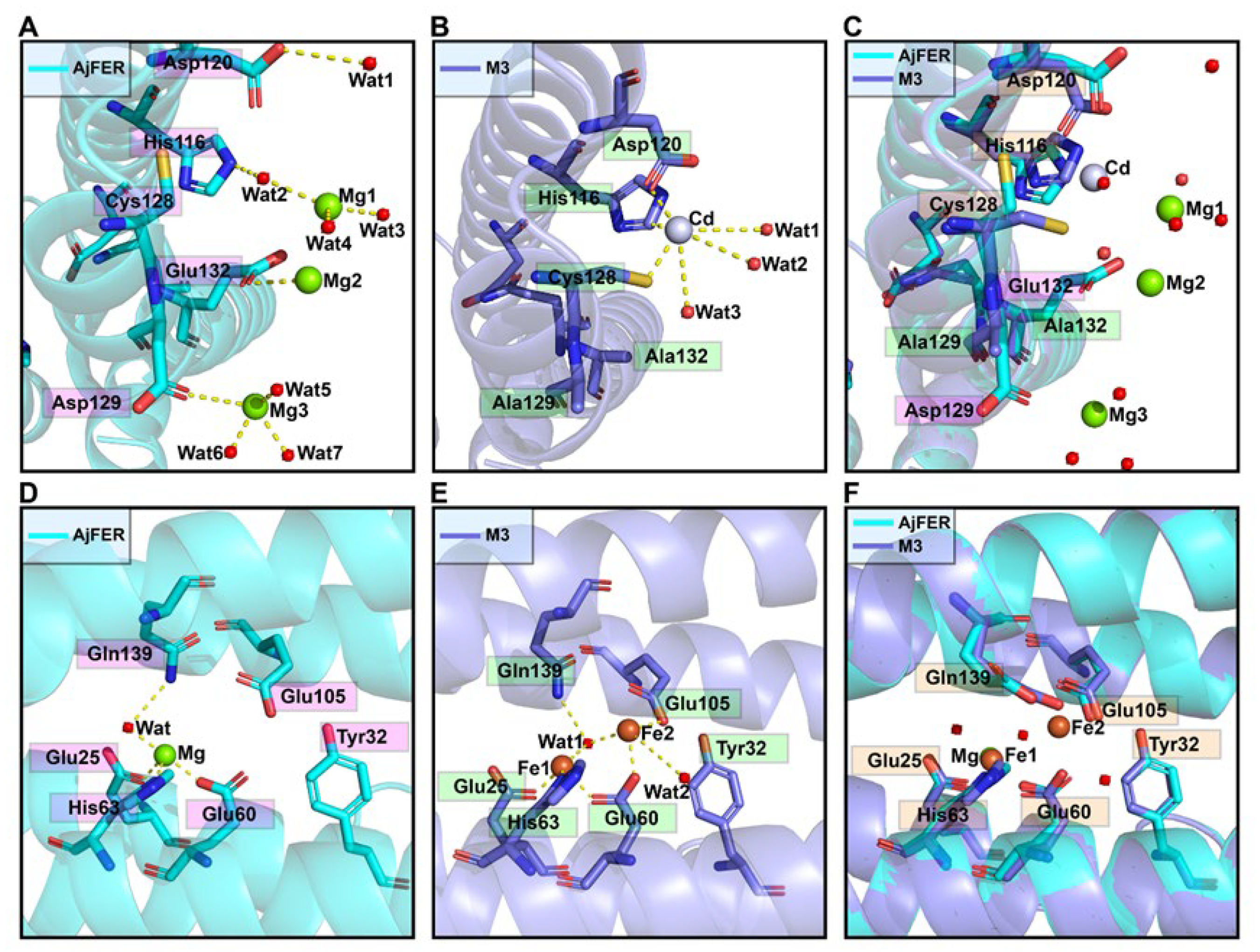
Being mostly consistent with the structural features of the D127A/E130A/S131A ferritin variant in BfMF [
39], the inherent hour-glass shape of the three-fold channel in the M3 protein was even wider than that in the previous holo-AjFER protein because of substitutions with relatively smaller alanine residues (
Figure 3D,E). The diameters of the three three-fold amino acid rings within the three-fold channel of the AjFER and M3 proteins varied significantly, with the inner ring of the D129A residue ranging from ~6.3 to ~9.8 Å, the middle ring of the E132A residue ranging from ~6.0 to ~7.4 Å, and the outer ring of the His116 and Cys128 residues ranging from ~7.5 to ~7.9 Å. Notably, the conformation of the Cys128 residue from the three-fold channel in the M3 protein, corresponding to the Cys126 residue in BfMF [
43], was profoundly affected by a single-point mutation (
Figure 4A–C). In addition, based on very strong positive electron densities presented in averaged
Fo–
Fc maps and the coordination environment of cadmium, three Cd
2+ ions were observed in the three-fold channel of the M3 protein, and each Cd
2+ ion was coordinated with the residues His116, Asp120, and Cys128, as well as three water molecules. A metal ion binding to the Cys128 residue (forming the Cd–S–Cys charge transfer complex) at the three-fold channel of ferritin revealed that metal ion coordination to sulfur can complement ionic binding during Fe
2+ transit through ion channels [
43,
44]. Additionally, consistent with most ferritins from marine invertebrates [
7,
9,
14,
15,
45], two metal ion-binding sites (i.e., sites A and B) in the ferroxidase center were observed in the middle of four
α-helical bundles per subunit in the M3 protein (
Figure 4E). On the basis of the positive electron density map and the coordination environment of iron around the ferroxidase site, two iron ions were assigned in sites A and B of the ferroxidase center of the M3 protein. Taking into account the highly conversed residues at the ferroxidase center [
9], it can be assumed that both the AjFER and M3 proteins may have some oxidative functions that are similar to those of other marine invertebrates.
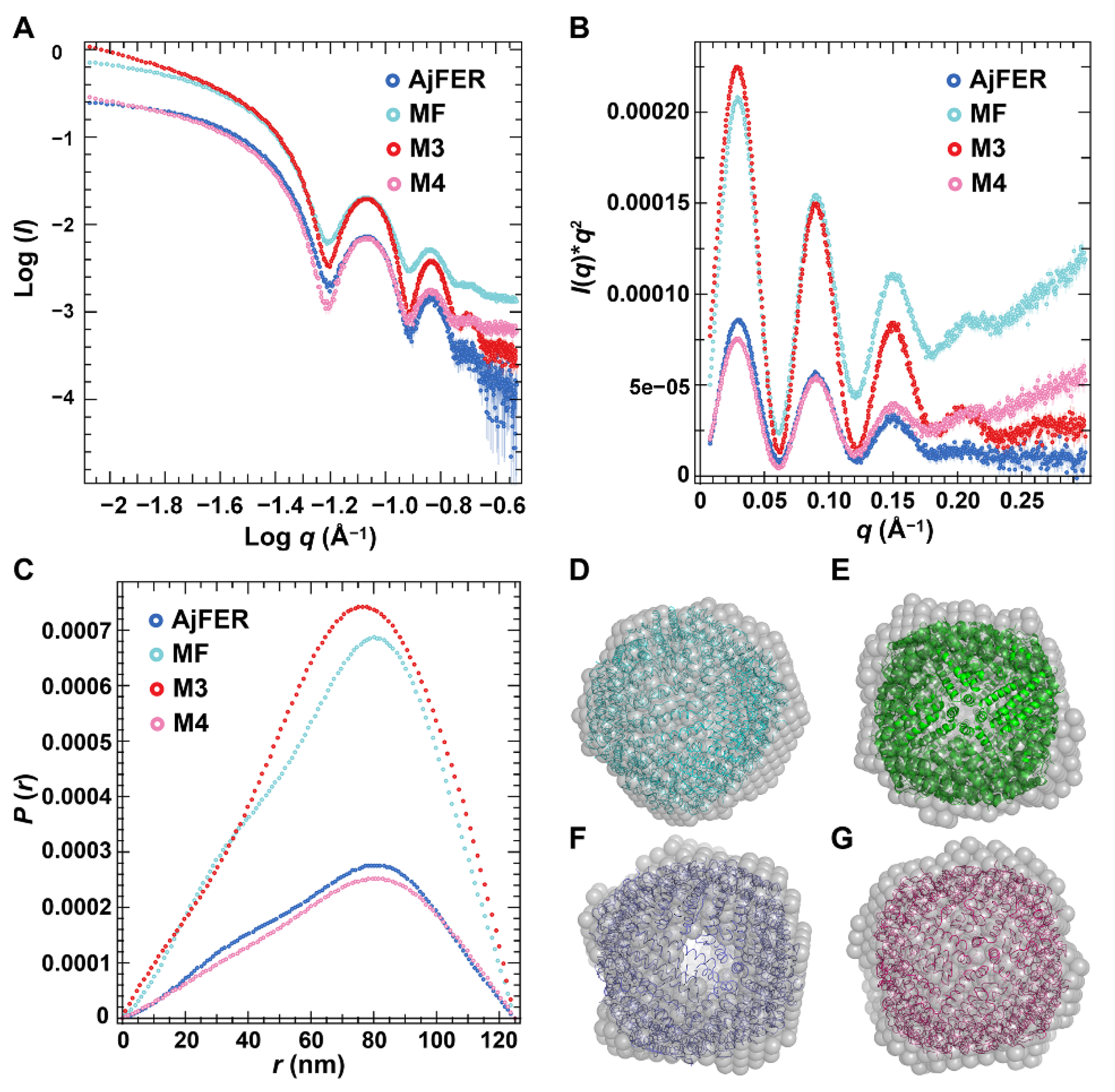
Since crystallographic methods may preferentially utilize the most stable conformation of the protein macromolecules and overlook some function-related differences in solution, the structures of AjFER and its variants were investigated using SAXS. The SAXS data revealed that the overall shape and size of the AjFER, MF, M3, and M4 proteins in solution were consistent with the crystal structures of the AjFER [
9] and M3 proteins (
Figure 2 and
Figure 5). Based on calculations using the SAXS data, some structural parameters in solution (e.g.,
Dmax,
Rg, and MWs) were similar and consistent with those from the crystal structures (
Table S3). In each case, the
Rg values, estimated from the Guinier plot and
p(r) distribution by GNOM, were in agreement with the Guinier analysis, varying between 52.61 and 53.81 Å (
Figure 5A–C). The
Dmax values for AjFER and its variants were distributed from 120 to 126 Å, which was very close to the diameter of the spherically symmetric 24-mer cage with a hollow core [
9]. Additionally, the shape of the
p(r) distribution curves and the Kratky plots indicated that all of the proteins were well folded and had a globular shape (
Figure 5A–C). Furthermore, the theoretical scattering curve generated from the AjFER model fit well to the experimental SAXS data (χ
2 of 52.7 using FoXS) (
Figure S3A). However, the results also showed that there were certain differences between the crystal and solution structures of the MF, M3, and M4 proteins (
Figure S3B–D). The results implied that the site-directed mutations of the key metal-binding sites had certain effects on the structure of AjFER in solution.
It is well documented that in addition to the iron storage function of AjFER from the sea cucumber (
S. monotuberculatus), AjFER also plays a dominant role in iron detoxification and the immune response [
8]. These unique characteristics of ferritin can not only maintain the cellular and organismic iron homeostasis process but also help to avoid the toxicity of ferrous iron. Similar to most ferritins from other marine invertebrates, the metal-binding sites in AjFER are commonly composed of a ferroxidase center, hydrophilic three-fold channel, and putative four-fold channel [
9]. Therefore, we further investigated the catalytic activity of the AjFER, MF, M3, and M4 proteins in converting Fe
2+ into Fe
3+ via UV absorption at 315 nm, aiming to elucidate the relationship between iron oxidation activity and the protein–metal binding sites (
Figure 6A). It is apparent that the D129A/E132A or E168A substitutions on each subunit of AjFER caused a significant decrease in the rate of Fe
2+ oxidation by oxygen, in contrast with AjFER. Notably, the amino acid substitutions E25A/E60A/E105A had already seriously affected the catalytic activity for Fe
2+ ions, possibly resulting in relatively low rates of iron oxidation, which was in agreement with the above results related to the iron contents (
Figure 6B). Such results indicated that the substitution of alanine residues for carboxylate ligands of carboxylate residues Asp and Glu most likely influenced the altered ability of the variants to capture and bind metal ions [
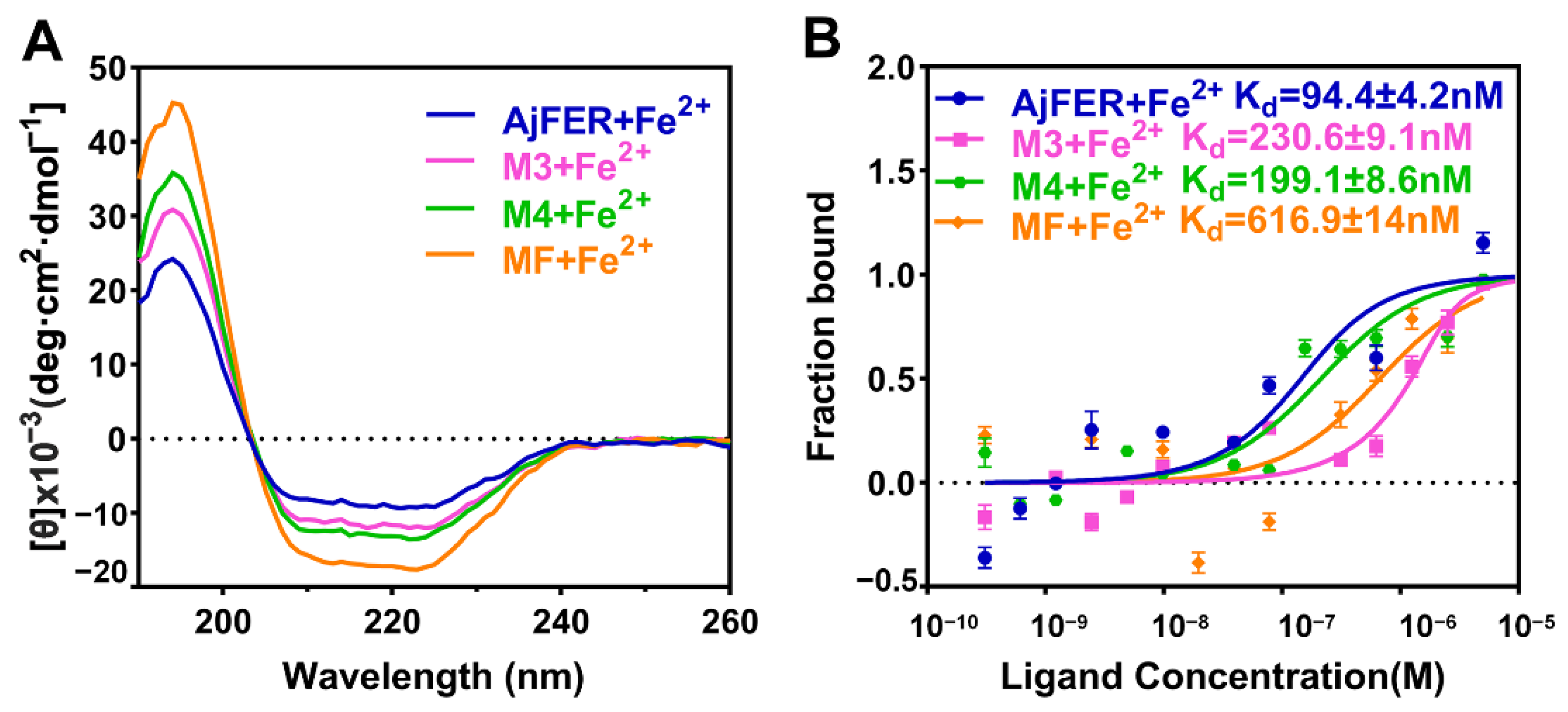
46]. Further, we determined the secondary structures and binding affinities after interaction between the AjFER, MF, M3, and M4 proteins with Fe
2+ ions by CD and MST analyses, respectively (
Figure 7A,B). The uptake of Fe
2+ ions within ferritins did not significantly affect the overall structure of these proteins, as revealed by the CD spectra (
Figure 7A). The MST results revealed significantly differential binding between AjFER and its variants to Fe
2+ ions, in which Fe
2+ had a higher affinity for AjFER (K
d value of 94.4 ± 4.2 nM) than for the other variants, and the order of the binding affinity was AjFER > M4 > M3 > MF (
Figure 7B). On the basis of these results, it can be roughly concluded that the residues Asp129 and Glu132 at the three-fold channel in AjFER are probably involved in Fe
2+ capture and its translocation to the internal cavity of the protein, from where Fe
2+ reaches the ferroxidase center, similar to that proposed for BfMF [
43]. Furthermore, our results confirm the ferroxidase center of AjFER indeed has a function in iron oxidation and the Glu168 residue in AjFER can act as a potential metal-ion binding site at the four-fold channel, similar to observations in previous studies [
9,
14]. In addition, the variants (MF, M3, and M4 proteins) exhibited lower unfolding temperatures than native AjFER, suggesting that mutations of the potential metal-binding sites in AjFER could have varying effects on its thermal stability and structural integrity. However, further research (e.g., using X-ray crystallography combined with site-directed mutagenesis) should provide further evidence regarding the mechanism of Fe
2+ entry and/or Fe
2+ binding to the ferroxidase center and the four-fold channel during iron ion transport in AjFER.




 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}