

Magnetic Resonance-Based Analytical Tools to Study Polyvinylpyrrolidone–Hydroxyapatite Composites

 , ,
, , 
 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
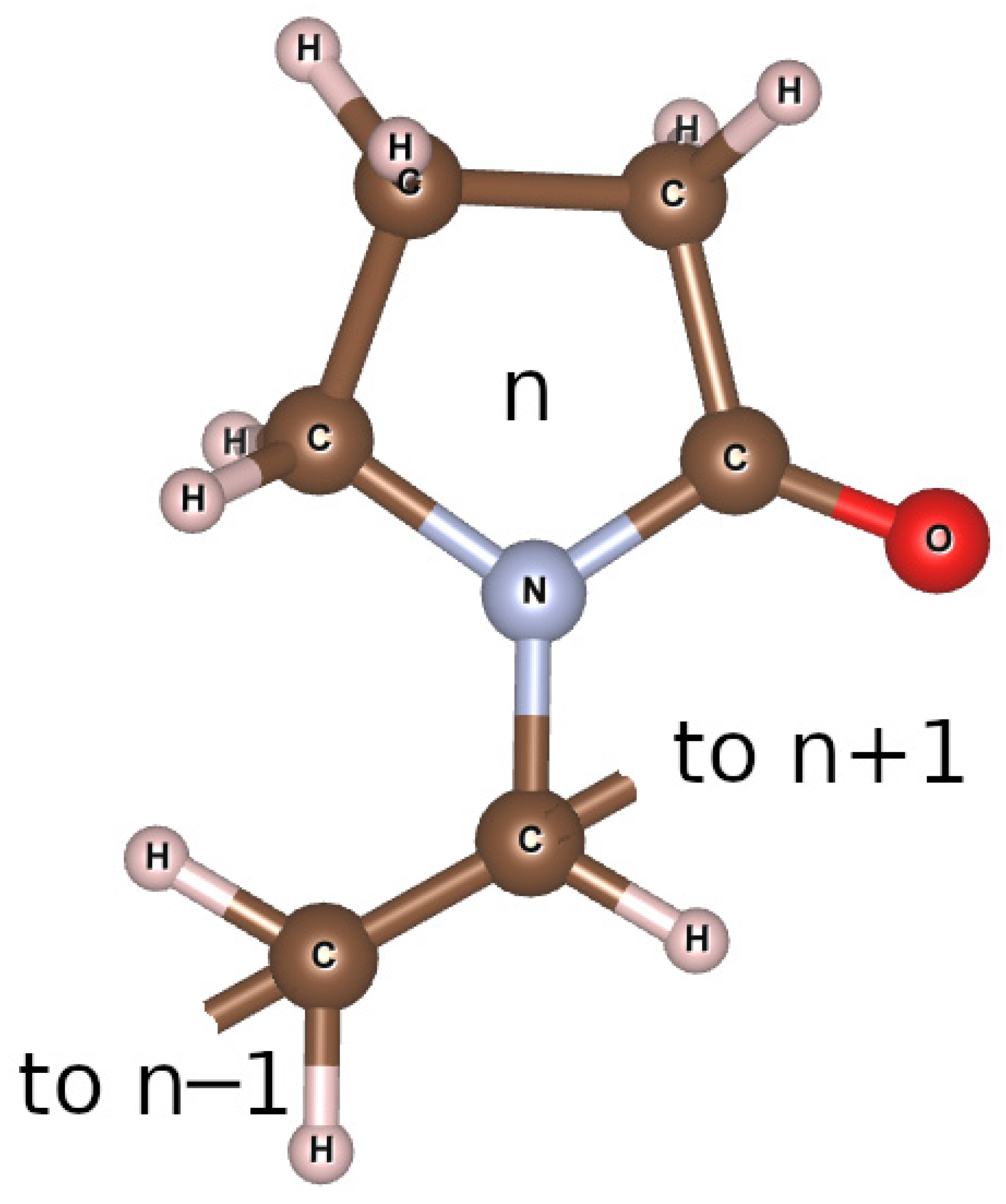
2.1. Synthesis of Composite Materials Based on Polyvinylpyrrolidone with Hydroxyapatite
2.2. EPR
2.3. Nuclear Magnetic Resonance
3. Results
3.1. SEM
3.2. EPR
3.3. NMR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Shifts of NMR Signals (ppm) | p1 | T2(1), ms | p2 | T2(2), ms | p3 | T2(3), ms |
|---|---|---|---|---|---|---|
| PVP-HA | ||||||
| 3 | 0.244 | 55.5 ± 0.5 | 0.189 | 4.2 ± 0.3 | 0.567 | 0.85 ± 0.05 |
4. Conclusions
- Using two microwave frequencies, the EPR parameters for the light- and radiation-induced paramagnetic centers in the PVP, HA and PVP-HA (such as components of g-factors and hyperfine constants A between electrons and 14N nuclei) were defined with high accuracy (Table 1). This shows the possibility of using EPR for the qualitative (synthesis, presence of impurities) and quantitative (concentration of impurities and defects) control of the initial materials (PVP, HA) and the final composite PVP-HA product.
- In the PVP-HA composite, the EPR spectra and electronic relaxation times of the radiation-induced paramagnetic centers were very close to those in HA. In the PVP-HA, the distribution of A components for the electron–14N interaction for NO32− radicals was larger than for the pure HA (Table 1). The PVP-HA composites did not contain light-induced radicals characteristic of PVP. This can be ascribed to the re-distribution of the electrical charges between PVP and HA. These results can be used for the control of the quality and success of the in situ synthesis of PVP-HA composites using the EPR techniques.
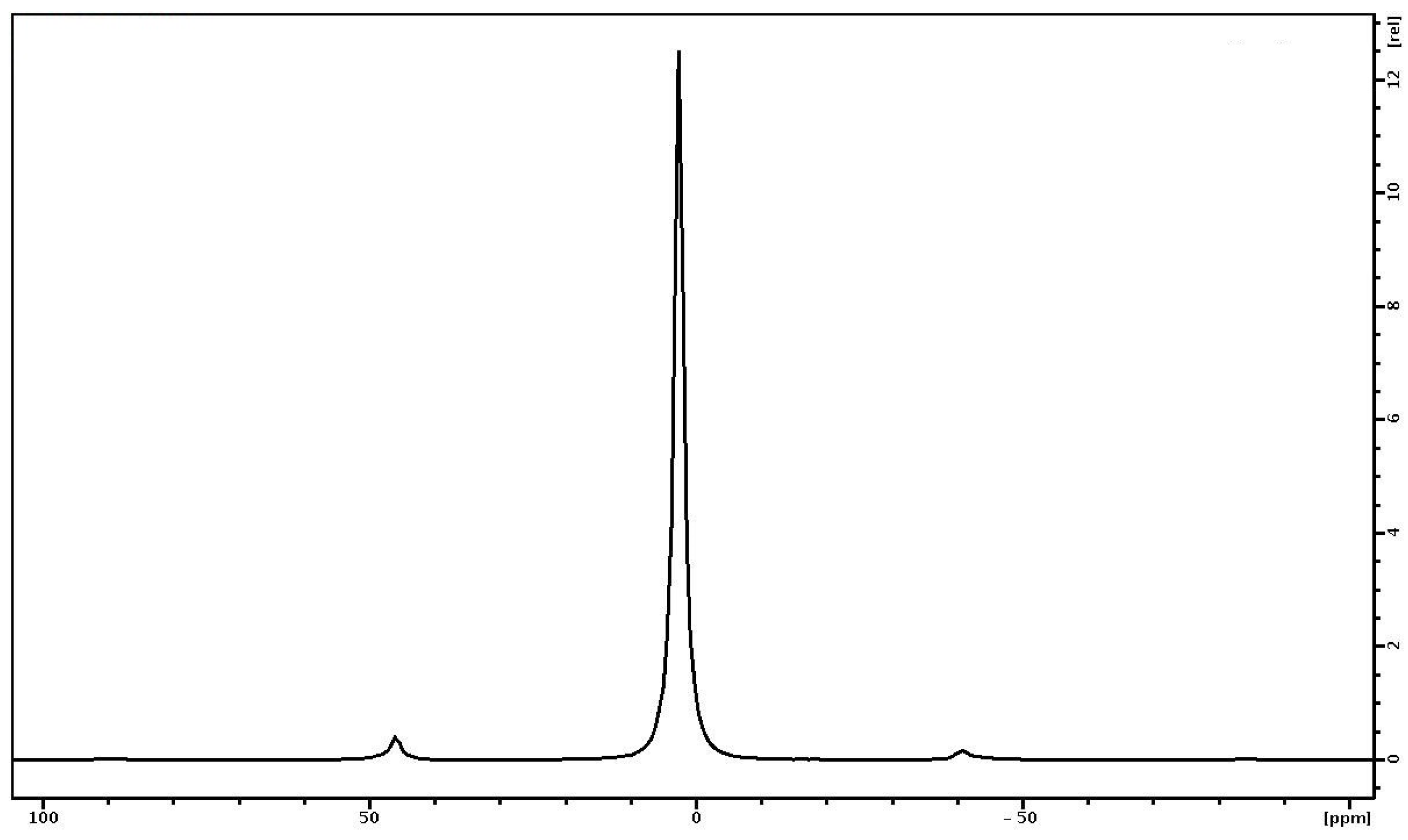
- In the 1H MAS NMR spectrum of the PVP-HA, the presence of two signals at 4.7 ppm and −2.15 ppm were attributed to “free” water and hydroxyl groups, and a single line attributed to 31P was registered.
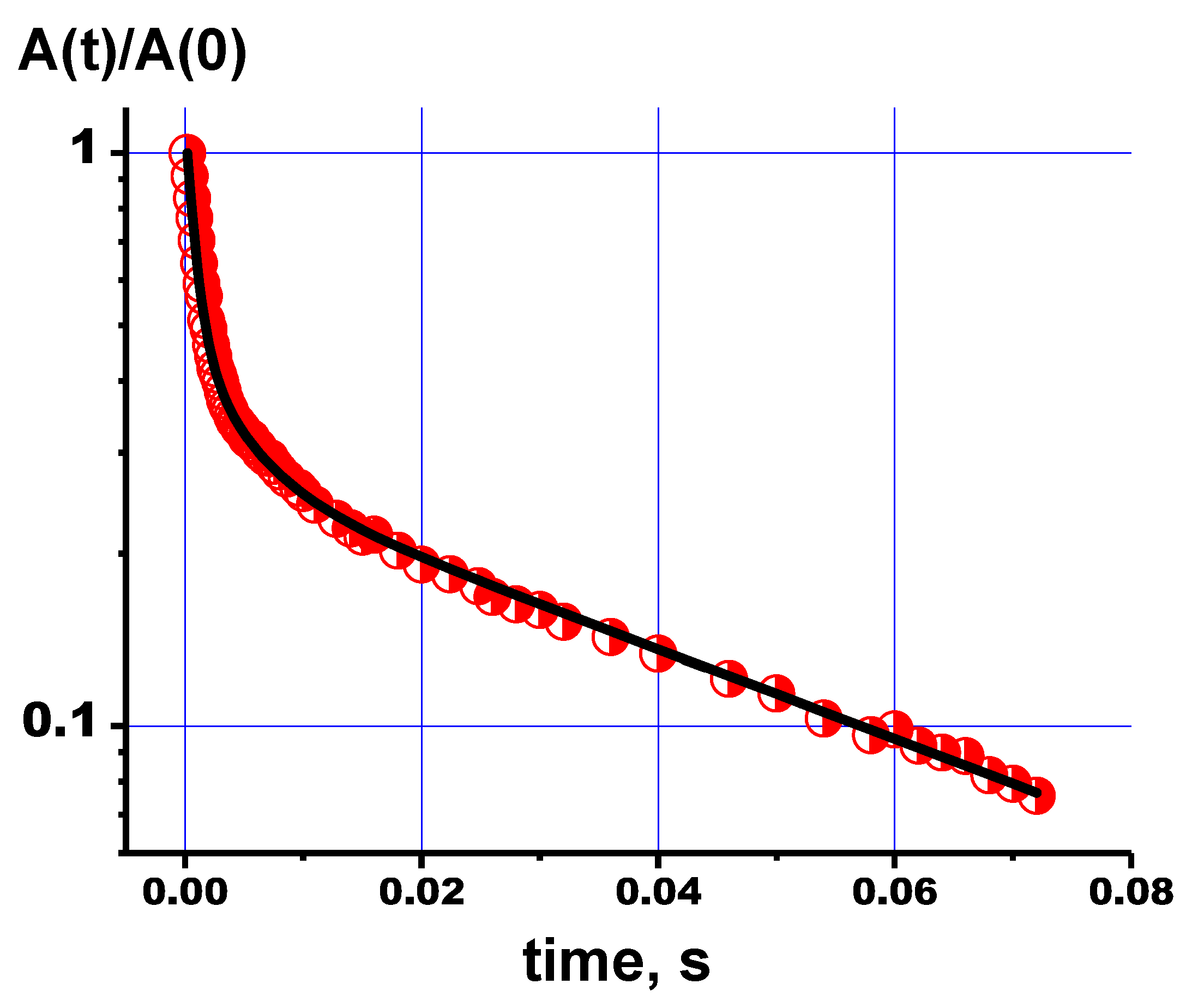
- The NMR relaxation measurements for 1H and 31P showed that the relaxation decays are multicomponent processes that can be described by three components of the transverse relaxation times. Multicomponent relaxation decay can be ascribed to the presence of defects in the HA lattice. The obtained data can serve as a basis for future NMR applications in clinical MRI for the evaluation of skeleton quality.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vallet-Regi, M. Ceramics for Medical Applications. J. Chem. Soc. Dalton Trans. 2001, 97–108. [Google Scholar] [CrossRef]
- Afshar, A.; Ghorbani, M.; Ehsani, N.; Saeri, M.R.; Sorrell, C.C. Some important factors in the wet precipitation process of hydroxyapatite. Mater. Des. 2003, 24, 197–202. [Google Scholar] [CrossRef]
- Riman, R.E.; Suchaned, W.L.; Byrappa, K.; Chen, C.W.; Shuk, P. Solution synthesis of hydroxyapatite designer particulates. Solid State Ion. 2002, 151, 393–402. [Google Scholar] [CrossRef]
- Chen, J.D.; Wang, Y.J.; Wei, K.; Zhang, S.H.; Shi, X.T. Self-organization of hydroxyapatite nanorods through oriented attachment. Biomaterials 2007, 28, 2275–2280. [Google Scholar] [CrossRef]
- Mobasherpour, I.; Heshajin, M.S.; Kazemzadeh, A.; Zakeri, M. Synthesis of nanocrystalline hydroxyapatite by using precipitation method. J. Alloys Compd. 2007, 430, 330–333. [Google Scholar] [CrossRef]
- Barinov, S.M. Calcium phosphate-based ceramic and composite materials for medicine. Russ. Chem. Rev. 2010, 79, 13. [Google Scholar] [CrossRef]
- Dziadek, M.; Stodolak-Zych, E.; Cholewa-Kowalska, K. Biodegradable ceramic-polymer composites for biomedical applications. A review. Mater. Sci. Eng. C 2017, 71, 1175–1191. [Google Scholar] [CrossRef]
- Vranceanu, M.D.; Saban, R.; Antoniac, I.; Albu, M.; Miculescu, F. Development and characterization of novel porous collagen based biocomposite for bone tissue regeneration. UPB Sci. Bull. Ser. B 2012, 74, 145–156. [Google Scholar]
- Armentano, I.; Dottori, M.; Fortunati, E.; Mattioli, S.; Kenny, J.M. Biodegradable polymer matrix nanocomposites for tissue engineering: A review. Polym. Degrad. Stab. 2010, 95, 2126–2146. [Google Scholar] [CrossRef]
- Nair, L.; Laurencin, C. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Mitra, I.; Bose, S. 3D printing for bone regeneration. Curr. Osteoporos. Rep. 2020, 18, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Lindner, M.; Zhang, W.; Koczur, K.; Kirsten, A.; Telle, R.; Fischer, H. 3D printing of bone substitute implants using calcium phosphate and bioactive glasses. J. Eur. Ceram. Soc. 2010, 30, 2563–2567. [Google Scholar] [CrossRef]
- Teodorescu, M.; Bercea, M. Poly (vinylpyrrolidone)—A versatile polymer for biomedical and beyond medical applications. Polym. Plast. Technol. Eng. 2015, 54, 923–943. [Google Scholar] [CrossRef]
- Haaf, F.; Sanner, A.; Straub, F. Polymers of N-vinylpyrrolidone: Synthesis, characterization and uses. Polym. J. 1985, 17, 143–152. [Google Scholar] [CrossRef]
- Anderson, C.C.; Rodriguez, F.; Thurston, D.A. Crosslinking aqueous poly(vinyl pyrrolidone) solutions by persulfate. J. Appl. Polym. Sci. 1979, 23, 2453–2462. [Google Scholar] [CrossRef]
- Raimi-Abraham, B.T.; Mahalingam, S.; Edirisinghe, M.; Craig, D.Q.M. Generation of poly (N-vinylpyrrolidone) nanofibres using pressurized gyration. Mater. Sci. Eng. C 2014, 39, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Morejón, L.; Mendizábal, E.; Delgado, J.A.; Davidenko, N.; López-Dellamary, F.; Manríquez, R.; Ginebra, M.P.; Gil, F.J.; Planell, J.A. Synthesis and characterization of poly (methyl methacrylate-styrene) copolymeric beads for bone cements. Lat. Am. Appl. Res. 2005, 35, 175–182. [Google Scholar]
- Zhang, Y.; Lu, J. A Mild and Efficient Biomimetic Synthesis of Rodlike Hydroxyapatite Particles with a High Aspect Ratio Using Polyvinylpyrrolidone as Capping Agent. Cryst. Growth Des. 2008, 8, 2101–2107. [Google Scholar] [CrossRef]
- Jemli, Y.E.; Abdelouahdi, K.; Minh, D.P.; Barakat, A.; Solhy, A. Synthesis and Characterization of Hydroxyapatite and Hydroxyapatite-Based Catalysts. In Design and Applications of Hydroxyapatite-Based Catalysts; Wiley: Hoboken, NJ, USA, 2022; pp. 19–72. [Google Scholar]
- Schöller, K.; Ethirajan, A.; Zeller, A.; Landfester, K. Biomimetic Route to Calcium Phosphate Coated Polymeric Nanoparticles: Influence of Different Functional Groups and pH. Macromol. Chem. Phys. 2011, 212, 1165–1175. [Google Scholar] [CrossRef]
- Langroudi, M.M.; Saravani, M.G.; Nouri, A. Surfactant-Assisted Synthesis of Polyvinylpyrrolidone-Hydroxyapatite Composites as a Bone Filler. J. Appl. Appl. Biomater. Funct. Mater. 2017, 15, 334–340. [Google Scholar] [CrossRef]
- Guesmi, Y.; Agougui, H.; Jabli, M.; Alsharabasy, A.M. Bioactive composites of hydroxyapatite/polyvinylpyrrolidone for bone regeneration applications. Chem. Eng. Commun. 2018, 206, 279–288. [Google Scholar] [CrossRef]
- Chen, J.; Peng, Q.; Peng, X.; Zhang, H.; Zen, H. Probing and manipulating noncovalent interactions in functional polymeric systems. Chem. Rev. 2022, 122, 14594–14678. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry. Science 1993, 260, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Noncovalent Interactions in Biochemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 3–17. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 2011; pp. 223–250. [Google Scholar]
- Murzakhanov, F.F.; Grishin, P.O.; Goldberg, M.A.; Yavkin, B.V.; Mamin, G.V.; Orlinskii, S.B.; Komlev, V.S. Radiation-induced stable radicals in calcium phosphates: Results of multifrequency EPR, EDNMR, ESEEM, and ENDOR studies. Appl. Sci. 2021, 11, 7727. [Google Scholar] [CrossRef]
- Shurtakova, D.V.; Yavkin, B.V.; Mamin, G.V.; Orlinskii, S.B.; Sirotinkin, V.P.; Fedotov, A.Y.; Shinkarev, A.; Antuzevics, A.; Smirnov, I.V.; Tovtin, V.I.; et al. X-ray diffraction and multifrequency EPR study of radiation-induced room temperature stable radicals in octacalcium phosphate. Radiat. Res. 2021, 195, 200–210. [Google Scholar] [CrossRef]
- Vorona, I.P.; Nosenko, V.V.; Baran, N.P.; Ishchenko, S.S.; Lemishko, S.V.; Zatovsky, I.V.; Strutynska, N.Y. EPR study of radiation-induced defects in carbonate-containing hydroxyapatite annealed at high temperature. Radiat. Meas. 2016, 87, 49–55. [Google Scholar] [CrossRef]
- Gilinskaya, L.G. Organic radicals in natural apatites according to EPR data: Potential genetic and paleoclimatic indicators. J. Struct. Chem. 2010, 51, 471–481. [Google Scholar] [CrossRef]
- Fisher, B.V.; Morgan, R.E.; Phillips, G.O.; Wardale, H.W. Radiation damage in calcium phosphates and collagen: An interpretation of ESR spectra. Radiat. Res. 1971, 46, 229–235. [Google Scholar] [CrossRef]
- Cevc, P.; Schara, M.; Ravnik, Č. Electron paramagnetic resonance study of irradiated tooth enamel. In Radiation Research; Radiation Research Society: Lawrence, UK, 1972; Volume 51, pp. 581–589. [Google Scholar]
- Vanhaelewyn, G.C.A.M.; Sadlo, J.; Matthys, P.F.A.E.; Callens, F.J. Comparative X-and Q-band EPR study of radiation-induced radicals in tooth enamel. Radiat. Res. 2002, 158, 615–625. [Google Scholar] [CrossRef]
- Vanhaelewyn, G.C.A.M.; Morent, R.A.; Callens, F.J.; Matthys, P.F.A.E. X-and Q-band electron paramagnetic resonance of CO2− in hydroxyapatite single crystals. Radiat. Res. 2000, 154, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Forysenkova, A.A.; Ivanova, V.A.; Fadeeva, I.V.; Mamin, G.V.; Rau, J. 1H NMR and EPR Spectroscopies Investigation of Alginate Cross-Linking by Divalent Ions. Materials 2023, 16, 2832. [Google Scholar] [CrossRef] [PubMed]
- Gervais, C.; Bonhomme, C.; Laurencin, D. Recent directions in the solid-state NMR study of synthetic and natural calcium phosphates. Solid State Nucl. Magn. Reson. 2020, 107, 101663. [Google Scholar] [CrossRef]
- Soldati, E.; Rossi, F.; Vicente, J.; Guenoun, D.; Pithioux, M.; Iotti, S.; Malucelli, E.; Bendahan, D. Survey of MRI usefulness for the clinical assessment of bone microstructure. Int. J. Mol. Sci. 2021, 22, 2509. [Google Scholar] [CrossRef] [PubMed]
- Jäger, C.; Welzel, T.; Meyer-Zaika, W. EppleM A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Manatunga, D.C.; de Silva, R.M.; Nalin de Silva, K.M.; de Silva, N.; Premalal EV, A. Metal and polymer-mediated synthesis of porous crystalline hydroxyapatite nanocomposites for environmental remediation. R. Soc. Open Sci. 2018, 5, 171557. [Google Scholar] [CrossRef]
- Hu, Y.-Y.; Liu, X.P.; Ma, X.; Rawal, A.; Prozorov, T.; Akinc, M.; Mallapragada, S.K.; Schmidt-Rohr, K. Biomimetic self-assembling copolymer− hydroxyapatite nanocomposites with the nanocrystal size controlled by citrate. Chem. Mater. 2011, 23, 2481–2490. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Trofimchuk, E.S.; Forysenkova, A.A.; Ahmed, A.I.; Gnezdilov, O.I.; Davydova, G.A.; Kozlova, S.G.; Antoniac, A.; Rau, J.V. Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications. Polymers 2021, 13, 3989. [Google Scholar] [CrossRef]
- Rai, R.K.; Sinha, N. Dehydration-induced structural changes in the collagen–hydroxyapatite interface in bone by high-resolution solid-state NMR spectroscopy. J. Phys. Chem. C 2011, 115, 14219–14227. [Google Scholar] [CrossRef]
- Gabbasov, B.; Gafurov, M.; Starshova, A.; Shurtakova, D.; Murzakhanov, F.; Mamin, G.; Orlinskii, S. Conventional, Pulsed and High-Field Electron Paramagnetic Reso¬nance for Studying Metal Impurities in Calcium Phosphates of Biogenic and Synthetic Origins. J. Magn. Magn. Mater. 2019, 470, 109–117. [Google Scholar] [CrossRef]
- Stoll, S. CW-EPR Spectral Simulations. Solid State Methods Enzymol. 2015, 563, 121–142. [Google Scholar]
- Vorona, I.P.; Ishchenko, S.S.; Baran, N.P.; Rudko, V.V.; Zatovskiĭ, I.V.; Gorodilova, N.A.; Povarchuk, V.Y. NO3 2-centers in synthetic hydroxyapatite. Phys. Solid State 2010, 52, 2364–2368. [Google Scholar] [CrossRef]
- Nosenko, V.V.; Vorona, I.P.; Ishchenko, S.S.; Baran, N.P.; Zatovsky, I.V.; Gorodilova, N.A.; Povarchuk, V.Y. Effect of pre-annealing on NO32-centers in synthetic hydroxyapatite. Radiat. Meas. 2012, 47, 970–973. [Google Scholar] [CrossRef]
- Fattibene, P.; Callens, F. EPR dosimetry with tooth enamel: A review. Appl. Radiat. Isot. 2010, 68, 2033–2116. [Google Scholar] [CrossRef] [PubMed]
- Radtsig, V.A. modern achievements in mechanical enzymative processing of plant raw materials. Khim. Fiz. 2004, 23, 70–109. (In Russian) [Google Scholar]
- Fadeeva, I.V.; Forysenkova, A.A.; Trofimchuk, E.S.; Gafurov, M.R.; Ahmed, A.I.; Davidova, G.A.; Antonova, O.S.; Barinov, S.M. Porous matrixes based on polyvinylpyrrolidone containing calcium phosphates for medical application. Russ. Chem. Bull. 2022, 71, 543–548. [Google Scholar] [CrossRef]
- Laurencin, D.; Almora-Barrios, N.; de Leeuw, N.H.; Gervais, C.; Bonhomme, C.; Mauri, F.; Chrzanowski, W.; Knowles, J.C.; Newport, R.J.; Wong, A.; et al. Magnesium incorporation into hydroxyapatite. Biomaterials 2011, 32, 1826–1837. [Google Scholar] [CrossRef]
- James, P.; Yesinowski Hellmut Eckert, J. Hydrogen Environments in Calcium Phosphates: NMR at High Spinning Speeds. Am. Chem. Soc. 1987, 109, 6274–6282. [Google Scholar]










| Sample | g⊥ | g|| | A⫠ (MHz) | A|| (MHz) | ΔA⫠ (MHz) | ΔA|| (MHz) |
|---|---|---|---|---|---|---|
| PVP | 2.0022 | 2.0026 | 38 ± 8 | 106 ± 10 | - | - |
| HA | 2.0011 | 2.0052 | 92.4 ± 0.5 | 186 ± 1 | 7 ± 1 | 12 ± 1 |
| PVP-HA | 2.0011 | 2.0051 | 93.5 ± 0.5 | 190 ± 2 | 13 ± 1 | 18 ± 1 |
| T1e (μs) | T2 (μs) | |
|---|---|---|
| HA | 28.5 | 3 |
| PVP-HA | 27.6 | 3.2 |
| Sample | 2.4 ppm | 4.7 ppm | −2.15 ppm |
|---|---|---|---|
| PVP | 0.67 ± 0.02 s−1 | - | - |
| PVP-HA | - | 0.49 ± 0.02 s−1 | 0.55 ± 0.02 s−1 |
| Chemical Shifts of NMR Signals (ppm) | p1 | T2(1), ms | p2 | T2(2), ms | p3 | T2(3), ms |
|---|---|---|---|---|---|---|
| PVP | ||||||
| 2.4 | 0.0015 | 29.6 ± 0.5 | 0.0067 | 3.6 ± 0.1 | 0.992 | 0.59 ± 0.02 |
| PVP-HA | ||||||
| 4.7 | 0.04 | 5.42 ± 0.2 | 0.18 | 1.67 ± 0.05 | 0.78 | 0.58 ± 0.02 |
| −2.15 | 0.346 | 29.2 ± 0.5 | 0.33 | 6.02 ± 0.05 | 0.334 | 0.46 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, A.; Mamin, G.; Gnezdilov, O.; Fadeeva, I.; Antonova, O.; Forysenkova, A.; Antoniac, I.V.; Rau, J.V.; Gafurov, M. Magnetic Resonance-Based Analytical Tools to Study Polyvinylpyrrolidone–Hydroxyapatite Composites. Polymers 2023, 15, 4445. https://doi.org/10.3390/polym15224445
Petrova A, Mamin G, Gnezdilov O, Fadeeva I, Antonova O, Forysenkova A, Antoniac IV, Rau JV, Gafurov M. Magnetic Resonance-Based Analytical Tools to Study Polyvinylpyrrolidone–Hydroxyapatite Composites. Polymers. 2023; 15(22):4445. https://doi.org/10.3390/polym15224445
Chicago/Turabian StylePetrova, Alina, Georgy Mamin, Oleg Gnezdilov, Inna Fadeeva, Olga Antonova, Anna Forysenkova, Iulian V. Antoniac, Julietta V. Rau, and Marat Gafurov. 2023. "Magnetic Resonance-Based Analytical Tools to Study Polyvinylpyrrolidone–Hydroxyapatite Composites" Polymers 15, no. 22: 4445. https://doi.org/10.3390/polym15224445
APA StylePetrova, A., Mamin, G., Gnezdilov, O., Fadeeva, I., Antonova, O., Forysenkova, A., Antoniac, I. V., Rau, J. V., & Gafurov, M. (2023). Magnetic Resonance-Based Analytical Tools to Study Polyvinylpyrrolidone–Hydroxyapatite Composites. Polymers, 15(22), 4445. https://doi.org/10.3390/polym15224445