
Modification of Covalent Triazine-Based Frameworks for Photocatalytic Hydrogen Generation


Abstract
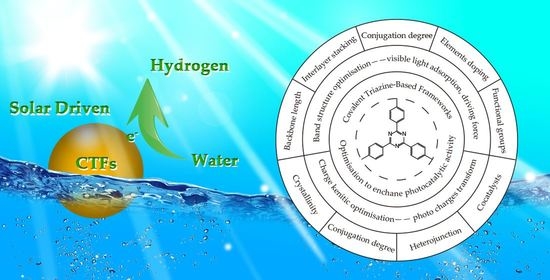
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Synthesis of CTFs
2.1. Ionothermal Synthesis
2.2. Microwave-Assisted Synthesis
2.3. Dehydration Polycondensation
2.4. Solution Polymerization
3. Optimization of CTFs for Photocatalytic Hydrogen Generation
3.1. Optimization of Band Structure to Broaden the Visible Light Absorption and Enhance the Driving Force for Hydrogen Generation
3.1.1. Optimize the Length of the Backbone
3.1.2. Control the Interlayer Stacking
3.1.3. Control the Degree of Polymerization
3.1.4. Element Doping
3.1.5. Functional Group Substitution
3.2. Control of Charge Dynamic to Enhance the Electron Transfer and Separation
3.2.1. Enhance the Degree of Crystallinity
3.2.2. Control the Degree of Conjugation
3.2.3. Heterojunction Construction
3.2.4. Cocatalyst Deposition
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic Water Splitting with a Quantum Efficiency of Almost Unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Domen, K. Reaction Systems for Solar Hydrogen Production via Water Splitting with Particulate Semiconductor Photocatalysts. Nat. Catal. 2019, 2, 387–399. [Google Scholar] [CrossRef]
- Song, X.; Wei, G.; Sun, J.; Peng, C.; Yin, J.; Zhang, X.; Jiang, Y.; Fei, H. Overall Photocatalytic Water Splitting by an Organolead Iodide Crystalline Material. Nat. Catal. 2020, 3, 1027–1033. [Google Scholar] [CrossRef]
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulfide Photocatalyst for Visible-light-driven Overall Water Splitting. Nat. Mater. 2019, 18, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zheng, Y.; Kobielusz, M.; Wang, Y.; Bai, Z.; Macyk, W.; Wang, X.; Tang, J. Recent Advances in Visible Light-driven Water Oxidation and Reduction in Suspension Systems. Mater. Today 2018, 21, 897–924. [Google Scholar] [CrossRef]
- Wang, Y.; Suzuki, H.; Xie, J.; Tomita, O.; Martin, D.J.; Higashi, M.; Kong, D.; Abe, R.; Tang, J. Mimicking Natural Photosynthesis: Solar to Renewable H2 Fuel Synthesis by Z-Scheme Water Splitting Systems. Chem. Rev. 2018, 118, 5201–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Vogel, A.; Sachs, M.; Sprick, R.S.; Wilbraham, L.; Moniz, S.J.A.; Godin, R.; Zwijnenburg, M.A.; Durrant, J.R.; Cooper, A.I.; et al. Current Understanding and Challenges of Solar-driven Hydrogen Generation Using Polymeric Photocatalysts. Nat. Energy 2019, 4, 746–760. [Google Scholar] [CrossRef]
- Stegbauer, L.; Schwinghammer, K.; Lotsch, B.V. A Hydrazone-based Covalent Organic Framework for Photocatalytic Hydrogen Production. Chem. Sci. 2014, 5, 2789–2793. [Google Scholar] [CrossRef] [Green Version]
- Ishi-i, T.; Yaguma, K.; Thiemann, T.; Yashima, M.; Ueno, K.; Mataka, S. High Electron Drift Mobility in an Amorphous Film of 2,4,6,-Tris[4-(1-naphthyl)phenyl]-1,3,5-triazine. Chem. Lett. 2004, 33, 1244–1245. [Google Scholar] [CrossRef]
- Xie, J.; Shevlin, S.A.; Ruan, Q.; Moniz, S.J.A.; Liu, Y.; Liu, X.; Li, Y.; Lau, C.C.; Guo, Z.X.; Tang, J. Efficient Visible Light-driven Water Oxidation and Proton Reduction by an Ordered Covalent Triazine-based Framework. Energy Environ. Sci. 2018, 11, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Chen, T.; Ma, L.; Wang, G.; Hu, W.; Zou, Z.; Wen, K.; Yang, H. Covalent Triazine-Based Polymers with Controllable Band Alignment Matched with BiVO4 to Boost Photogeneration of Holes for Water Splitting. Chem. Mater. 2019, 31, 8062–8068. [Google Scholar] [CrossRef]
- Kong, D.; Han, X.; Xie, J.; Ruan, Q.; Windle, C.D.; Gadipelli, S.; Shen, K.; Bai, Z.; Guo, Z.; Tang, J. Tunable Covalent Triazine-Based Frameworks (CTF-0) for Visible-Light-Driven Hydrogen and Oxygen Generation from Water Splitting. ACS Catal. 2019, 9, 7697–7707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Sun, X.; Xu, X.; Zhang, C.; He, X. Donor-acceptor Type Triazine-based Conjugated Porous Polymer for Visible-light-driven Photocatalytic Hydrogen Evolution. Appl. Catal. B Environ. 2019, 257, 117935. [Google Scholar] [CrossRef]
- Meier, C.B.; Clowes, R.; Berardo, E.; Jelfs, K.E.; Zwijnenburg, M.A.; Sprick, R.S.; Cooper, A.I. Structurally Diverse Covalent Triazine-Based Framework Materials for Photocatalytic Hydrogen Evolution from Water. Chem. Mater. 2019, 31, 8830–8838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cheng, G.; Guo, L.; Wang, N.; Tan, B.; Jin, S. Strong-Base-Assisted Synthesis of a Crystalline Covalent Triazine Framework with High Hydrophilicity via Benzylamine Monomer for Photocatalytic Water Splitting. Angew. Chem. Int. Ed. 2020, 59, 6007–6014. [Google Scholar] [CrossRef]
- Bhunia, A.; Vasylyeva, V.; Janiak, C. From a Supramolecular Tetranitrile to a Porous Covalent Triazine-based Framework with High Gas Uptake Capacities. Chem. Commun. 2013, 49, 3961. [Google Scholar] [CrossRef]
- Kuhn, P.; Antonietti, M.; Thomas, A. Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis. Angew. Chem. Int. Ed. 2008, 47, 3450–3453. [Google Scholar] [CrossRef]
- Kuhn, P.; Thomas, A.; Antonietti, M. Toward Tailorable Porous Organic Polymer Networks: A High-Temperature Dynamic Polymerization Scheme Based on Aromatic Nitriles. Macromolecules 2009, 42, 319–326. [Google Scholar] [CrossRef]
- Bojdys, M.J.; Jeromenok, J.; Thomas, A.; Antonietti, M. Rational Extension of the Family of Layered, Covalent, Triazine-Based Frameworks with Regular Porosity. Adv. Mater. 2010, 22, 2202–2205. [Google Scholar] [CrossRef]
- Schwinghammer, K.; Hug, S.; Mesch, M.B.; Senker, J.; Lotsch, B.V. Phenyl-triazine Oligomers for Light-driven Hydrogen Evolution. Energy Environ. Sci. 2015, 8, 3345–3353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liang, F.; Li, C.; Qiu, L.; Yuan, Y.; Peng, F.; Jiang, X.; Xie, A.; Shen, Y.; Zhu, J. Microwave-enhanced Synthesis of Magnetic Porous Covalent Triazine-based Framework Composites for Fast Separation of Organic Dye from Aqueous Solution. J. Hazard. Mater. 2011, 186, 984–990. [Google Scholar] [CrossRef]
- Ren, S.; Bojdys, M.J.; Dawson, R.; Laybourn, A.; Khimyak, Y.Z.; Adams, D.J.; Cooper, A.I. Porous, Fluorescent, Covalent Triazine-Based Frameworks via Room-Temperature and Microwave-Assisted Synthesis. Adv. Mater. 2012, 24, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, L.; Wang, X.; Guo, L.; Cheng, G.; Zhang, C.; Jin, S.; Tan, B.; Cooper, A. Covalent Triazine Frameworks via a Low-Temperature Polycondensation Approach. Angew. Chem. 2017, 129, 14337–14341. [Google Scholar] [CrossRef]
- Yu, S.; Mahmood, J.; Noh, H.; Seo, J.; Jung, S.; Shin, S.; Im, Y.; Jeon, I.; Baek, J. Direct Synthesis of a Covalent Triazine-Based Framework from Aromatic Amides. Angew. Chem. Int. Ed. 2018, 57, 8438–8442. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Fang, W.; Li, L.; Wang, J.; Liang, S.; He, Y.; Liu, M.; Wu, L. Covalent Triazine-Based Frameworks as Visible Light Photocatalysts for the Splitting of Water. Macromol. Rapid. Comm. 2015, 36, 1799–1805. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Cho, S.; Lee, Y.; Ahn, W. Microporous Covalent Triazine Polymers: Efficient Friedel–Crafts Synthesis and Adsorption/Storage of CO2 and CH4. J. Mater. Chem. A 2015, 3, 6792–6797. [Google Scholar] [CrossRef]
- Dey, S.; Bhunia, A.; Esquivel, D.; Janiak, C. Covalent Triazine-based Frameworks (CTFs) from Triptycene and Fluorene Motifs for CO2 Adsorption. J. Mater. Chem. A 2016, 4, 6259–6263. [Google Scholar] [CrossRef] [Green Version]
- Geng, T.; Zhang, W.; Zhu, Z.; Chen, G.; Ma, L.; Ye, S.; Niu, Q. A Covalent Triazine-based Framework from Tetraphenylthiophene and 2,4,6-trichloro-1,3,5-triazine Motifs for Sensing o-nitrophenol and Effective I2 Uptake. Polym. Chem. 2018, 9, 777–784. [Google Scholar] [CrossRef]
- Meier, C.B.; Sprick, R.S.; Monti, A.; Guiglion, P.; Lee, J.M.; Zwijnenburg, M.A.; Cooper, A.I. Structure-property Relationships for Covalent Triazine-based Frameworks: The Effect of Spacer Length on Photocatalytic Hydrogen Evolution from Water. Polymer 2017, 126, 283–290. [Google Scholar] [CrossRef]
- Johns, I.B.; McElhill, E.A.; Smith, J.O. Thermal Stability of Organic Compounds. Ind. Eng. Chem. Prod. Res. Dev. 1962, 1, 2–6. [Google Scholar] [CrossRef]
- Liu, J.; Lyu, P.; Zhang, Y.; Nachtigall, P.; Xu, Y. New Layered Triazine Framework/Exfoliated 2D Polymer with Superior Sodium-Storage Properties. Adv. Mater. 2018, 30, 1705401. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Liang, Y.; Xu, Y. Rapid, Ordered Polymerization of Crystalline Semiconducting Covalent Triazine Frameworks. Angew. Chem. Int. Ed. 2022, 4, e202113926. [Google Scholar]
- Kong, D.; Xie, J.; Guo, Z.; Yang, D.; Tang, J. Stable Complete Water Splitting by Covalent Triazine-based Framework CTF-0. Chemcatchem 2020, 12, 2708–2712. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, P.; Zhao, J. 2D Covalent Triazine Framework: A New Class of Organic Photocatalyst for Water Splitting. J. Mater. Chem. A 2015, 3, 7750–7758. [Google Scholar] [CrossRef]
- Li, L.; Fang, W.; Zhang, P.; Bi, J.; He, Y.; Wang, J.; Su, W. Sulfur-doped Covalent Triazine-based Frameworks for Enhanced Photocatalytic Hydrogen Evolution from Water under Visible Light. J. Mater. Chem. A 2016, 4, 12402–12406. [Google Scholar] [CrossRef]
- Cheng, Z.; Fang, W.; Zhao, T.; Fang, S.; Bi, J.; Liang, S.; Li, L.; Yu, Y.; Wu, L. Efficient Visible-Light-Driven Photocatalytic Hydrogen Evolution on Phosphorus-Doped Covalent Triazine-Based Frameworks. ACS Appl. Mater. Interfaces 2018, 10, 41415–41421. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zheng, K.; Lin, G.; Fang, S.; Li, L.; Bi, J.; Shen, J.; Wu, L. Constructing a Novel Family of Halogen-doped Covalent Triazine-based Frameworks as Efficient Metal-free Photocatalysts for Hydrogen Production. Nanoscale Adv. 2019, 1, 2674–2680. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wu, M.; Guo, T.; Zheng, L.; Wang, D.; Mu, Y.; Xing, Q.; Zou, J. Chlorine-mediated Photocatalytic Hydrogen Production Based on Triazine Covalent Organic Framework. Appl. Catal. B Environ. 2020, 272, 118989. [Google Scholar] [CrossRef]
- Kong, D.; Han, X.; Shevlin, S.A.; Windle, C.; Warner, J.H.; Guo, Z.; Tang, J. A Metal-Free Oxygenated Covalent Triazine 2-D Photocatalyst Works Effectively from the Ultraviolet to Near-Infrared Spectrum for Water Oxidation Apart from Water Reduction. ACS Appl. Energy Mater. 2020, 3, 8960–8968. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Niu, Y.; Xu, H.; Li, Q.; Razzaque, S.; Huang, Q.; Jin, S.; Tan, B. Engineering Heteroatoms with Atomic Precision in Donor–acceptor Covalent Triazine Frameworks to Boost Photocatalytic Hydrogen Production. J. Mater. Chem. A 2018, 6, 19775–19781. [Google Scholar] [CrossRef]
- Wang, D.; Li, X.; Zheng, L.; Qin, L.; Li, S.; Ye, P.; Li, Y.; Zou, J. Size-controlled Synthesis of CdS Nanoparticles Confined on Covalent Triazine-based Frameworks for Durable Photocatalytic Hydrogen Evolution under Visible Light. Nanoscale 2018, 10, 19509–19516. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shi, Y.; Xu, W.; Wang, Y.; Zhang, Y.; Wang, Y.; Wu, Y.; Wu, N.; Sun, Y.; Du, Y.; et al. Ultra-thin Deaminated Tri-s-triazine-based Crystalline Nanosheets with High Photocatalytic Hydrogen Evolution Performance. J. Alloys Compd. 2020, 827, 154307. [Google Scholar] [CrossRef]
- Jiang, Q.; Sun, L.; Bi, J.; Liang, S.; Li, L.; Yu, Y.; Wu, L. MoS2 Quantum Dots-Modified Covalent Triazine-Based Frameworks for Enhanced Photocatalytic Hydrogen Evolution. Chemsuschem 2018, 11, 1108–1113. [Google Scholar] [CrossRef]
- Huang, W.; He, Q.; Hu, Y.; Li, Y. Molecular Heterostructures of Covalent Triazine Frameworks for Enhanced Photocatalytic Hydrogen Production. Angew. Chem. 2019, 131, 8768–8772. [Google Scholar] [CrossRef]
- Guo, L.; Niu, Y.; Razzaque, S.; Tan, B.; Jin, S. Design of D–A1–A2 Covalent Triazine Frameworks via Copolymerization for Photocatalytic Hydrogen Evolution. ACS Catal. 2019, 9, 9438–9445. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, D.; Wu, S.; Jiang, X.; Zhang, J.; Xing, Q.; Zou, J.; Luo, S. Unveiling localized Pt–P–N bonding states constructed on covalent triazine-based frameworks for boosting photocatalytic hydrogen evolution. J. Mater. Chem. A 2020, 8, 25425–25430. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, Y.; Wang, L.; Tan, M.; Xiao, Y.; Gao, B.; Lin, B. Metal-free 2D/2D Heterostructured Photocatalyst of Black Phosphorus/Covalent Triazine-based Frameworks for Water Splitting and Pollutant Degradation. Sustain. Energy Fuels 2020, 4, 3739–3746. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Liu, J.; Wang, K.; Jin, S.; Tan, B. Palladium as a Superior Cocatalyst to Platinum for Hydrogen Evolution Using Covalent Triazine Frameworks as a Support. ACS Appl. Mater. Int. 2020, 12, 12774–12782. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, H.; Luo, W.; Sun, T.; Xu, Y. Ultrathin Crystalline Covalent-Triazine-Framework Nanosheets with Electron Donor Groups for Synergistically Enhanced Photocatalytic Water Splitting. Angew. Chem. Int. Ed. 2021, 60, 25381–25390. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, P.; Huang, G.; Chen, Q.; Bi, J.; Wu, L. Band Gap Tuning of Covalent Triazine-Based Frameworks through Iron Doping for Visible-Light-Driven Photocatalytic Hydrogen Evolution. Chemsuschem 2021, 14, 3850–3857. [Google Scholar] [CrossRef]
- Ye, H.; Gong, N.; Cao, Y.; Fan, X.; Song, X.; Li, H.; Wang, C.; Mei, Y.; Zhu, Y. Insights into the Role of Protonation in Covalent Triazine Framework-Based Photocatalytic Hydrogen Evolution. Chem. Mater. 2022, 34, 1481–1490. [Google Scholar] [CrossRef]
- Lan, Z.A.; Chi, X.; Wu, M.; Zhang, X.; Chen, X.; Zhang, G.; Wang, X. Molecular Design of Covalent Triazine Frameworks with Anisotropic Charge Migration for Photocatalytic Hydrogen Production. Small 2022, 2200129. [Google Scholar] [CrossRef]
- Abutaha, A.; Kumar, P.; Yildirim, E.; Shi, W.; Yang, S.; Wu, G.; Hippalgaonkar, K. Correlating charge and thermoelectric transport to paracrystallinity in conducting polymers. Nat. Commun. 2020, 11, 1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Z.A.; Wu, M.; Fang, Z.; Zhang, Y.; Chen, X.; Zhang, G.; Wang, X. Ionothermal Synthesis of Covalent Triazine Frameworks in a NaCl-KCl-ZnCl2 Eutectic Salt for the Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 2022, e202201482. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Huang, X.; Tao, L.; Bi, Y. Direct observation of dynamic interfacial bonding and charge transfer in metal-free photocatalysts for efficient hydrogen evolution. Appl. Catal. B Environ. 2021, 283, 119633. [Google Scholar] [CrossRef]
- Xu, N.; Cai, B.; Li, Q.; Liu, Y.; Tang, J.; Wang, K.; Xu, B.; Fan, Y. The noble-metal-free Ni2P/CTF composites for efficient photocatalytic hydrogen evolution under visible-light irradiation. J. Alloys Compd. 2021, 871, 159565. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, P.; Chen, Q.; Fang, S.; Huang, G.; Bi, J.; Wu, L. Visible-light-driven photocatalyst based upon metal-free covalent triazine-based frameworks for enhanced hydrogen production. Catal. Sci. Technol. 2021, 11, 1188–1874. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.C.; Yang, Q.; Li, X.Y.; Yi, F.; Liu, K.W.; Cao, H.M.; Wang, C.J.; Yan, H.J. Graphene Oxide-Assisted Covalent Triazine Framework for Boosting Photocatalytic H2 Evolution. Chem. Eur. J. 2021, 27, 13059–13066. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, J.; Fang, Z.; Wang, H. Modification of Covalent Triazine-Based Frameworks for Photocatalytic Hydrogen Generation. Polymers 2022, 14, 1363. https://doi.org/10.3390/polym14071363
Xie J, Fang Z, Wang H. Modification of Covalent Triazine-Based Frameworks for Photocatalytic Hydrogen Generation. Polymers. 2022; 14(7):1363. https://doi.org/10.3390/polym14071363
Chicago/Turabian StyleXie, Jijia, Zhiping Fang, and Hui Wang. 2022. "Modification of Covalent Triazine-Based Frameworks for Photocatalytic Hydrogen Generation" Polymers 14, no. 7: 1363. https://doi.org/10.3390/polym14071363
APA StyleXie, J., Fang, Z., & Wang, H. (2022). Modification of Covalent Triazine-Based Frameworks for Photocatalytic Hydrogen Generation. Polymers, 14(7), 1363. https://doi.org/10.3390/polym14071363