1. Introduction
Steel and steel-made infrastructures are critical for the expansion and operation of industry and global economies. Short-term forecasts for steel demand project further growth, with the industrialization of developing nations being part of the driving force [
1,
2]. Steel is susceptible to corrosion failures, particularly in aggressive environments, and such failures pose a serious and expensive problem, with estimated costs reaching over 3% of global gross domestic product (GDP) [
3,
4,
5,
6]. Effective corrosion prevention and management methods can be divided into four broad categories: alteration of the environment, modification of the steel composition, adjustment of procedures, and application of protective coatings [
7]. Inorganic or organic coatings are efficient approaches to protect metallic components, including steel, from the detrimental action of aggressive environments, such as chemical and mechanical damage, moisture, and biological degradation, thus preventing and contributing to the efficient management of corrosion failures [
3].
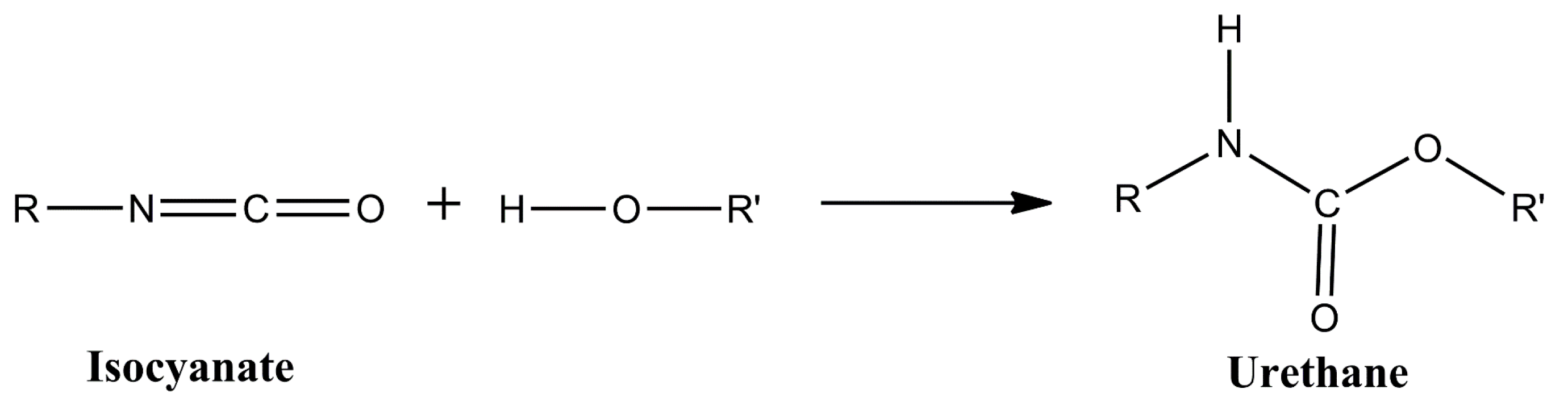
Polyurethane (PU) coatings are widely used for the corrosion protection of steel. PU consists of polymer chains or networks containing urethane groups, which are formed from the reaction of an isocyanate and a hydroxyl group (
Figure 1).
They are generally composed of soft and hard segments in a single polymeric matrix and are usually known to possess excellent adhesion, abrasion resistance, and toughness, along with a high corrosion protection ability and chemical resistance [
8,
9].
PUs are commonly produced from the step-growth polymerization of diisocyanates or polyisocyanates with glycol, or polyol compounds, without the liberation of low-molecular-weight by-products. A cross-linked polymer is formed if one or both reactants are multifunctional. The soft segments are usually relative to the polyol-derived chains, while the hard segments derive from the highly reactive isocyanate compounds. Commercial PU synthesis often uses petroleum-based raw materials, not complying with the trendy “green chemistry” and sustainability requirements, raising health and environmental concerns.
The main driving forces for the replacement of common fossil-based raw materials with bio-based feedstocks derive from the tendency for the depletion of fossil fuel feedstocks, increasing demands of energy at the global scale, and other environmental concerns, which aim at the transition to a more circular economy and the reduction of carbon footprint [
6,
8,
10,
11]. Moreover, the possibility to impart new functionalities to PU coatings also promotes the development of biopolyurethane (BioPU) chemistries, where bio-based feedstock raw materials replace common fossil-based raw materials. At present, BioPUs are being synthesized from vegetable oils obtained from various plant feedstocks such as castor, cotton, rapeseed, palm, and soybean [
8,
9,
11,
12,
13,
14,
15,
16,
17], as well as from lignin and lignin derivatives [
18,
19,
20,
21,
22,
23,
24]. Lignin and lignin derivatives are commonly obtained as subproducts of the paper and pulp industry, among other bio-renewable sources [
25,
26].
Plant biomass is mainly composed of organic polymers, namely, cellulose, hemicellulose, and lignin. It has the advantages of being an inexpensive raw material in addition to possessing high renewability, sustainability, and accessibility. Many processes, either thermochemical, biochemical, or mechanical, can be used to break down the biomass’ complex structure into smaller molecules and manageable fractions. Thermochemical conversion processes include pyrolysis, gasification, and liquefaction [
27,
28,
29,
30]. The thermochemical liquefaction of lignocellulosic biomass is a process that consists of the depolymerization and solubilization of biomass at moderate temperatures in a suitable solvent, usually polyhydric alcohols. The process has been widely investigated and has been shown to be a great tool to add value to biomass subproducts and wastes [
27,
28,
31,
32,
33,
34,
35,
36,
37,
38]. In the process of thermochemical liquefaction, lignin is degraded into phenolic compounds, while cellulose and hemicellulose are hydrolyzed into monosaccharides such as glucose and xylose, and further to acids, aldehydes, ketones [
29,
35]. Thermochemical liquefaction is heavily influenced by several factors, the most important being biomass composition, temperature, type of solvent, residence time, the ratio of biomass to solvent, and the presence of a catalyst [
27,
29,
34]. When it comes to solvents, simple alcohols, such as methanol, ethanol, propanol, and butanol, give higher conversions, but have lower boiling points, requiring high-pressure reactors [
27]. Longer-chain alcohols such as glycerol, ethylene glycol, 2-ethyl hexanol, or polyethylene glycol are widely used [
29,
34,
35]. The effect of the solvent in the thermochemical liquefaction of lignocellulosic biomass has been widely studied; however, the reaction pathways and the solvent influence are many times still unclear, mostly due to the complexity and variability of the biomass and process conditions. High-polar and -protic solvents have been shown to have higher efficiency. This can be, in part, due to the products of biomass decomposition being rich in similar functional groups, allowing good solubility. The solvent capacity to act as hydrogen donor also influences the efficiency of the process. It is stated that the presence of a hydrogen donor solvent may facilitate the hydrocracking of large molecules as well as the stabilization of free radicals and molecular rearrangements [
39,
40]. A low mass ratio of biomass to solvent, commonly 1:3 to 1:5, contributes to high reaction yield as the products are more readily extracted to the solvent medium [
32,
34]. The depolymerization reactions usually occur in the presence of a catalyst, with Brønsted–Lowry acids being commonly used. Among these, p-toluenesulfonic acid has been reported to display high conversion rates without leading to secondary condensation reactions (by-product formation) and repolymerization reactions [
41].
Despite the recent focus on developing BioPU from biomass feedstocks, and on the potential of thermochemical liquefaction to turn biomass subproducts and residues into polyols, relevant studies on the use of these thermochemical liquefaction polyols in BioPU coatings are relatively scarce. Most of these studies apply the liquified biomass-derived polyols to the production of PU foams [
42,
43,
44,
45,
46,
47,
48] and adhesives [
49,
50,
51], and are not focused on coatings, as the present paper is. Recently, Gosz et al. [
52,
53] developed BioPU from biopolyols derived from the thermochemical liquefaction of liquified oak and alder wood sawdust, reporting an optimum liquefaction temperature of 150 °C for a period of 6 h. It was stated that the obtained PUs possess a high Young’s module, up to 839 MPa, and beneficial properties for coating wood decks. Olszewski et al. [
54] suggested that PU wood composites could be a green alternative for the construction, furniture, or automotive industries if biopolyols from the thermochemical liquefaction of wood shavings are used as raw materials. In their work, liquefaction was employed at a temperature of 150 °C for 6 h utilizing three solvents: glycerol, poly(ethylene glycol), and a 1:1 mixture of both. The obtained biopolyols were mixed with wood shavings, which were used as filler, before adding polymeric methylene diphenyl diisocyanate (pMDI). The mixture of all components was subjected to a hydraulic pressing machine. The resulting PU composite was said to possess improved characteristics, such as hardness, impact strength, flexural strength, and Young’s module.
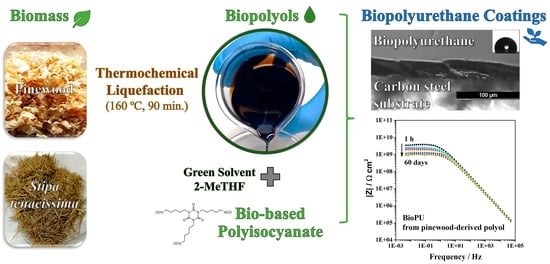
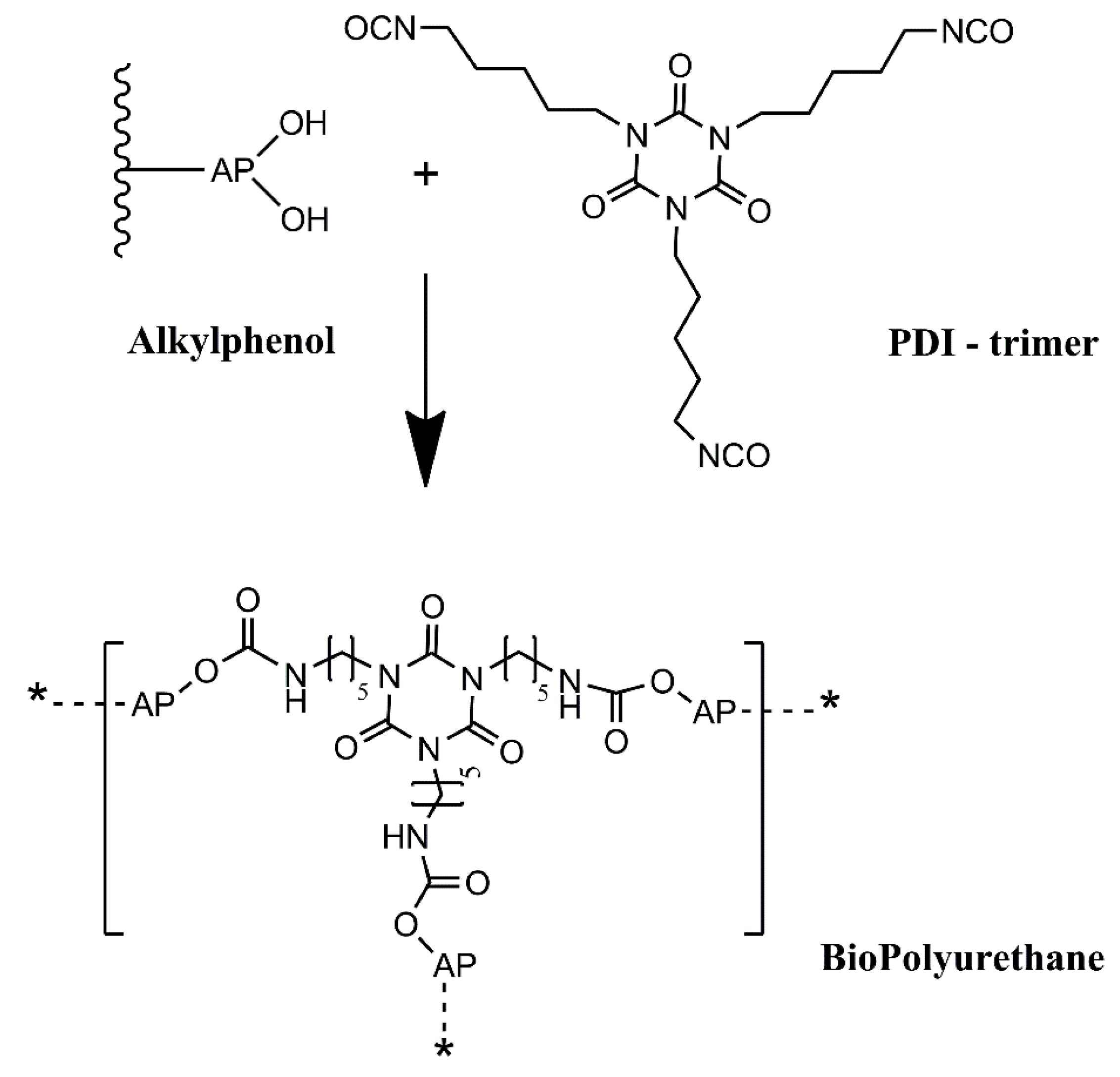
In this work, we intend to develop an alternative green path, where bio-based raw materials, either at the isocyanate and polyol component level, are employed to produce polyurethane coatings for corrosion protection. This will add value to biomass residues, contributing to a more circular economy, while alleviating the dependency on fossil-based raw materials. Thus, this work reports a novel BioPU-based coating for corrosion protection of steel, where petroleum-derived polyols and diisocyanates are fully replaced by bio-based feedstock raw materials, obtained from the thermochemical liquefaction of pinewood and
Stipa tenacissima biomasses, and a new green commercially available bio-based pentamethylene diisocyanate (PDI) trimer, Desmodur
® Eco N7300 (
Figure 2). This compound was the first commercial bio-based PDI isocyanurate trimer, launched by Covestro in 2015, in which approximately 68% of the carbon content is bio-based. The application of residues of pinewood biomass shows that they can be employed for much more than fuel for burning furnaces. At the same time, the utilization of
Stipa tenacissima not only illustrates the flexibility of the process, as this fibrous grass nature contrasts with the wood residues utilized, but it also provides economic valorization to this biomass, which is endemic to the Mediterranean regions, south of the Iberian Peninsula and in the north of Africa. It is mainly used in the handicraft industry or for the production of paper, and currently has a low economic value. A green solvent, 2-methyl tetrahydrofuran (2-MTHF), was also employed to further promote a sustainable coating. The solvent 2-MTHF can be derived from bio-renewable sources, utilizing reagents such as furfural or levulinic acid for its synthesis. Moreover, it is reported that 2-MTHF holds high biodegradability, being abiotically degraded by sunlight and air, making it a possible alternative solvent for environmentally friendly, green synthesis processes [
55,
56].
The resulting BioPU base coatings reveal good anti-corrosion properties, as demonstrated by electrochemical impedance spectroscopy, and hold potential for further modification with bio-based anti-corrosion to improve the corrosion protection of metallic infrastructures.
2. Materials and Methods
2.1. Materials
Pinewood shaves purchased from Versele-Laga (Deinze, Belgium), and Stipa tenacissima grass (referred to as Stipa throughout this paper), kindly provided by a Tunisian company, were used as raw materials for the thermochemical liquefaction process. The solvents 2-ethyl hexanol (2EH; ≥90 wt.%), propylene glycol (PG; ≥90 wt.%), 2-methyl tetrahydrofuran (2-MTHF; ≥99.5 wt.%) and deuterated chloroform (≥99.8 atom % D), as well as the catalyst p-toluenesulfonic acid (PTSA; ≥98 wt.%), were purchased from Sigma-Aldrich (Sigma-Aldrich Ltd., St. Louis, MO, USA). N,N-dimethylformamide (DMF; >99.5 wt.%) was obtained from Carlo Herba (Carlo Erba Reagents S.A.S., Val De Reuil, France). Desmodur® Eco N7300, a bio-based aliphatic 1,5-pentamethylene polyisocyanate, was obtained from Covestro (Covestro AG, Leverkusen, Germany). It has an NCO content of about 21.9%, a viscosity at 23 °C of ca. 9500 mPa.s, less than 0.3% of monomeric PDI and an equivalent weight (EW) of ca. 195. The 0.5 M and 0.1 M KOH solutions used in the determination of the polyols’ acid and hydroxyl values were purchased from Honeywell (Charlotte, NC, USA), and the solvent tetrahydrofuran (THF; 99.8 wt.%) was purchased from Fisher Scientific (Thermo Fisher Scientific Inc., Waltham, MA, USA). The DC01 carbon steel plates used were kindly provided by Voestalpine AG (Linz, Austria).
2.2. Thermochemical Liquefaction of Biomass into Polyol
Before the thermochemical liquefaction reaction, Stipa samples were dried in an oven at 60 °C, until no changes in weight were visible, and then ground in a Retsch©SM 2000 blade mill (Retsch GmbH, Haan, Germany) with a 4 mm sieve. Pinewood shaves were used as obtained.
Biomass, solvent, either 2EH or PG, (1:5 wt. of biomass to solvent), and PTSA catalyst (3 wt.%) were fed to a reactor and allowed to react at a moderate temperature of 160 °C for 90 min, with mechanical stirring in a nitrogen (N
2) inert atmosphere. A Dean–Stark apparatus was used to capture moisture from the biomass and water formed during the reaction. After the set time, the reactor was allowed to cool down at room temperature, after which the contents were vacuum filtered. The solid biomass residue was separately washed with organic solvent and thoroughly dried at 60 °C until no meaningful weight change was observed, after which it was collected for further analysis. The process was adapted from other works from the group, described elsewhere [
27,
37,
38]. The biomass conversion was calculated according to Equation (1):
where
mresidue is the mass (g) of solid biomass residues at the end of the reaction, and
mbiomass is the mass (g) of biomass fed to the reactor, on a dry basis.
The liquified biomass obtained from the reaction was subjected to a process of liquid–liquid extraction with water, in a proportion of 1:3 (v/v), and the liquified biomass organic fraction was retrieved. The organic fraction was further submitted to vacuum distillation to recover all the solvent from the mixture and obtain the biopolyol.
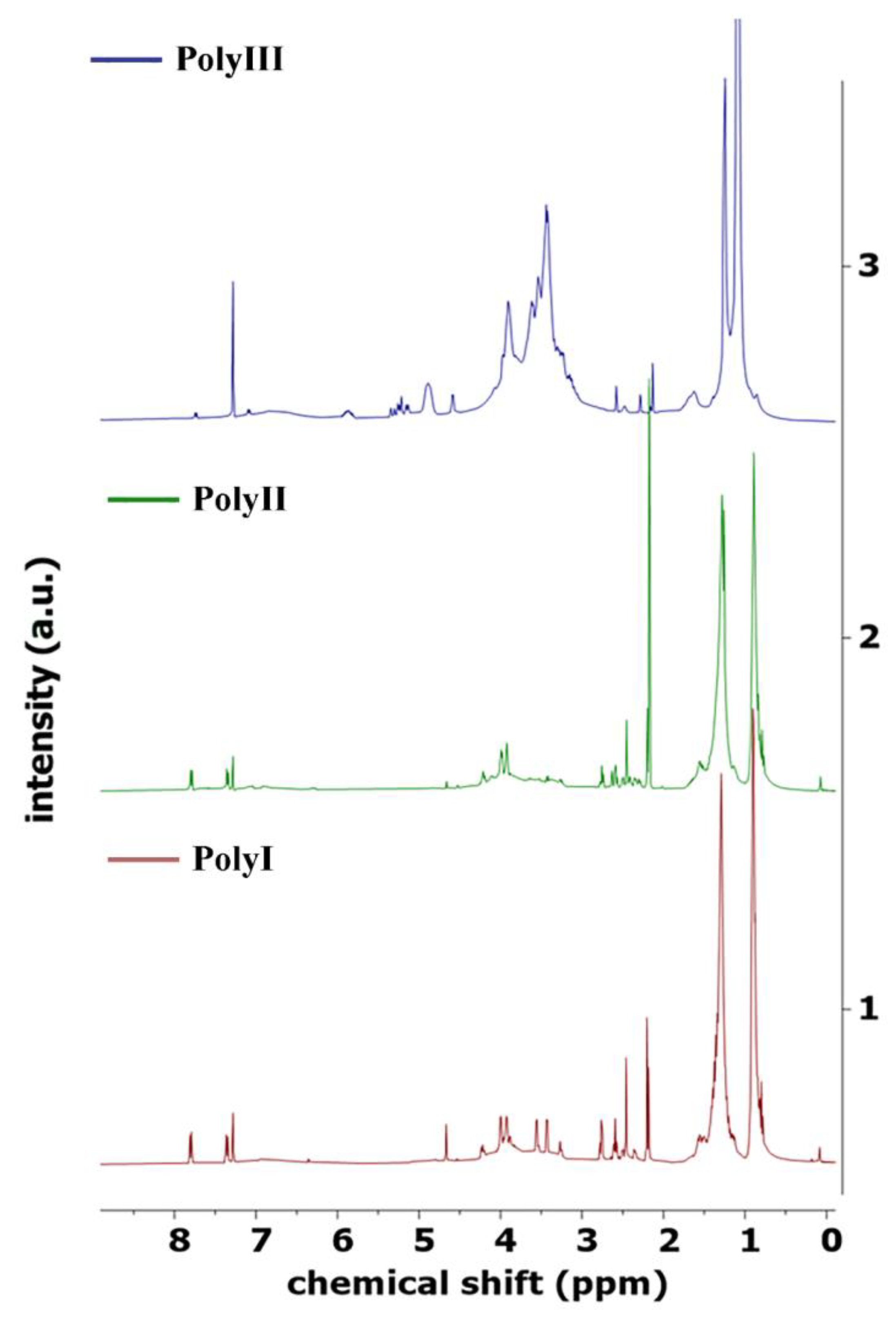
Using the above procedures, two distinct biopolyols, named PolyI and PolyII, were produced using 2EH as the solvent for thermochemical liquefaction, and pinewood and Stipa as biomasses, respectively. A third biopolyol, hereafter named PolyIII, was produced using pinewood biomass and a green solvent PG to replace the typically used 2EH solvent in the liquefaction process (
Table 1).
2.3. BioPolyurethane (BioPU) Synthesis and Coating Preparation
BioPUs were prepared by a one-pot method, where the biopolyol is mixed with the bio-based aliphatic polyisocyanate Desmodur® Eco N7300 (hereafter referred to as Eco N7300) at a 0.9 ratio of isocyanate (NCO) to hydroxyl (OH) value of polyol.
The polyol was dissolved in the green solvent 2-MTHF (50:50 wt.%), to allow for a good homogeneity of the reactants. The Eco N 7300 was added to the mixture, which was sonicated for 15 min at room temperature. Three distinct BioPU coatings with around 100 µm thickness, hereafter called BPUI, BPUII, and BPUIII, were manually deposited by the bar coating method on rectangular carbon steel substrates (surface area ~20 cm2) and allowed to dry at 50 °C over a period of at least 72 h. The substrates had been previously washed and sonicated in ethanol and acetone for 15 min each and dried with hot air.
2.4. Characterization Techniques
2.4.1. Biopolyols’ Acid Value (AV) and Hydroxyl Value (OHV)
The acid value (AV) was determined by the method based on ASTM D 947 and DIN 51558 standards, and 1 g of polyol (titrand) dissolved in 50 mL of THF was titrated with a 0.1 N KOH solution (titrant) with a phenolphthalein indicator.
Hydroxyl value (OHV) was determined by titration method based on ASTM D 1957 and ASTM E222-10 standards. Approximately 1 g of polyol was dissolved in 50 mL of tetrahydrofuran (THF) and acetylated by 10 mL of 12.5 wt.% acetic anhydride solution in THF for 10 min under stirring. After this, 2 mL of water was added to react with the remaining excess of acetic anhydride for 30 min, stopping the acetylation process. The OH groups turned acetic acid, with a thymolphthalein indicator, were titrated using a 0.5 N KOH solution (titrant). A blank solution following the same procedure, without adding titrand, was prepared at the same time.
After the determination of the turning point of the titrations, the following formulas were applied:
where AV is the Acid value (mg KOH g
−1) and the OHV is the Hydroxyl value (mg KOH g
−1). The
[C]titrant is the concentration of titrant (N, normality) utilized and
MMKOH is the molecular mass of KOH (56.1 g mol
−1). The
Vblank is the quantity of titrant used in the titration of the blank solution (mL) and
VKOH is the quantity of titrant used in the titration (mL). The
mtitrand is the mass of the titrated sample weighted (g).
2.4.2. Physico-Chemical Characterization
The elemental composition of the biomass feedstock and respective polyols prepared were analyzed concerning their carbon (
C), hydrogen (
H), nitrogen (
N), and sulfur (
S) content, by an EMA 502 Elemental Micro Analyzer (VELP Scientifica Srl, Usmate Velate, Italy). The oxygen (
O) content was estimated according to (Equation (4)):
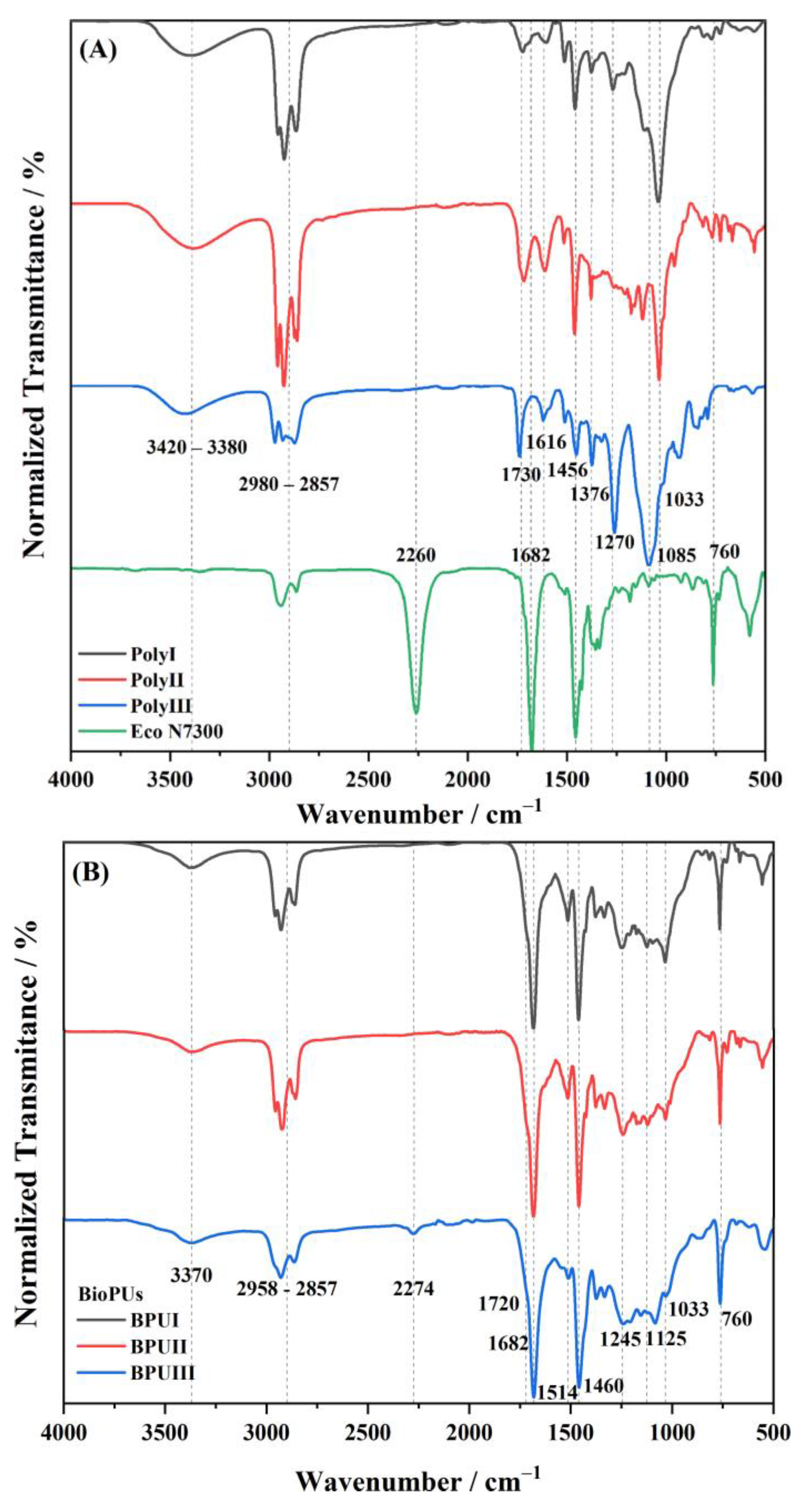
An Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR) analysis of the polyols and BioPU were performed in a Spectrum Two from PerkinElmer equipped with a UATR accessory (PerkinElmer Inc., Waltham, MA, USA). The spectra were obtained using 32 scans of data accumulation at a resolution of 4 cm−1, in a wavenumber range of 4000 to 400 cm−1.
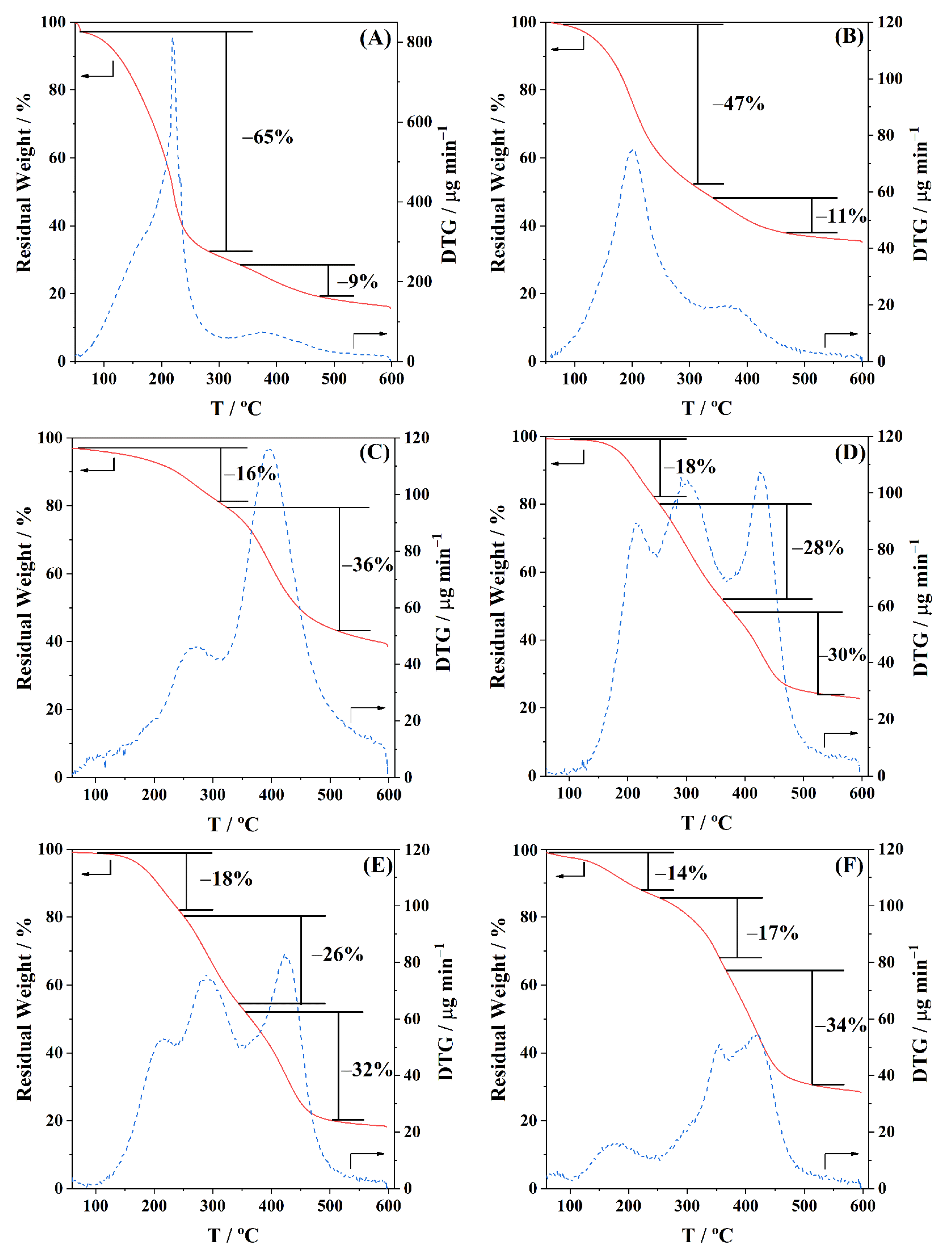
The thermal stability of the polyols and BioPU was analyzed by thermogravimetric analysis (TGA), using aluminium (Al) pans (ø 5.2 mm, h 2.5 mm), performed on a Hitachi-STA7200 (Hitachi High-Tech Corporation, Tokyo, Japan), from room temperature to 600 °C, at a heating rate of 10 °C min−1, under a 100 mL min−1 flow rate of nitrogen (N2; ≥99.99 wt.%) gas.
The 1H NMR spectra of the biopolyols were acquired in a Bruker AVANCE III RNM (Bruker Inc., Billerica, MA, USA), running at 400 MHz, with deuterated chloroform as solvent.
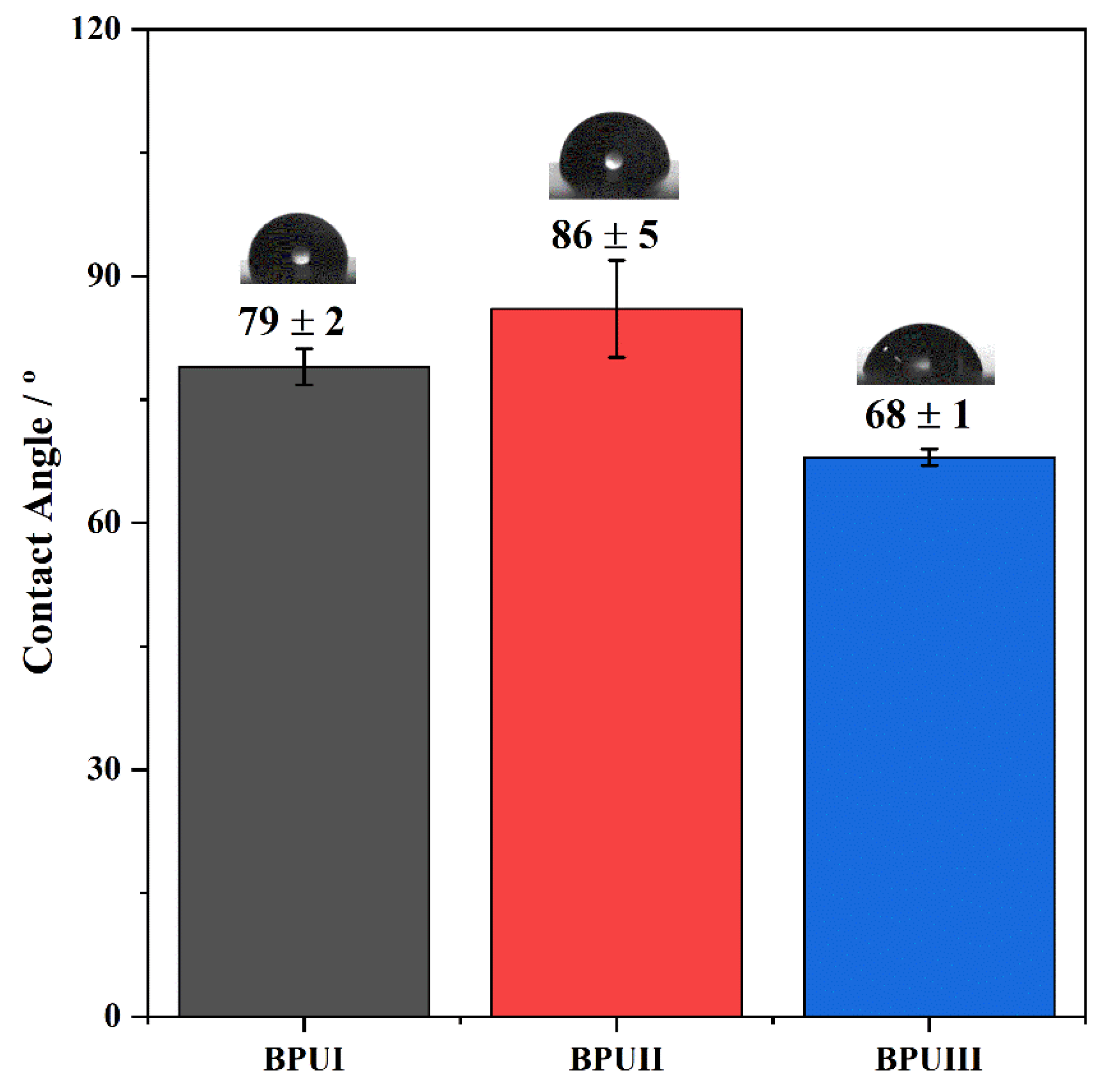
The hydrophobicity of the coatings was evaluated by measuring the optical contact angle (OCA) of water on dry BioPU samples through the sessile drop method. Static OCA measurements on the BioPU films were performed using a JAI CV-A50 (JAI Ltd., Copenhagen, Denmark) video camera attached to a Wild M3Z (Leica Microsystems, Wetzlar, Germany) microscope connected to a Data Translation DT3155 frame grabber and a micrometric syringe to dispense Millipore water droplets (2 to 3 μL) at room temperature. The acquisition and analysis of the images were performed using ADSA-P software (Axisymmetric Drop Shape Analysis Profile). A minimum of 10 measurements were taken in different regions of the coating surface.
Adhesion tests based on ASTM D4541 were performed on the BioPU coatings on carbon steel substrate (surface area ca. 20 cm2) by employing a PosiTest AT-A adhesion pull-off tester (DeFelsko Corporation, Ogdensburg, NY, USA). The tester measures the pulling force needed to detach a 20 mm diameter aluminium dolly adhered to the PU film, employing epoxy adhesive.
The Shore hardness of the biopolyurethanes was manually measured by X.F Shore A and D durometers, following the standard ASTM 2240. A minimum of 10 measurements were made, for about 3 s each, at room temperature.
Swelling tests were conducted on samples of BioPU blocks of ca. 100 mg in weight, in both solvents—Millipore water and DMF. The samples were weighed and submerged in individual containers with 20 mL of solvent at room temperature. The samples were taken out, dried on a paper towel, and weighed periodically over the course of 72 h. The swelling rate (
SR) (Equation (5)) was calculated from the weight of the dried polymer (
) and swollen polymer (
).
The density of each polymer was estimated by the water displacement method at room temperature.
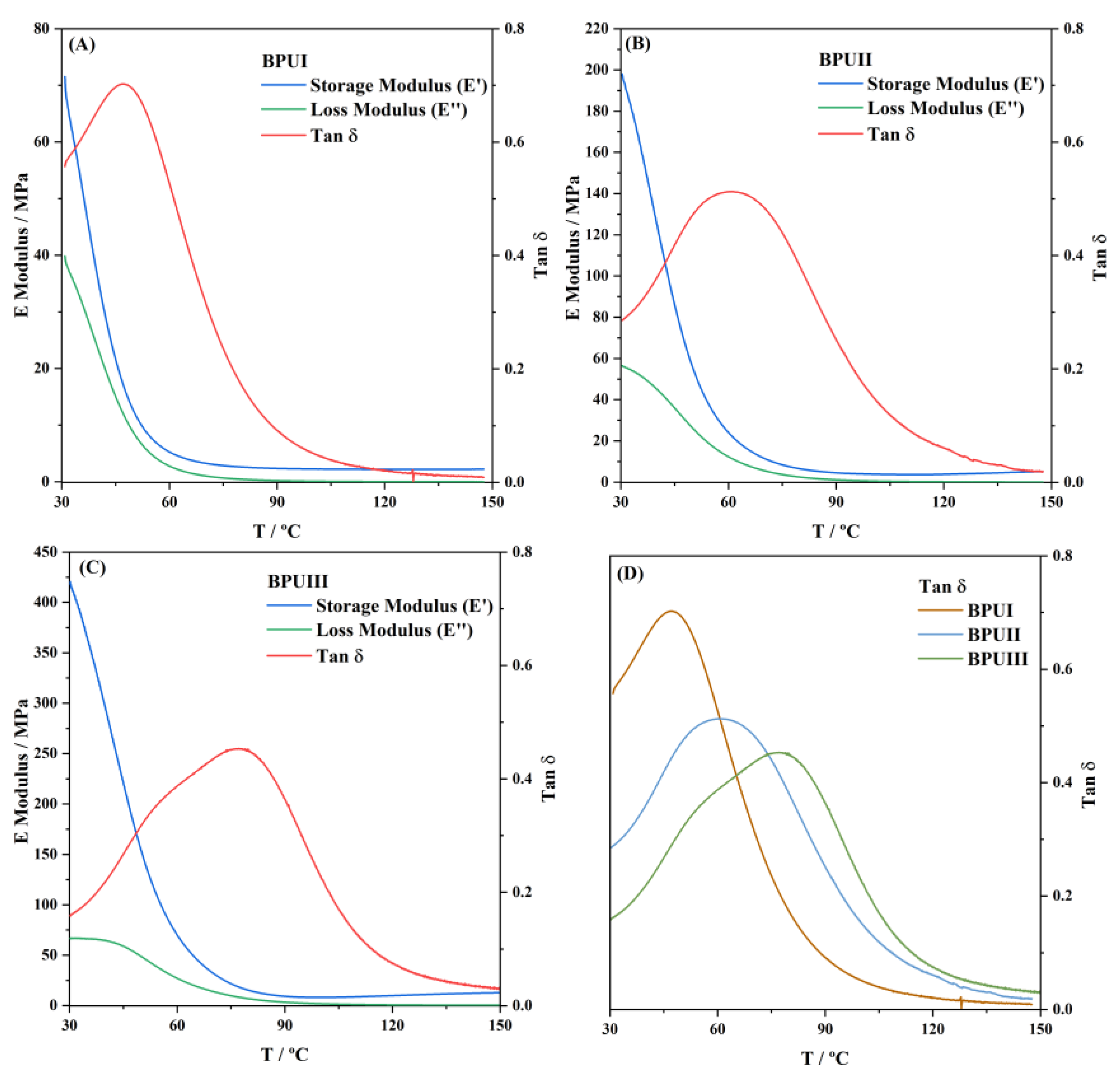
Dynamic mechanical analyses (DMA) were carried out on a DMA Q800 device (TA Instruments, New Castle, DE, USA). Multi-frequency strain of samples (20 × 14 × 3 mm) was performed with a heating rate of 2 °C·min
−1 from 30 to 150 °C, operating at a frequency of 1 Hz and an amplitude of 15 µm. The cross-linking density (
νx) of all the BioPUs was estimated according to the kinetic theory of rubber elasticity (Equation (6)) [
57]. The molecular weight of the chain between cross-linking points (
Mx), inversely proportional to
, was calculated according to Equation (7):
where
T′ is the temperature of samples in the rubbery plateau region at Tg + 50 °C, and
G′
T′ is the corresponding storage modulus at
T′.
R is the ideal gas constant (8.314 m
3 Pa K
−1 mol
−1).
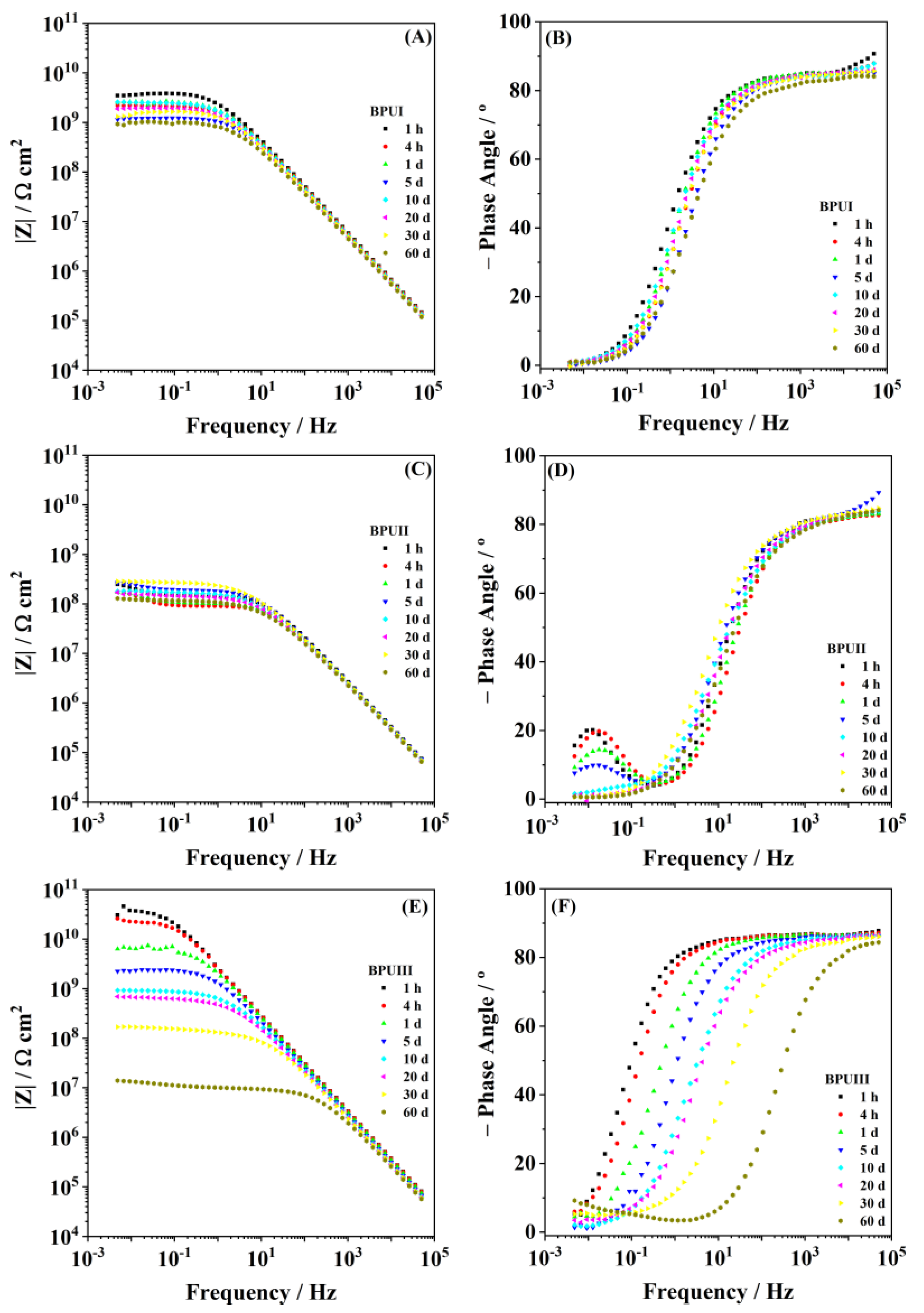
2.4.3. Electrochemical Impedance Spectroscopy (EIS)
Electrochemical impedance spectroscopy (EIS) was used to study the barrier properties of the coating applied on steel immersed in NaCl 0.05 M. A Gamry potentiostat Reference REF 600 (Gamry Instruments, Pennsylvania, USA) apparatus was used for the measurements, carried out on a conventional three-electrode cell configuration inside a Faraday cage. A saturated calomel electrode (SCE; model HI5412, Hanna Instruments, Woonsocket, RI, USA) was used as the reference electrode, a platinum coil as the counter electrode, and the coated DC01 carbon steel, with a total exposed area of around 3.46 cm2, was used as the working electrode. The EIS measurements were carried out at open circuit potential under a 10 mV (r.m.s.) sinusoidal perturbation and in the frequency range from 50 kHz to 5 mHz. ZView® software (ScribnerAssociates, Southern Pines, NC, USA) was used to treat the obtained EIS data.
4. Conclusions
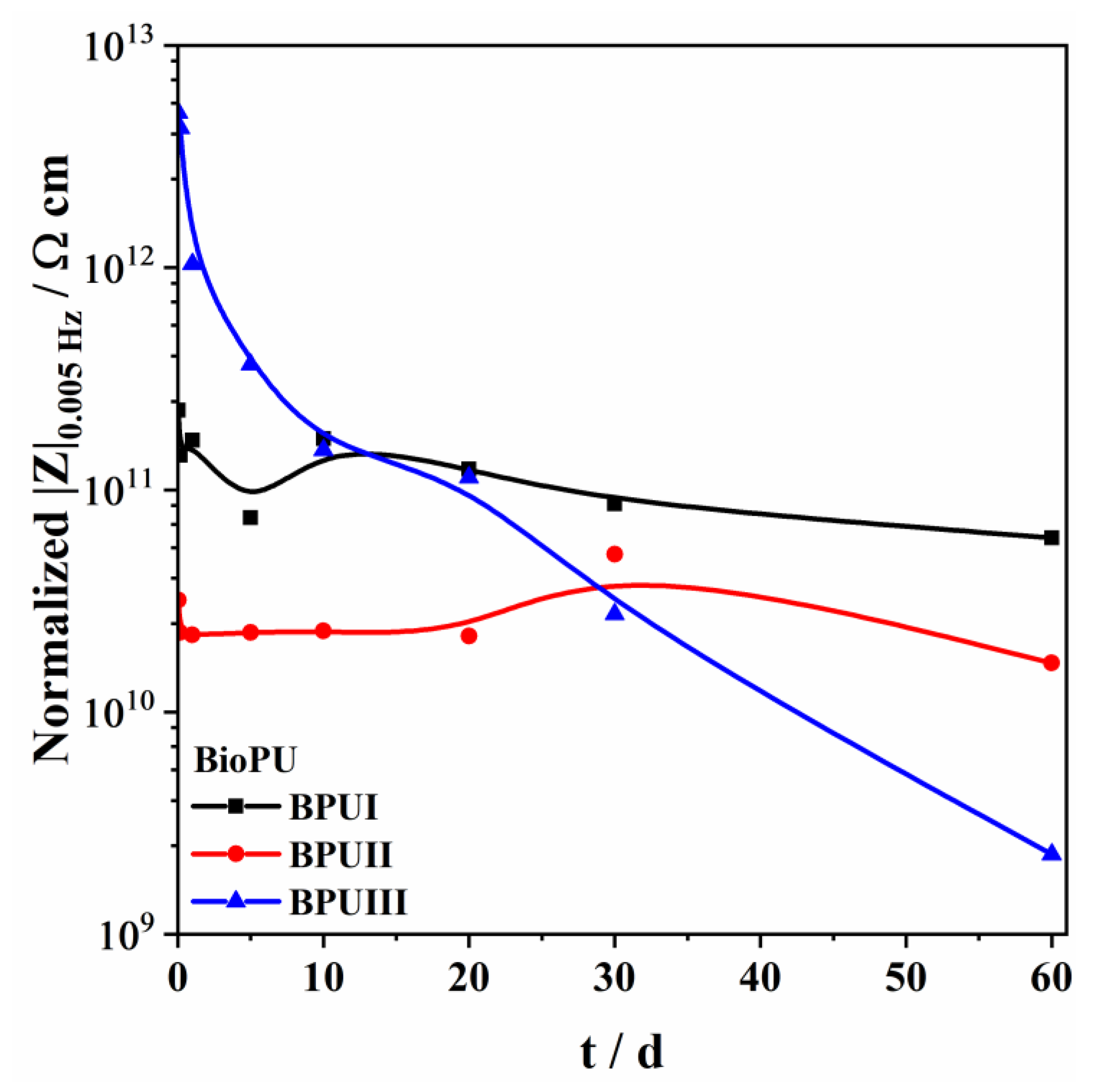
Three bio-based polyols were prepared from the thermochemical liquefaction of two biomass feedstocks, namely pinewood and Stipa tenacissima. The thermochemical liquefaction process resulted in a conversion from biomass to biopolyol from 72 to 79 wt.%, depending on the biomass feedstock and solvents employed. Pinewood biomass and propylene glycol solvent resulted in larger conversion rates to biopolyol (PolyIII), and larger OHV (160 mg KOH g−1). The polyols were also characterized by elemental analysis (EA), NMR analysis, ATR-FTIR analysis, and TGA, revealing the presence of phenolic and aliphatic compounds with hydroxyl (OH) functional groups. Biopolyols obtained when 2-ethyl hexanol was employed as the solvent (PolyI and PolyII) have similar chemical structures, despite the difference in the initial feedstock. According to the TGA results, the biopolyol from pinewood and propylene glycol, PolyIII, may possess lignin derivatives with higher molecular weight, or more cross-linked structures, since it exhibits higher thermal resistance and char residue at 600 °C. The polyols obtained were considered suitable for the preparation of BioPU coatings. Hydrophobic and good quality coatings were produced from the reaction of each of the developed polyols with a bio-based polyisocyanate (Desmodur® Eco N7300), in an NCO/OH ratio of 0.9, and manually deposited by bar coating on carbon steel substrates. The formation of urethane moieties and the complete or extensive reaction of isocyanate species was confirmed by ATR-FTIR spectroscopy. Noteworthily, the coating prepared with PolyIII (BPUIII) still reveals the presence of free isocyanate (NCO) species, which may indicate some limited reactivity between the OH and NCO species. This coating also exhibits lower adhesive strength, in line with a higher cross-linking density and hardness. On the other hand, BioPU coatings derived from PolyI and PolyII (BioPUI and BioPUII) are the ones with higher adhesive strength, up to 2.2 MPa, indicating relatively good compatibility with the carbon steel substrate. Regarding corrosion resistance, assessed by electrochemical impedance spectroscopy (EIS), all coatings display an impedance modulus in the low-frequency region—|Z|0.005 Hz, above 1 × 107 Ω cm2—with BPUI, BPUII, and BPUIII presenting values in the order of 9.3 × 108, 1.3 × 108, and 1.4 × 107 Ω cm2, respectively, after a period of 60 days in 0.05 M NaCl solution. The most effective coating is the BPUI since no signs of corrosion onset could be observed in the phase angle plot.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









