
Anomalous Dynamics in Macromolecular Liquids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
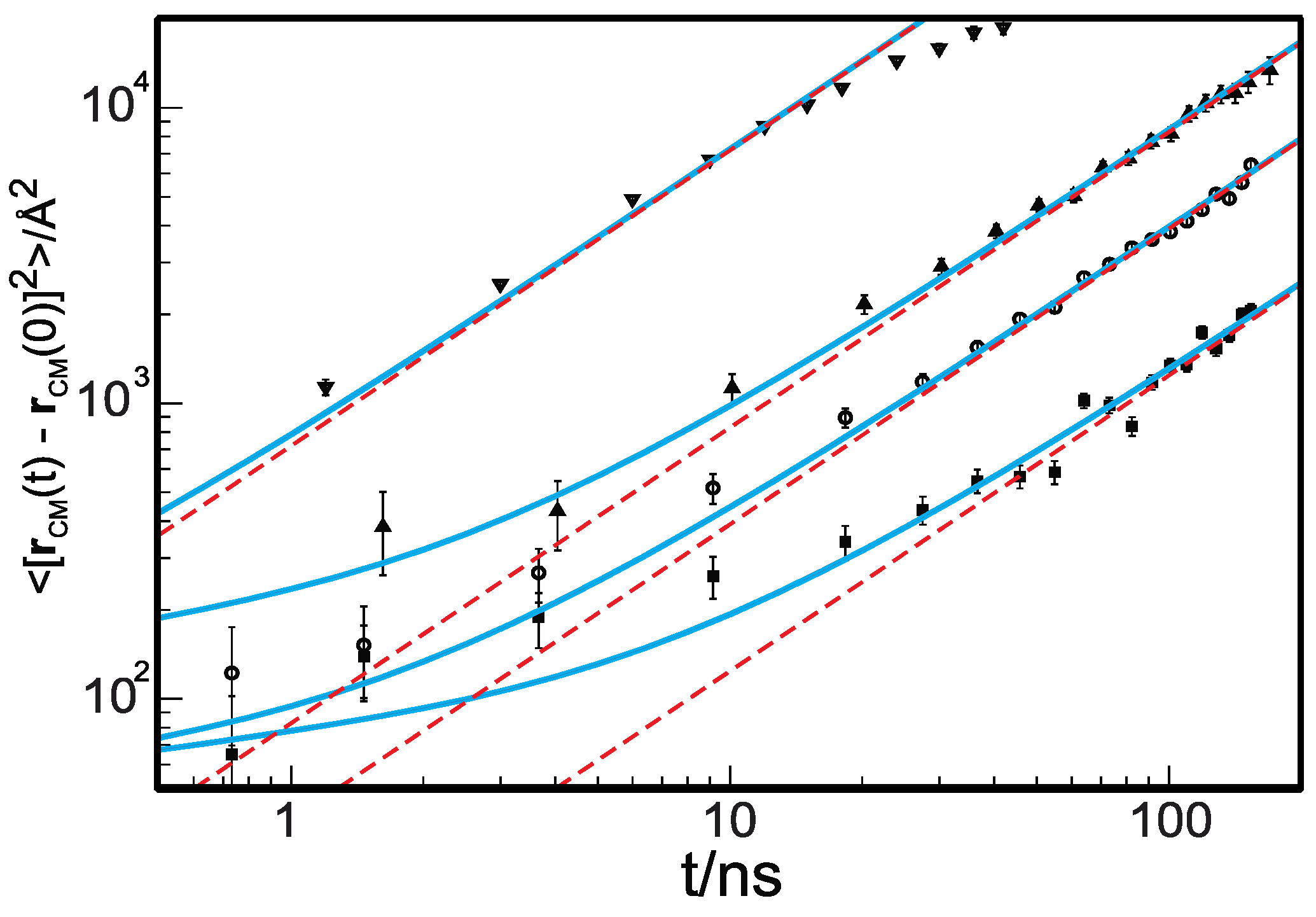
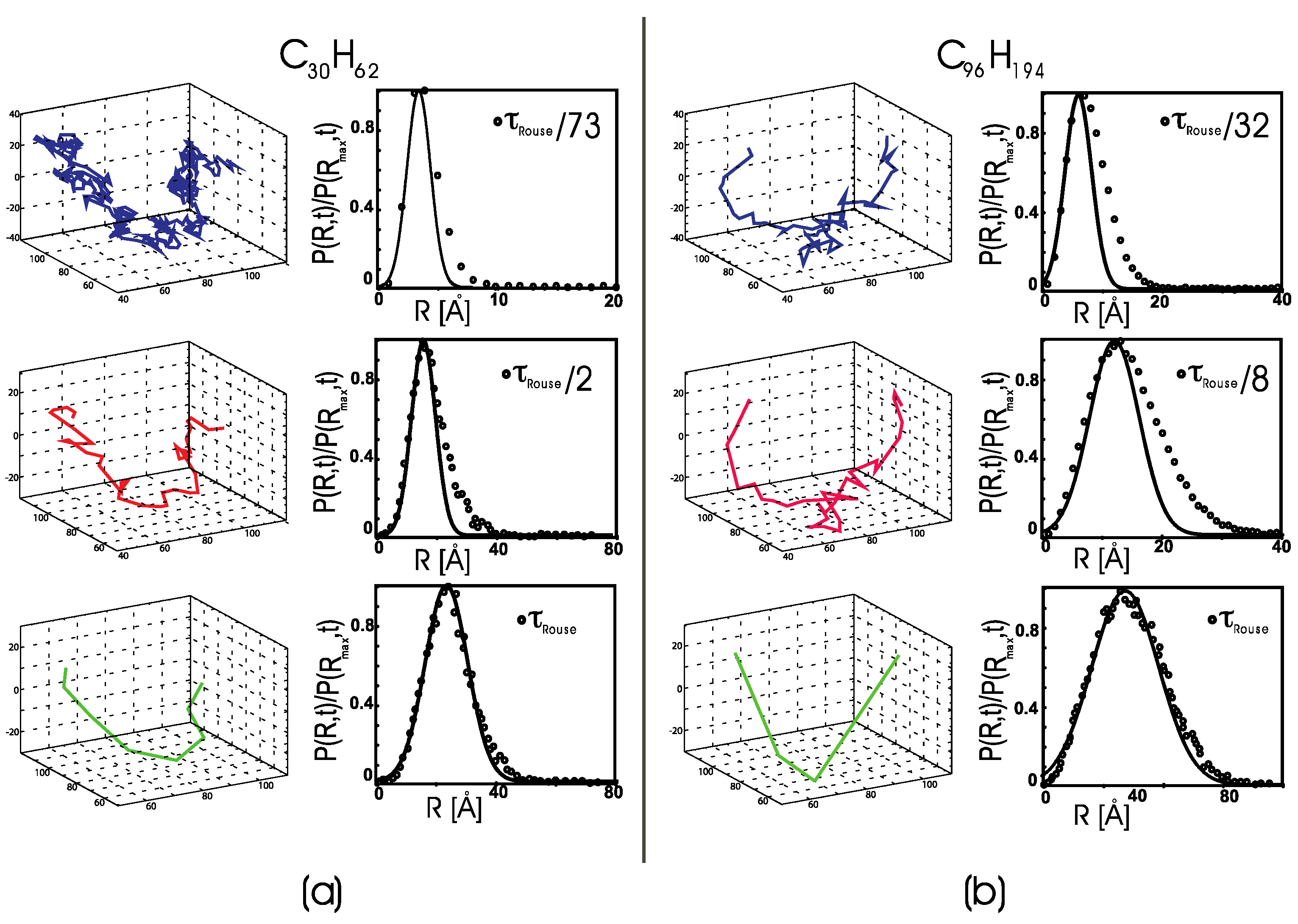
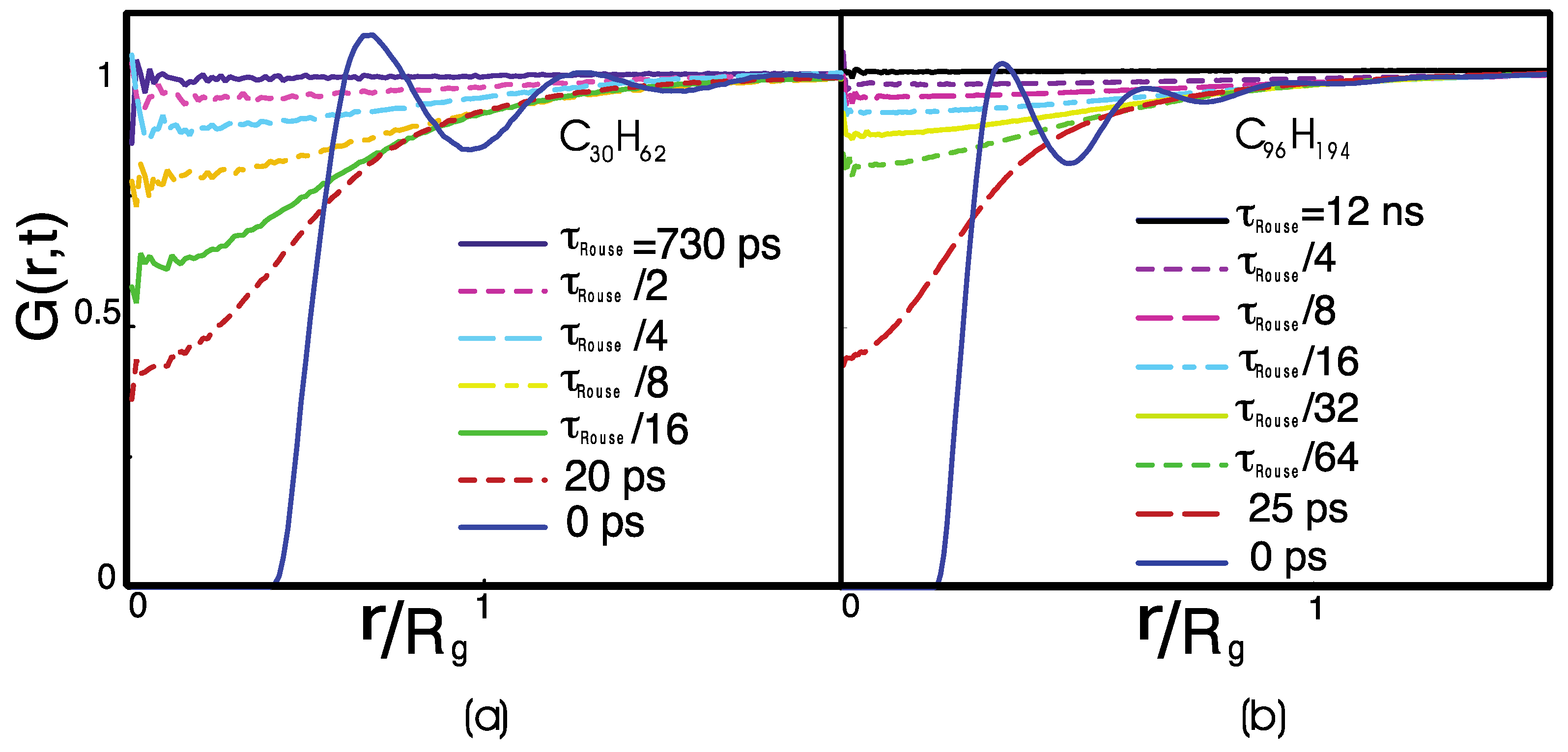
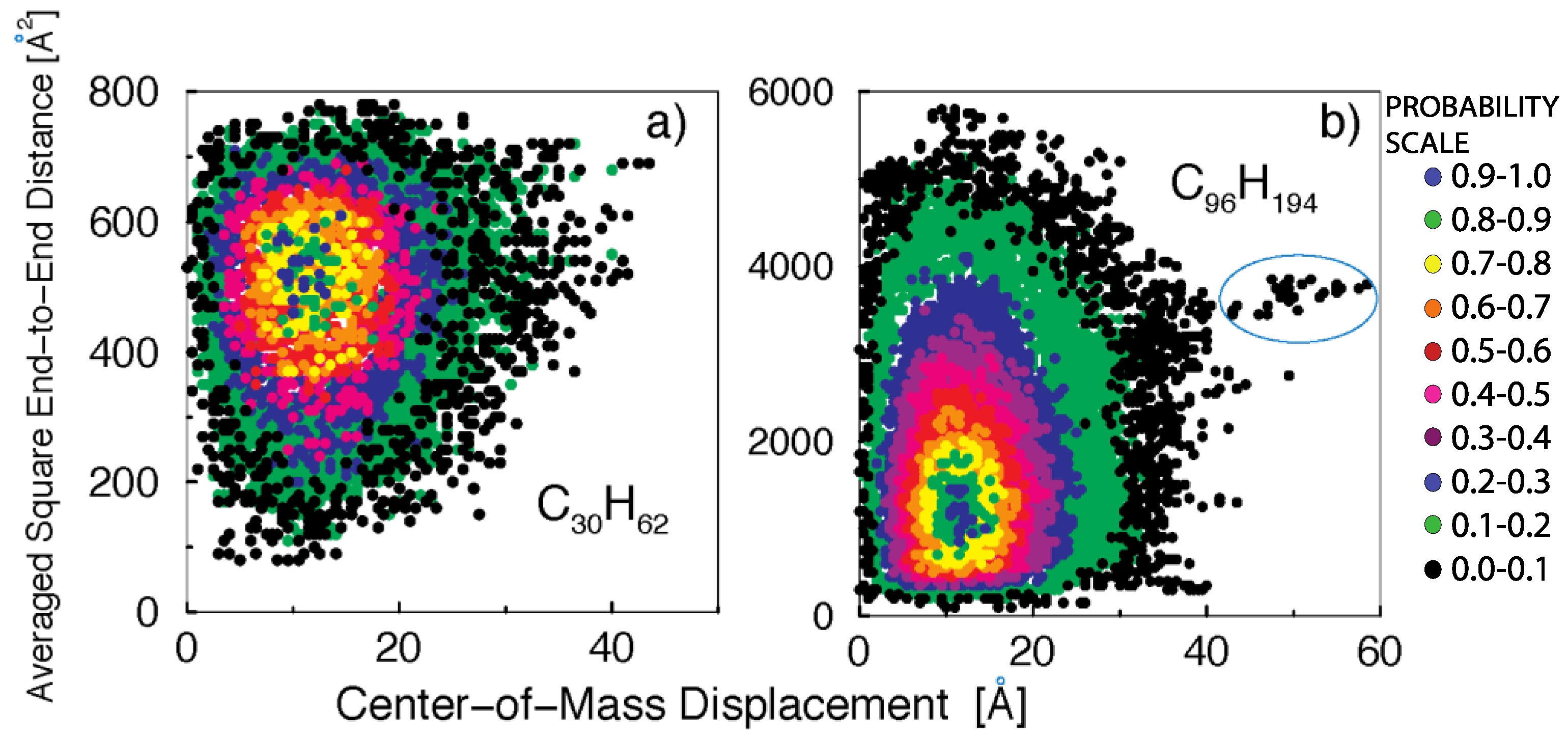
3. Results
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press—Oxford University Press: New York, NY, USA, 1988. [Google Scholar]
- Beyerle, E.R.; Guenza, M.G. Kinetics analysis of ubiquitin local fluctuations with Markov state modeling of the LE4PD normal modes. J. Chem. Phys. 2019, 151, 164119. [Google Scholar] [CrossRef] [PubMed]
- Copperman, J.; Guenza, M.G. Coarse-Grained Langevin Equation for Protein Dynamics: Global Anisotropy and a Mode Approach to Local Complexity. J. Phys. Chem. B 2014, 119, 9195–9211. [Google Scholar] [CrossRef] [PubMed]
- Copperman, J.; Dinpajooh, M.; Beyerle, E.R.; Guenza, M.G. Universality and Specificity in Protein Fluctuation Dynamics. Phys. Rev. Lett. 2017, 119, 158101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostov, K.S.; Freed, K.F. Long-Time Dynamics of Met-Enkephalin: Comparison of Theory with Brownian Dynamics Simulations. Biophys. J. 1999, 76, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-C.; Schurr, J.M. Dynamic light-scattering studies of internal motions in DNA. I. Applicability of the Rouse-Zimm model. Biopolymers 1978, 17, 425–461. [Google Scholar] [CrossRef]
- Socol, M.; Wang, R.; Jost, D.; Carrivain, P.; Vaillant, C.; Le Cam, E.; Dahirel, V.; Normand, C.; Bystricky, K.; Victor, J.-M.; et al. Rouse model with transient intramolecular contacts on a timescale of seconds recapitulates folding and fluctuation of yeast chromosomes. Nucleic Acids Res. 2019, 47, 6195–6207. [Google Scholar] [CrossRef] [Green Version]
- Rolls, E.; Togashi, Y.; Erban, R. Varying the Resolution of the Rouse Model on Temporal and Spatial Scales: Application to Multiscale Modeling of DNA Dynamics. Multiscale Model. Simul. 2017, 15, 1672–1693. [Google Scholar] [CrossRef]
- Knowles, M.K.; Guenza, M.; Capaldi, R.A.; Marcus, A.H. Cytoskeletal-assisted dynamics of the mitochondrial reticulum in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14772–14777. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids; Academic Press: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Schweizer, K.S. Microscopic theory of the dynamics of polymeric liquids: General formulation of a mode–mode-coupling approach. J. Chem. Phys. 1989, 91, 5802–5821. [Google Scholar] [CrossRef]
- Schweizer, K.S. Mode-coupling theory of the dynamics of polymer liquids: Qualitative predictions for flexible chain and ring melts. J. Chem. Phys. 1989, 91, 5822–5839. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Fuchs, M.; Szamel, G.; Guenza, M.; Tang, H. Polymer-mode-coupling theory of the slow dynamics of entangled macromolecular fluids. Macromol. Theory Simul. 1997, 6, 1037–1117. [Google Scholar] [CrossRef] [Green Version]
- Paul, W.; Smith, G.D.; Yoon, D.Y.; Farago, B.; Rathgeber, S.; Zirkel, A.; Willner, L.; Richter, D. Chain Motion in an Unentangled Polyethylene Melt: A Critical Test of the Rouse Model by Molecular Dynamics Simulations and Neutron Spin Echo Spectroscopy. Phys. Rev. Lett. 1998, 80, 2346–2349. [Google Scholar] [CrossRef]
- Bixon, M.; Zwanzig, R. Optimized Rouse–Zimm theory for stiff polymers. J. Chem. Phys. 1978, 68, 1896–1902. [Google Scholar] [CrossRef]
- Copperman, J.; Guenza, M. Predicting protein dynamics from structural ensembles. J. Chem. Phys. 2015, 143, 243131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyerle, E.R.; Guenza, M.G. Comparison between slow anisotropic LE4PD fluctuations and the principal component analysis modes of ubiquitin. J. Chem. Phys. 2021, 154, 124111. [Google Scholar] [CrossRef]
- Copperman, J.; Guenza, M. Mode localization in the cooperative dynamics of protein recognition. J. Chem. Phys. 2016, 145, 015101. [Google Scholar] [CrossRef] [Green Version]
- Beyerle, E.R.; Guenza, M.G. Identifying the leading dynamics of ubiquitin: A comparison between the tICA and the LE4PD slow fluctuations in amino acids’ position. J. Chem. Phys. 2021, 155, 244108. [Google Scholar] [CrossRef]
- Hall, J.; Guenza, M.G. Construction of Langevin Equations in Body-Fixed Frames, (in preparation).
- Skolnick, J.; Yaris, R. Phenomenological theory of the dynamics of polymer melts. II. Viscoelastic properties. J. Chem. Phys. 1988, 88, 1418–1442. [Google Scholar] [CrossRef]
- Douglas, J.F.; Hubbard, J.B. Semiempirical theory of relaxation: Concentrated polymer solution dynamics. Macromolecules 1991, 24, 3163–3177. [Google Scholar] [CrossRef]
- Paul, W.; Yoon, D.Y.; Smith, G.D. An optimized united atom model for simulations of polymethylene melts. J. Chem. Phys. 1995, 103, 1702–1709. [Google Scholar] [CrossRef]
- Koliński, A.; Skolnick, J.; Yaris, R. Monte Carlo studies on the long time dynamic properties of dense cubic lattice multichain systems. II. Probe polymer in a matrix of different degrees of polymerization. J. Chem. Phys. 1987, 86, 7174–7180. [Google Scholar] [CrossRef]
- Jaramillo, E.; Wu, D.T.; Grest, G.S.; Curro, J.G. Anomalous mixing behavior of polyisobutylene/polypropylene blends: Molecular dynamics simulation study. J. Chem. Phys. 2004, 120, 8883–8886. [Google Scholar] [CrossRef] [PubMed]
- Heine, D.; Wu, D.T.; Curro, J.G.; Grest, G.S. Role of intramolecular energy on polyolefin miscibility: Isotactic polypropylene/polyethylene blends. J. Chem. Phys. 2003, 118, 914–924. [Google Scholar] [CrossRef]
- Wu, X.-L.; Libchaber, A. Particle Diffusion in a Quasi-Two-Dimensional Bacterial Bath. Phys. Rev. Lett. 2000, 84, 3017–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, A.; Granek, R.; Elbaum, M. Enhanced Diffusion in Active Intracellular Transport. Phys. Rev. Lett. 2000, 85, 5655–5658. [Google Scholar] [CrossRef] [PubMed]
- Guenza, M. Cooperative Dynamics in Unentangled Polymer Fluids. Phys. Rev. Lett. 2001, 88, 025901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenza, M. Many chain correlated dynamics in polymer fluids. J. Chem. Phys. 1999, 110, 7574–7588. [Google Scholar] [CrossRef]
- Guenza, M. Intermolecular Effects in the Center-of-Mass Dynamics of Unentangled Polymer Fluids. Macromolecules 2002, 35, 2714–2722. [Google Scholar] [CrossRef] [Green Version]
- Mondello, M.; Grest, G.S. Viscosity calculations ofn-alkanes by equilibrium molecular dynamics. J. Chem. Phys. 1997, 106, 9327–9336. [Google Scholar] [CrossRef]
- Dinpajooh, M.; Guenza, M.G. On the Density Dependence of the Integral Equation Coarse-Graining Effective Potential. J. Phys. Chem. B 2017, 122, 3426–3440. [Google Scholar] [CrossRef]
- Dinpajooh, M.; Guenza, M. Thermodynamic consistency in the structure-based integral equation coarse-grained method. Polymer 2017, 117, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Dinpajooh, M.; Guenza, M.G. Coarse-graining simulation approaches for polymer melts: The effect of potential range on computational efficiency. Soft Matter 2018, 14, 7126–7144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinpajooh, M.; Guenza, M.G. Can pure polymer liquids be represented at two different resolutions simultaneously? J. Chem. Phys. 2019, 151, 061102. [Google Scholar] [CrossRef]
- Sambriski, E.J.; Yatsenko, G.; A Nemirovskaya, M.; Guenza, M.G. Bridging length scales in polymer melt relaxation for macromolecules with specific local structures. J. Phys. Condens. Matter 2007, 19. [Google Scholar] [CrossRef]
- Kopf, A.; Dünweg, B.; Paul, W. Dynamics of polymer “isotope” mixtures: Molecular dynamics simulation and Rouse model analysis. J. Chem. Phys. 1997, 107, 6945–6955. [Google Scholar] [CrossRef]
- Yamakawa, H. Modern Theory of Polymer Solutions; Harper & Row: New York, NY, USA, 1971. [Google Scholar]
- Smith, G.; Paul, W.; Monkenbusch, M.; Richter, D. A comparison of neutron scattering studies and computer simulations of polymer melts. Chem. Phys. 2000, 261, 61–74. [Google Scholar] [CrossRef]
- Zamponi, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Richter, D.; Falus, P.; Farago, B.; Guenza, M. Cooperative Dynamics in Homopolymer Melts: A Comparison of Theoretical Predictions with Neutron Spin Echo Experiments. J. Phys. Chem. B 2008, 112, 16220–16229. [Google Scholar] [CrossRef]
- Gaur, U.; Wunderlich, B. The Glass Transition Temperature of Polyethylene. Macromolecules 1980, 13, 445–446. [Google Scholar] [CrossRef]
- Weeks, E.R.; Crocker, J.C.; Levitt, A.C.; Schofield, A.; Weitz, D.A. Three-Dimensional Direct Imaging of Structural Relaxation Near the Colloidal Glass Transition. Science 2000, 287, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Bennemann, C.; Donati, C.; Baschnagel, J.; Glotzer, S.C. Growing range of correlated motion in a polymer melt on cooling towards the glass transition. Nature 1999, 399, 246–249. [Google Scholar] [CrossRef]
- de Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Schweizer, K.S.; Curro, J.G. PRISM Theory of the Structure, Thermodynamics, and Phase Transitions of Polymer Liquids and Alloys; Springer: Berlin/Heidelberg, Germany, 2006; pp. 319–377. [Google Scholar] [CrossRef]
- Guenza, M.G. Theoretical models for bridging timescales in polymer dynamics. J. Physics Condens. Matter 2007, 20. [Google Scholar] [CrossRef]
- Guenza, M.G. Localization of chain dynamics in entangled polymer melts. Phys. Rev. E 2014, 89, 052603. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, M.; Kruteva, M.; Monkenbusch, M.; Willner, L.; Wischnewski, A.; Hoffmann, I.; Richter, D. Cooperative Chain Dynamics of Tracer Chains in Highly Entangled Polyethylene Melts. Phys. Rev. Lett. 2021, 126, 187801. [Google Scholar] [CrossRef] [PubMed]
- Kruteva, M.; Zamponi, M.; Hoffmann, I.; Allgaier, J.; Monkenbusch, M.; Richter, D. Non-Gaussian and Cooperative Dynamics of Entanglement Strands in Polymer Melts. Macromolecules 2021. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guenza, M.G. Anomalous Dynamics in Macromolecular Liquids. Polymers 2022, 14, 856. https://doi.org/10.3390/polym14050856
Guenza MG. Anomalous Dynamics in Macromolecular Liquids. Polymers. 2022; 14(5):856. https://doi.org/10.3390/polym14050856
Chicago/Turabian StyleGuenza, Marina G. 2022. "Anomalous Dynamics in Macromolecular Liquids" Polymers 14, no. 5: 856. https://doi.org/10.3390/polym14050856
APA StyleGuenza, M. G. (2022). Anomalous Dynamics in Macromolecular Liquids. Polymers, 14(5), 856. https://doi.org/10.3390/polym14050856