
Addressing the Structural Organization of Silicone Alternatives in Formulations by Molecular Dynamics Simulations and a Novel Equilibration Protocol

 ,
, 

 and
and 
Abstract
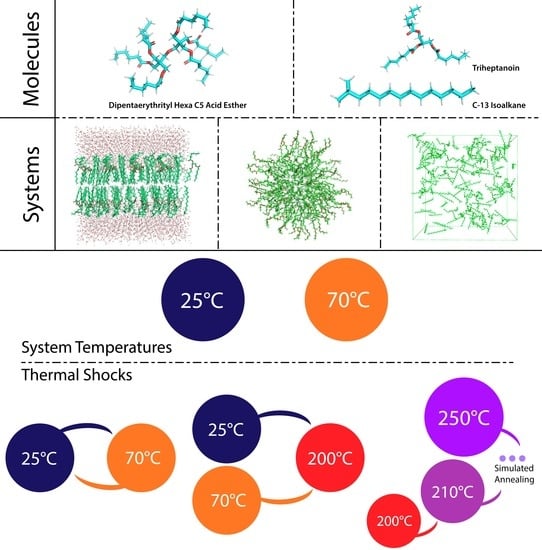
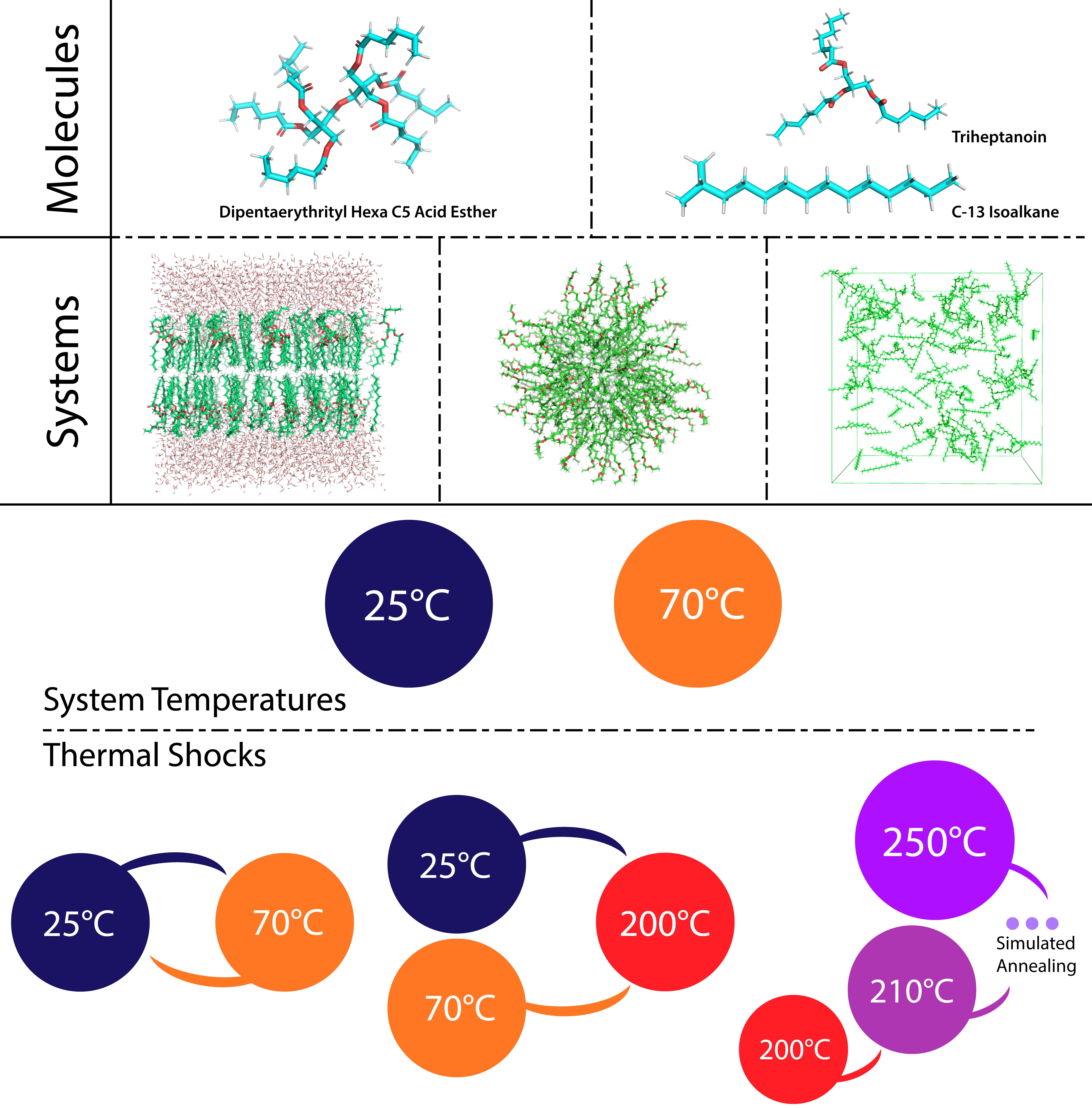
:
1. Introduction
2. Methodology
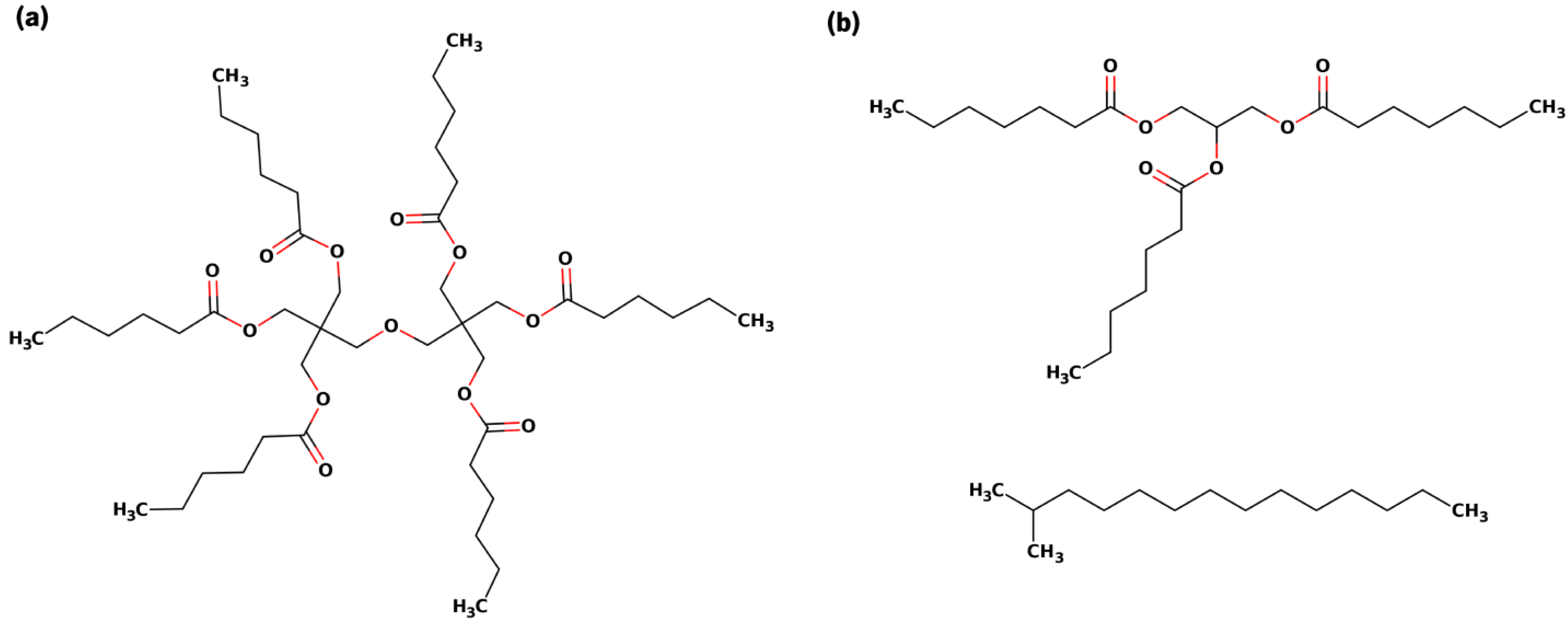
2.1. Molecules’ Design and Parametrization
2.2. Systems Design and Construction
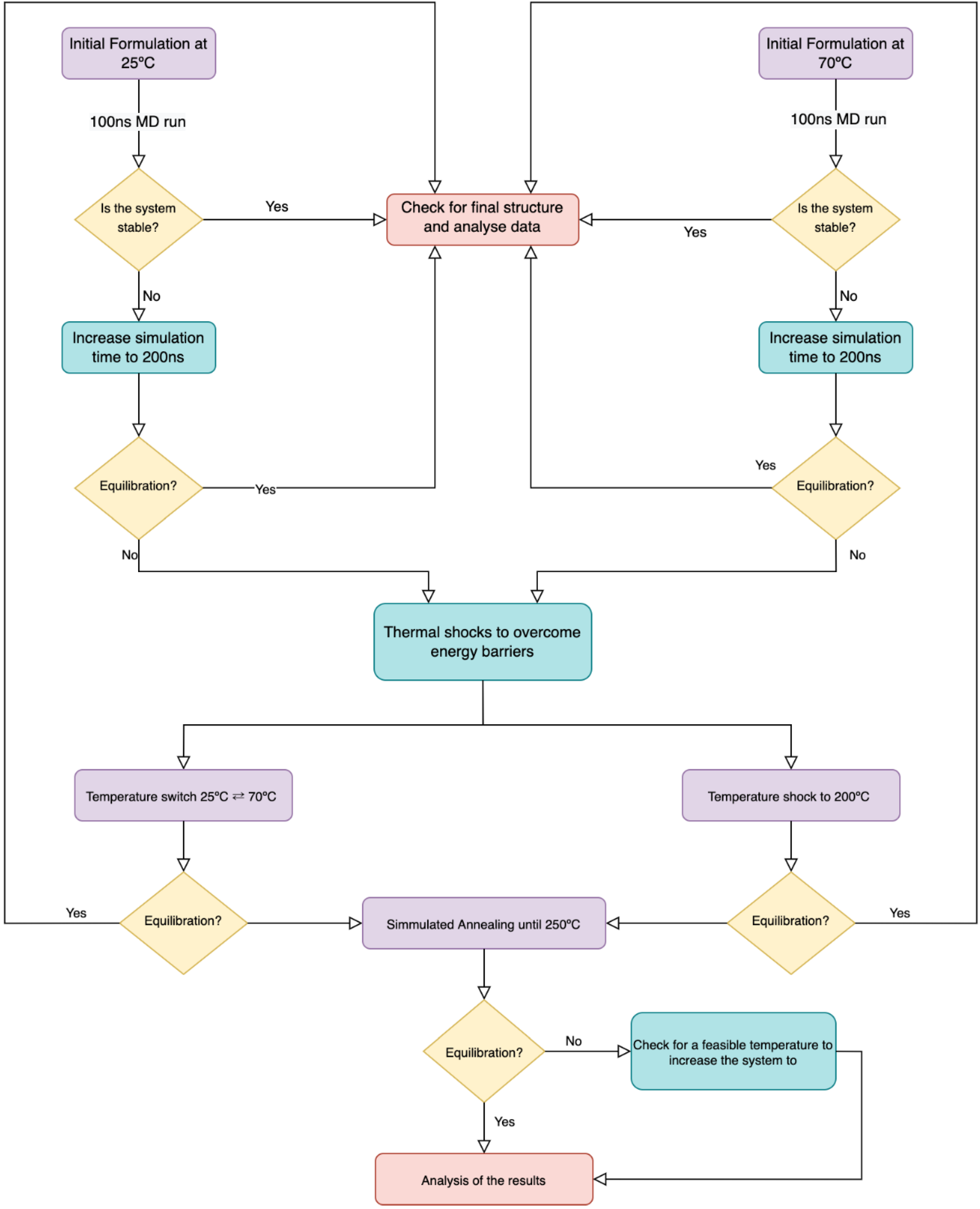
2.3. MD Simulations Options and Analysis
3. Results and Discussion
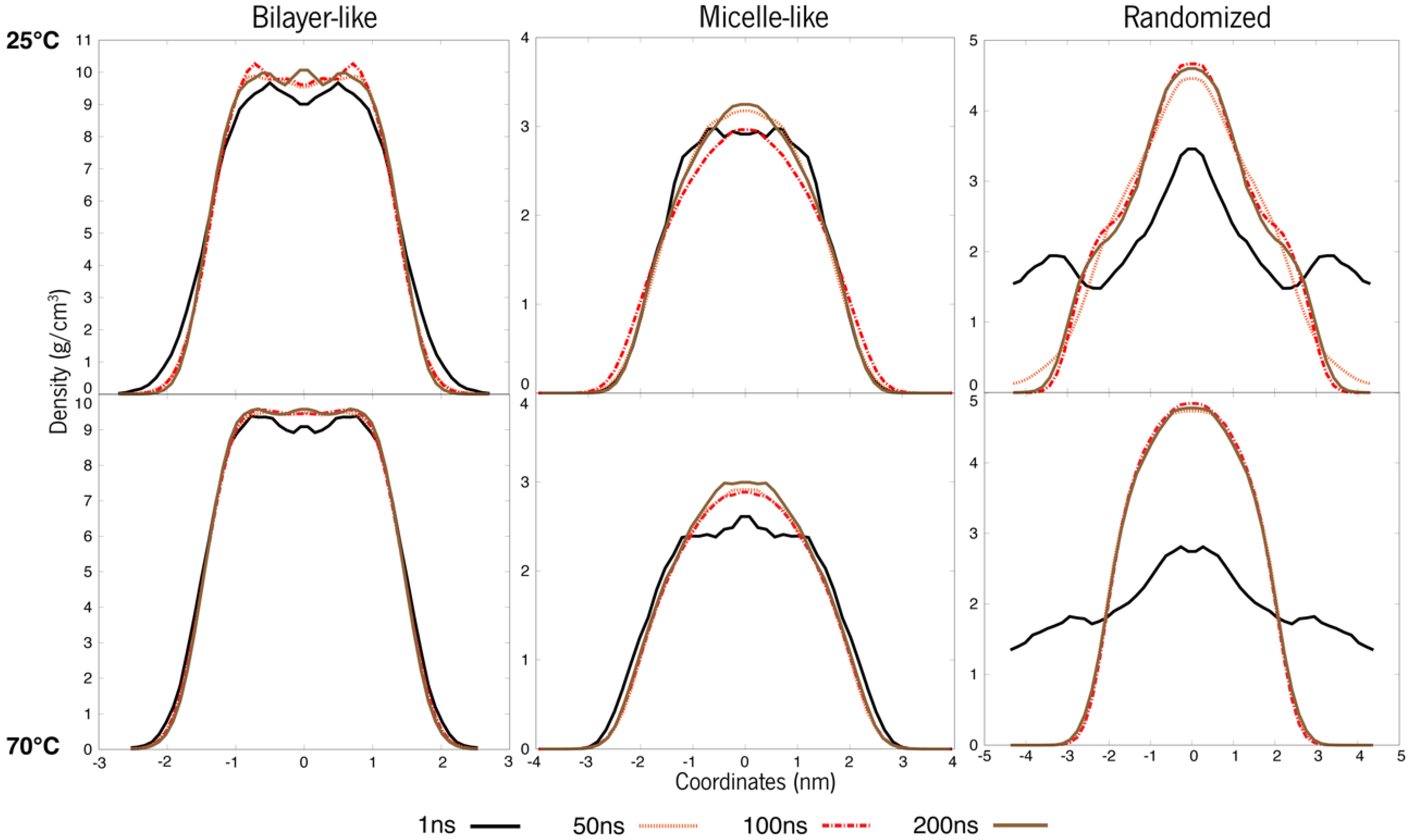
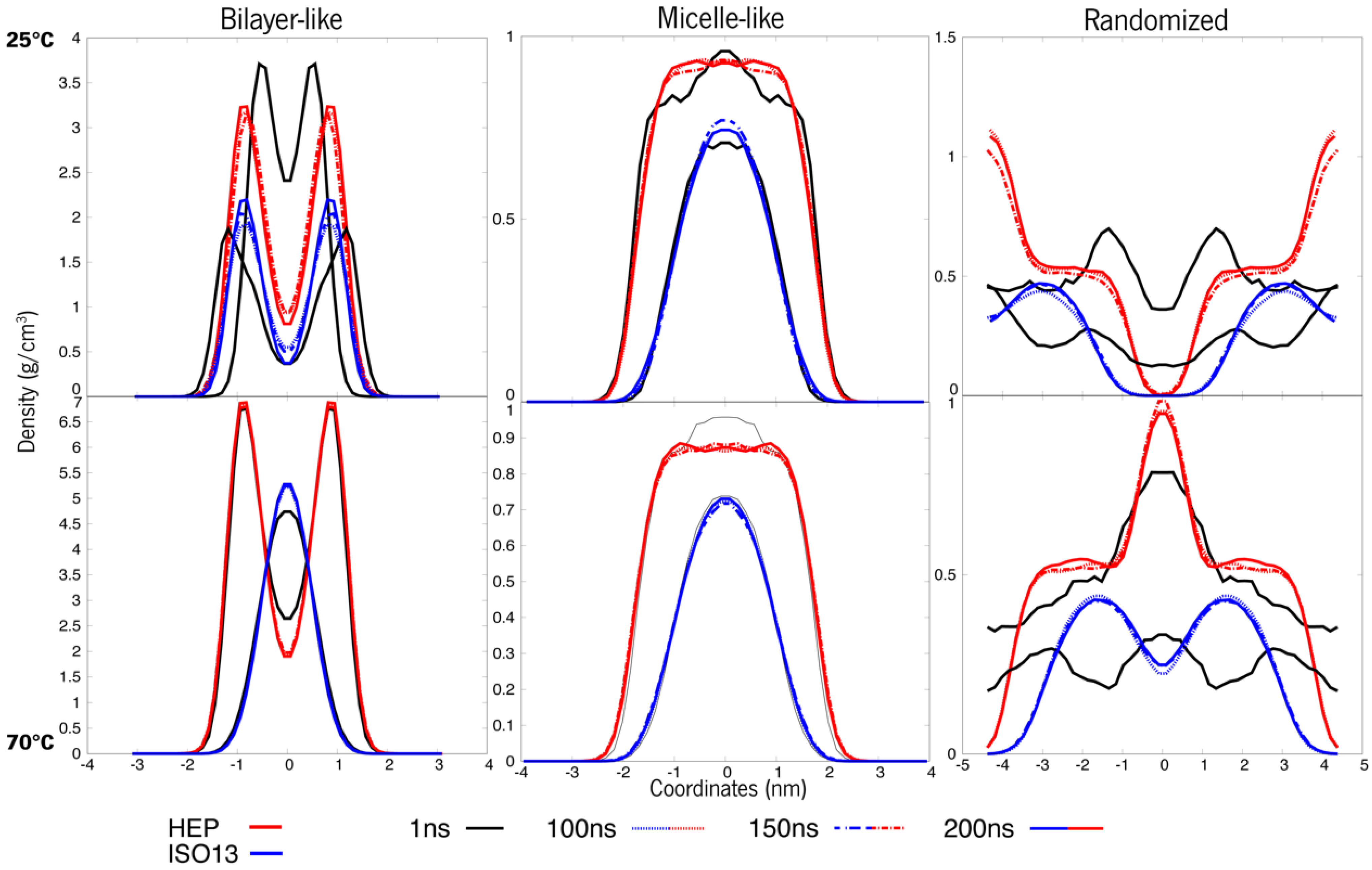
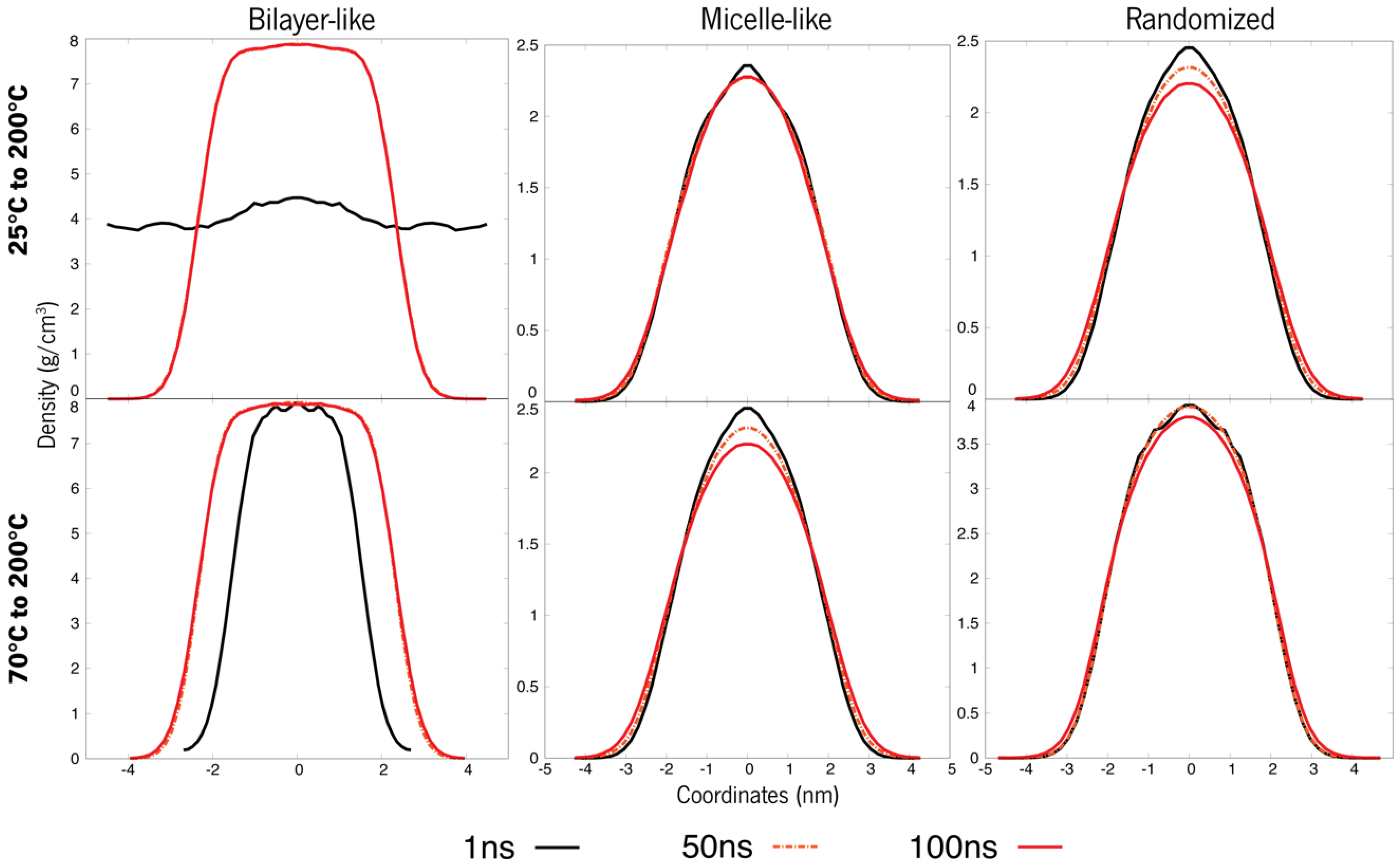
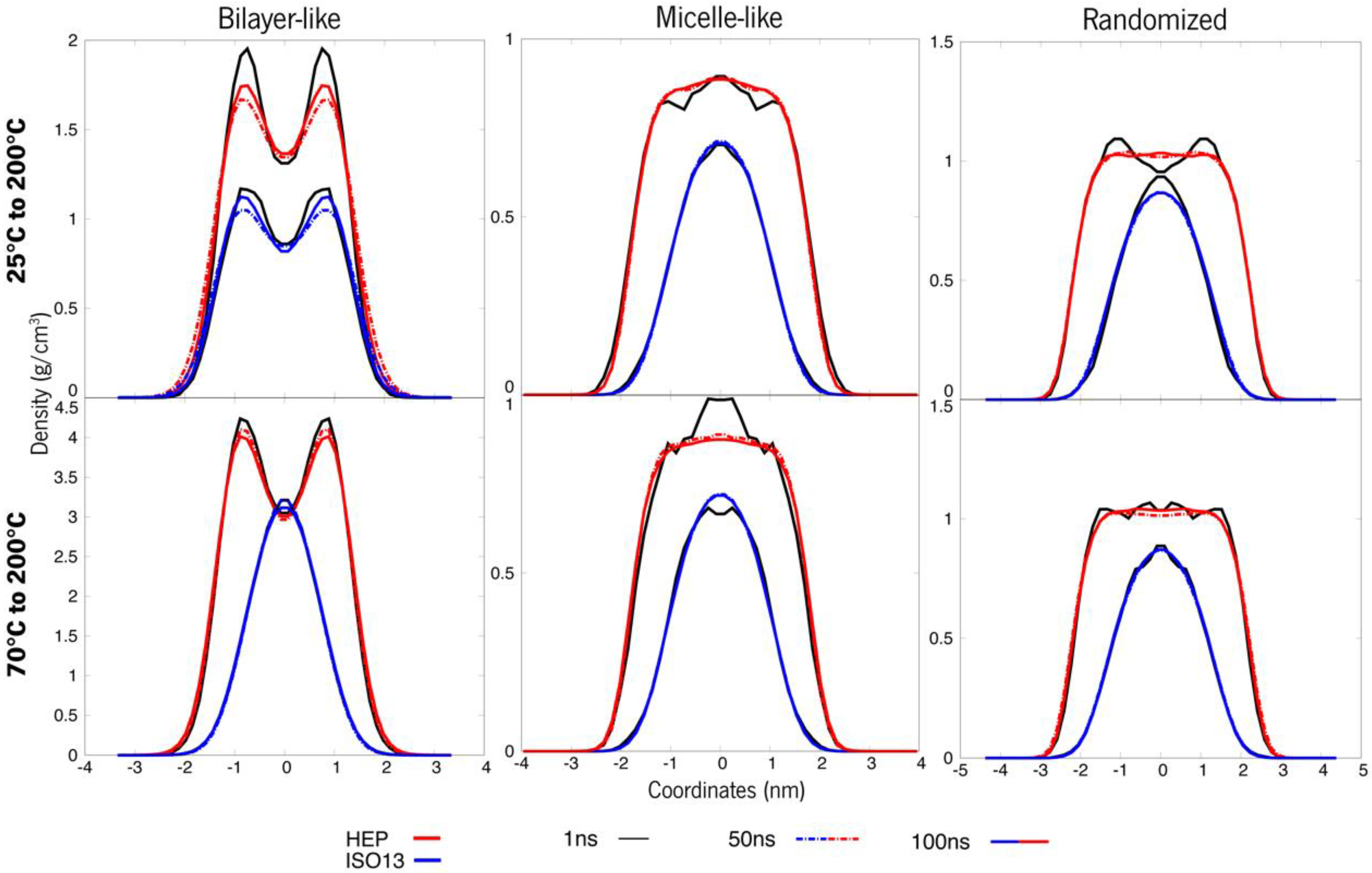
3.1. Systems at 25 °C and 70 °C
3.2. Temperature-Dependent Behaviour
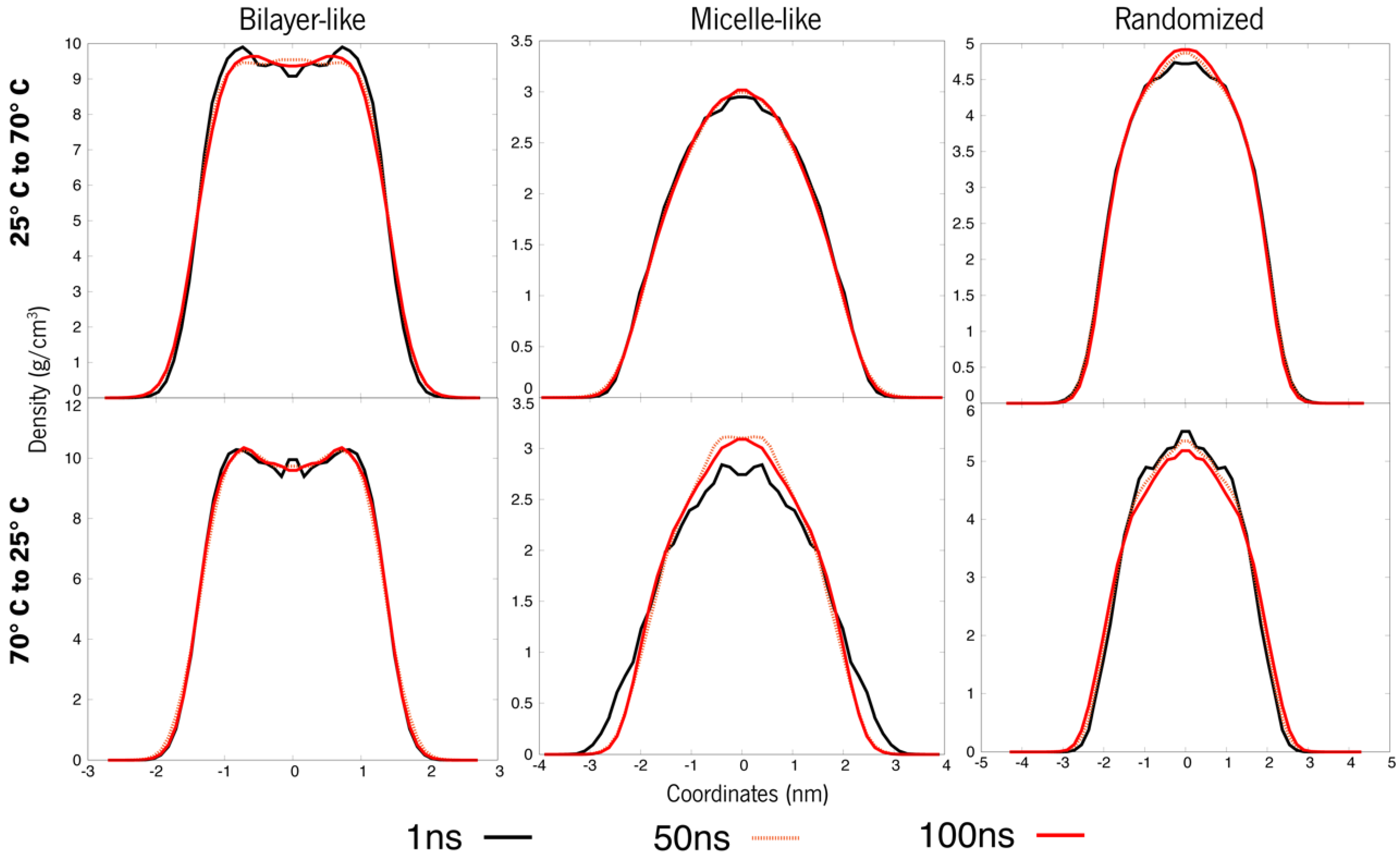
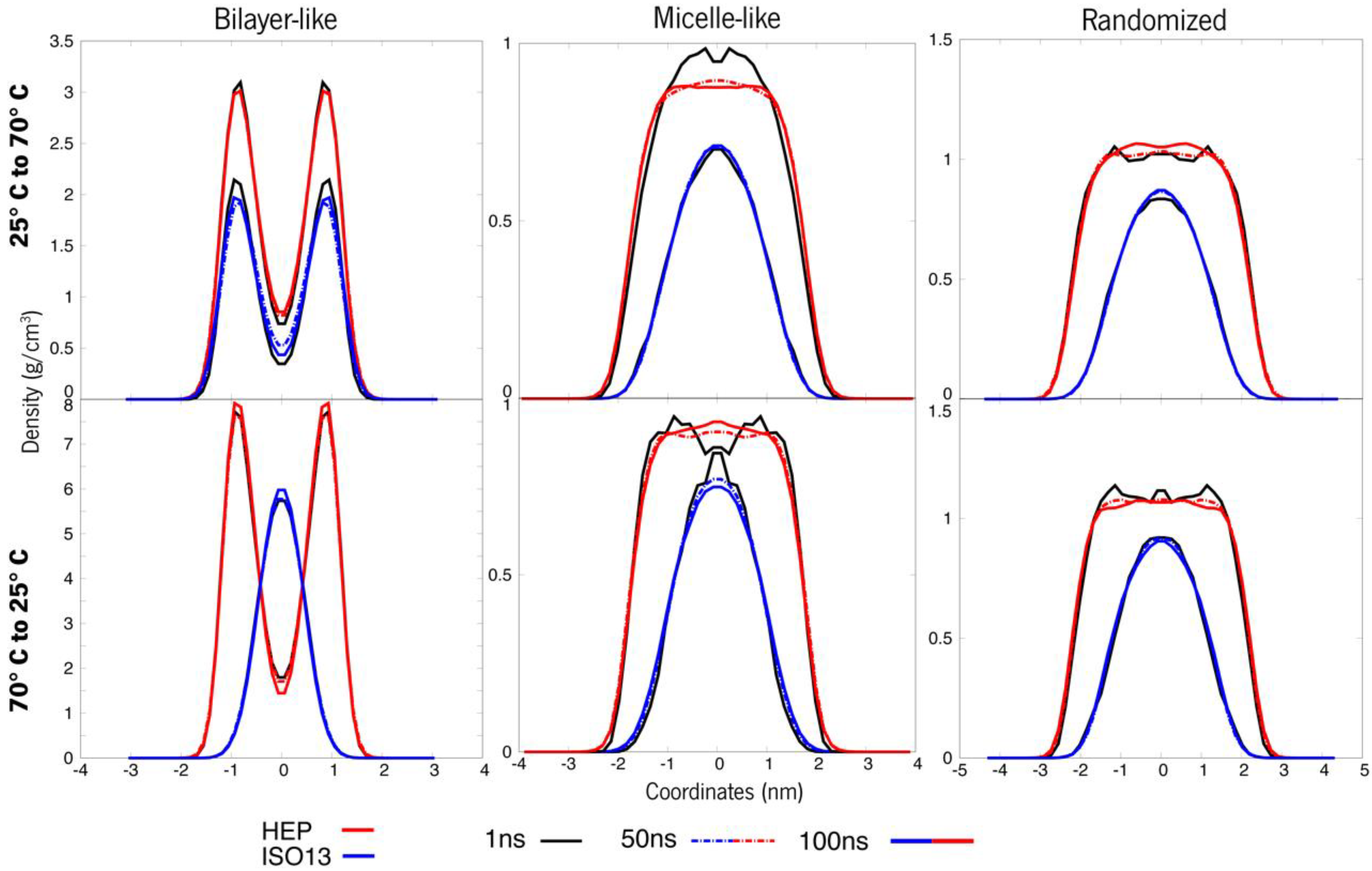
3.2.1. The Switch
3.2.2. The Shock
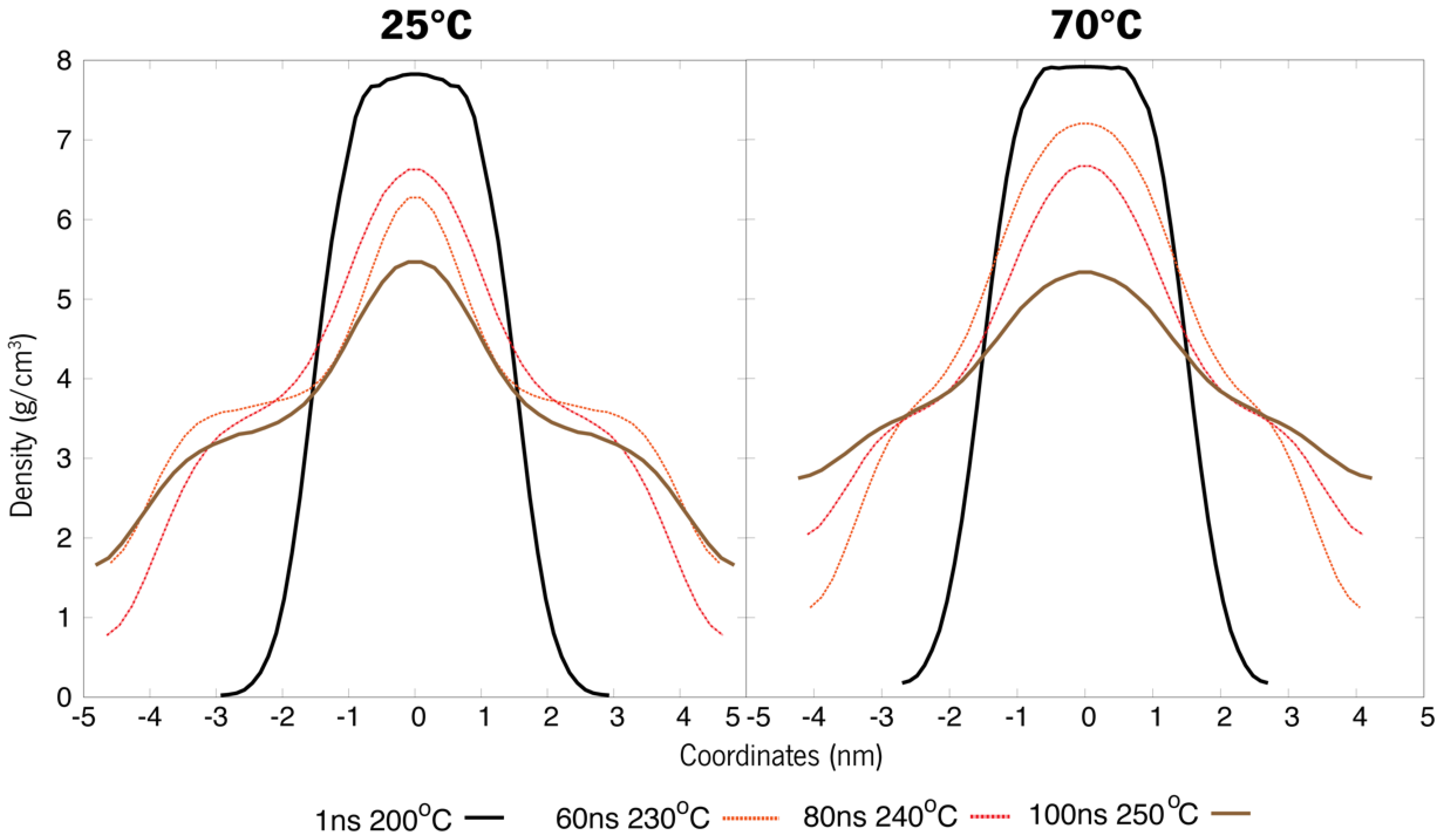
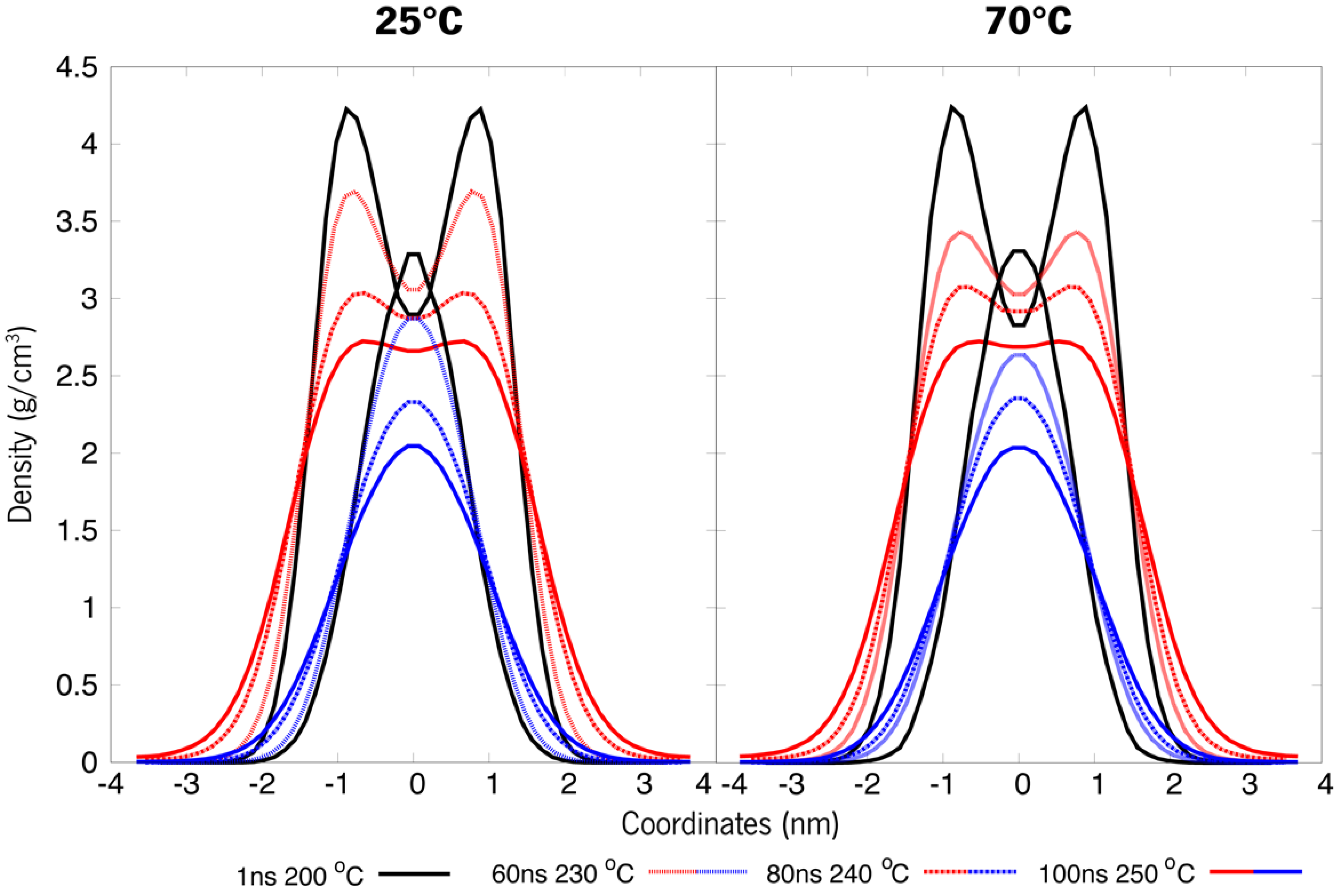
3.3. Simulated Annealing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bunker, A.; Róg, T. Mechanistic Understanding from Molecular Dynamics Simulation in Pharmaceutical Research 1: Drug Delivery. Front. Mol. Biosci. 2020, 7, 604770. [Google Scholar] [CrossRef] [PubMed]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Gupta, R.; Sridhar, D.B.; Rai, B. Molecular Dynamics Simulation Study of Permeation of Molecules through Skin Lipid Bilayer. J. Phys. Chem. B 2016, 120, 8987–8996. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Dwadasi, B.S.; Rai, B.; Mitragotri, S. Effect of Chemical Permeation Enhancers on Skin Permeability: In Silico Screening Using Molecular Dynamics Simulations. Sci. Rep. 2019, 9, 1456. [Google Scholar] [CrossRef] [PubMed]
- Erucar, I.; Keskin, S. Efficient Storage of Drug and Cosmetic Molecules in Biocompatible Metal Organic Frameworks: A Molecular Simulation Study. Ind. Eng. Chem. Res. 2016, 55, 1929–1939. [Google Scholar] [CrossRef]
- Lundborg, M.; Wennberg, C.; Lidmar, J.; Hess, B.; Lindahl, E.; Norlén, L. Skin permeability prediction with MD simulation sampling spatial and alchemical reaction coordinates. Biophys. J. 2022, 121, 3837–3849. [Google Scholar] [CrossRef]
- Gupta, K.M.; Das, S.; Chow, P.S. Molecular Dynamics Simulations to Elucidate Translocation and Permeation of Active from Lipid Nanoparticle to Skin: Complemented by Experiments. Nanoscale 2021, 13, 12916–12928. [Google Scholar] [CrossRef]
- Antunes, E.; Cavaco-Paulo, A. Stratum Corneum Lipid Matrix with Unusual Packing: A Molecular Dynamics Study. Colloids Surf. B Biointerfaces 2020, 190, 110928. [Google Scholar] [CrossRef]
- Antunes, E.; Cruz, C.F.; Azoia, N.G.; Cavaco-Paulo, A. Insights on the Mechanical Behavior of Keratin Fibrils. Int. J. Biol. Macromol. 2016, 89, 477–483. [Google Scholar] [CrossRef]
- Castro, T.G.; Silva, C.; Matamá, T.; Cavaco-Paulo, A. The Structural Properties of Odorants Modulate Their Association to Human Odorant Binding Protein. Biomolecules 2021, 11, 145. [Google Scholar] [CrossRef]
- Gonçalves, F.; Castro, T.G.; Nogueira, E.; Pires, R.; Silva, C.; Ribeiro, A.; Cavaco-Paulo, A. OBP Fused with Cell-Penetrating Peptides Promotes Liposomal Transduction. Colloids Surf. B Biointerfaces 2018, 161, 645–653. [Google Scholar] [CrossRef]
- Wilson, F.; Loughran, D. The Link between Function and Structure of Esters. Available online: https://www.aston-chemicals.com/assets/Publications/Publication%20-%20Aston%20-%20Personal%20Care%20mag%20Esters%20article%20Nov%2017.pdf (accessed on 23 January 2023).
- Biocatalytic Solutions to Cyclomethicones Problem in Cosmetics—Montiel—2019—Engineering in Life Sciences—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/elsc.201800194 (accessed on 23 January 2023).
- EWG Skin Deep® Cosmetics Database. Available online: http://www.ewg.org/skindeep/ (accessed on 28 July 2022).
- Mojsiewicz-Pieńkowska, K.; Jamrógiewicz, M.; Szymkowska, K.; Krenczkowska, D. Direct Human Contact with Siloxanes (Silicones)—Safety or Risk Part 1. Characteristics of Siloxanes (Silicones). Front. Pharmacol. 2016, 7, 132. [Google Scholar] [CrossRef]
- Farias, C.B.B.; Almeida, F.C.G.; Silva, I.A.; Souza, T.C.; Meira, H.M.; Soares da Silva, R.C.F.; Luna, J.M.; Santos, V.A.; Converti, A.; Banat, I.M.; et al. Production of Green Surfactants: Market Prospects. Electron. J. Biotechnol. 2021, 51, 28–39. [Google Scholar] [CrossRef]
- Badmus, S.O.; Amusa, H.K.; Oyehan, T.A.; Saleh, T.A. Environmental Risks and Toxicity of Surfactants: Overview of Analysis, Assessment, and Remediation Techniques. Environ. Sci. Pollut. Res. 2021, 28, 62085–62104. [Google Scholar] [CrossRef]
- Fain, J.H.; Gad, S. Silicones. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 270–272. ISBN 978-0-12-386455-0. [Google Scholar]
- Marchioretto, S.; Vervier, I.; Reeth, I.V.; Plotzke, K.; Johnson, B. The Power of Silicones in Cosmetic Applications: The Science behind the Performance. Available online: https://www.dow.com/documents/en-us/tech-art/27/27-32/27-3299-01-power-of-silicones-in-cosmetic-applications.pdf?linkId=100000103633685 (accessed on 23 January 2023).
- Ferrito, M.S.; Owen, M.J. Silicone Wettability and Its Significance in Beauty Products. Polym. Pers. Care Cosmet. 2013, 14, 219–232. [Google Scholar] [CrossRef]
- ChemAxon Ltd. MarvinSketch 17.16.0. Available online: http://www.chemaxon.com (accessed on 23 January 2023).
- Jorgensen, W.L.; Tirado-Rives, J. Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular Systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Molecular Modeling of Organic and Biomolecular Systems Using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef]
- Engelman, D.M.; Hillman, G.M. Molecular Organization of the Cholesteryl Ester Droplets in the Fatty Streaks of Human Aorta. J. Clin. Investig. 1976, 58, 997–1007. [Google Scholar] [CrossRef]
- Aramaki, K.; Kunieda, H.; Ishitobi, M. Self-Organization of Sucrose Fatty Acid Ester in Water. In Studies in Surface Science and Catalysis; Iwasawa, Y., Oyama, N., Kunieda, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 132, pp. 985–988. [Google Scholar]
- Knight, C.J.; Hub, J.S. MemGen: A General Web Server for the Setup of Lipid Membrane Simulation Systems. Bioinformatics 2015, 31, 2897–2899. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-Isobaric Molecular Dynamics Using Stochastic Velocity Rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [PubMed]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting Crystal Structures: The Parrinello-Rahman Method Revisited. Phys. Rev. Lett. 2003, 90, 4. [Google Scholar] [CrossRef]
- GROMACS Documentation, Release 2021.2.632. Available online: https://manual.gromacs.org/2021.2/index.html (accessed on 23 January 2023).
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M. Gromacs—A parallel computer for molecular-dynamics simulations. In Proceedings of the 4th International Conference on Computational Physics (PC 92), Prague, Czech Republic, 24–28 August 1993; pp. 252–256. [Google Scholar]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Kelley, C. Gnuplot, An Interactive Plotting Program (User Manual). Available online: http://www.gnuplot.info/ (accessed on 23 January 2023).
- Lai, K.; Zhang, Y.; Zheng, Y. Computer Simulation Study of Nanoparticle Interaction with a Lipid Membrane under Mechanical Stress. Phys. Chem. Chem. Phys. PCCP 2012, 15, 270–278. [Google Scholar] [CrossRef]
- Abdul Rahman, M.B.; Latif, M.A.M.; Basri, M.; Salleh, A.B.; Abdul Rahman, R.N.Z. Molecular Dynamics Simulation of Oleyl Oleate Swollen Micelles System. Mol. Simul. 2010, 36, 403–407. [Google Scholar] [CrossRef]
- Abdul Rahman, M.B.; Huan, Q.-Y.; Tejo, B.A.; Basri, M.; Salleh, A.B.; Rahman, R.N.Z.A. Self-Assembly Formation of Palm-Based Esters Nano-Emulsion: A Molecular Dynamics Study. Chem. Phys. Lett. 2009, 480, 220–224. [Google Scholar] [CrossRef]
- Sudibjo, A.; Spearot, D.E. Molecular Dynamics Simulation of Diffusion of Small Atmospheric Penetratesin Polydimethylsiloxane. Mol. Simul. 2011, 37, 115–122. [Google Scholar] [CrossRef]
- Menges, M.G.; Penelle, J.; Le Fevere de Ten Hove, C.; Jonas, A.M.; Schmidt-Rohr, K. Characterization of Long-Chain Aliphatic Polyesters: Crystalline and Supramolecular Structure of PE22,4 Elucidated by X-ray Scattering and Nuclear Magnetic Resonance. Macromolecules 2007, 40, 8714–8725. [Google Scholar] [CrossRef]
- Eren, Y.; Küçükdemiral, İ.B.; Üstoğlu, İ. Chapter 2—Introduction to Optimization. In Optimization in Renewable Energy Systems; Erdinç, O., Ed.; Butterworth-Heinemann: Boston, MA, USA, 2017; pp. 27–74. ISBN 978-0-08-101041-9. [Google Scholar]
- Sakae, Y.; Hiroyasu, T.; Miki, M.; Okamoto, Y. Protein Structure Predictions by Parallel Simulated Annealing Molecular Dynamics Using Genetic Crossover. J. Comput. Chem. 2011, 32, 1353–1360. [Google Scholar] [CrossRef]
- Petitjean, H.; Guiglion, P.; In, M.; Aldrich-Wright, J.R.; Castignolles, P.; Ganachaud, F.; Gaborieau, M. Supramolecular Organization of Protonated Aminosilicones in Water. J. Colloid Interface Sci. 2013, 408, 87–93. [Google Scholar] [CrossRef]
- Ganachaud, F.; Boileau, S.; Boury, B. Silicon Based Polymers: Advances in Synthesis and Supramolecular Organization; Springer: Dordrecht, The Netherlands, 2008; ISBN 978-1-4020-8528-4. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ERY | HEP | ISO13 | |
|---|---|---|---|
| facial moisturizer and treatment | 1 | 122 | 9 |
| moisturizer | - | 37 | 3 |
| serums & essences | 1 | 5 | 27 |
| conditioner | 1 | 2 | 7 |
| hair styling aide | 1 | 16 | 3 |
| hair treatment/serum | - | 20 | 7 |
| anti-ageing | - | 16 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, T.; Loureiro, A.; Noro, J.; Cavaco-Paulo, A.; Castro, T.G. Addressing the Structural Organization of Silicone Alternatives in Formulations by Molecular Dynamics Simulations and a Novel Equilibration Protocol. Polymers 2023, 15, 796. https://doi.org/10.3390/polym15040796
Ferreira T, Loureiro A, Noro J, Cavaco-Paulo A, Castro TG. Addressing the Structural Organization of Silicone Alternatives in Formulations by Molecular Dynamics Simulations and a Novel Equilibration Protocol. Polymers. 2023; 15(4):796. https://doi.org/10.3390/polym15040796
Chicago/Turabian StyleFerreira, Tiago, Ana Loureiro, Jennifer Noro, Artur Cavaco-Paulo, and Tarsila G. Castro. 2023. "Addressing the Structural Organization of Silicone Alternatives in Formulations by Molecular Dynamics Simulations and a Novel Equilibration Protocol" Polymers 15, no. 4: 796. https://doi.org/10.3390/polym15040796
APA StyleFerreira, T., Loureiro, A., Noro, J., Cavaco-Paulo, A., & Castro, T. G. (2023). Addressing the Structural Organization of Silicone Alternatives in Formulations by Molecular Dynamics Simulations and a Novel Equilibration Protocol. Polymers, 15(4), 796. https://doi.org/10.3390/polym15040796