
Phosphonate-Functionalized Polycarbonates Synthesis through Ring-Opening Polymerization and Alternative Approaches


Abstract
:1. Introduction
2. Experimental Section and Methods
2.1. Materials
2.2. Characterization
2.2.1. High-Resolution Mass Spectrometry (HRMS)
2.2.2. Fourier-Transformation Infrared (FT-IR)
2.2.3. Nuclear Magnetic Resonance (NMR)
2.2.4. Size Exclusion Chromatography (SEC)
2.3. Synthesis of 2,2,5-Trimethyl-1,3-dioxane-5-carboxylic Acid (Acetonide-bisMPA, 1)
2.4. Synthesis of 2-(Dimethoxyphosphoryl)ethyl 2,2,5-trimethyl-1,3-dioxane-5-carboxylate (2)
2.5. Synthesis of 2-(Dimethoxyphosphoryl)ethyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (3)
2.6. Synthesis of 2-(Dimethoxyphosphoryl)ethyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate (4)
2.7. Synthesis of Diethyl (3-Azidopropyl)phosphonate (Azido-phosphonate)
2.8. Ring-Opening Polymerization of 4
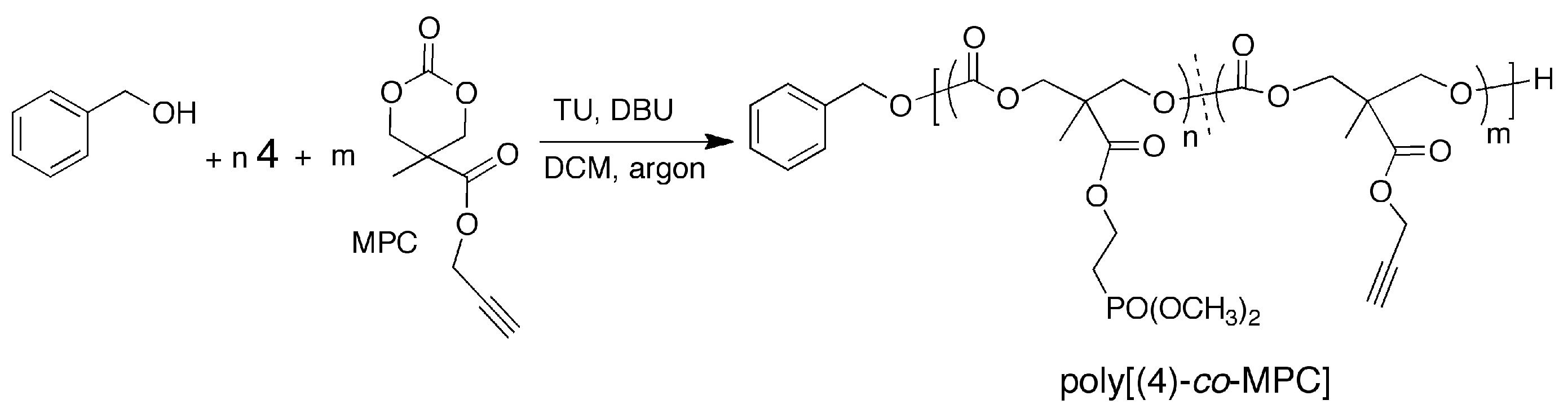
2.9. A Typical Copolymerization of 4 and MPC
2.10. Synthesis of Alkyne-Functionalized Polycarbonate
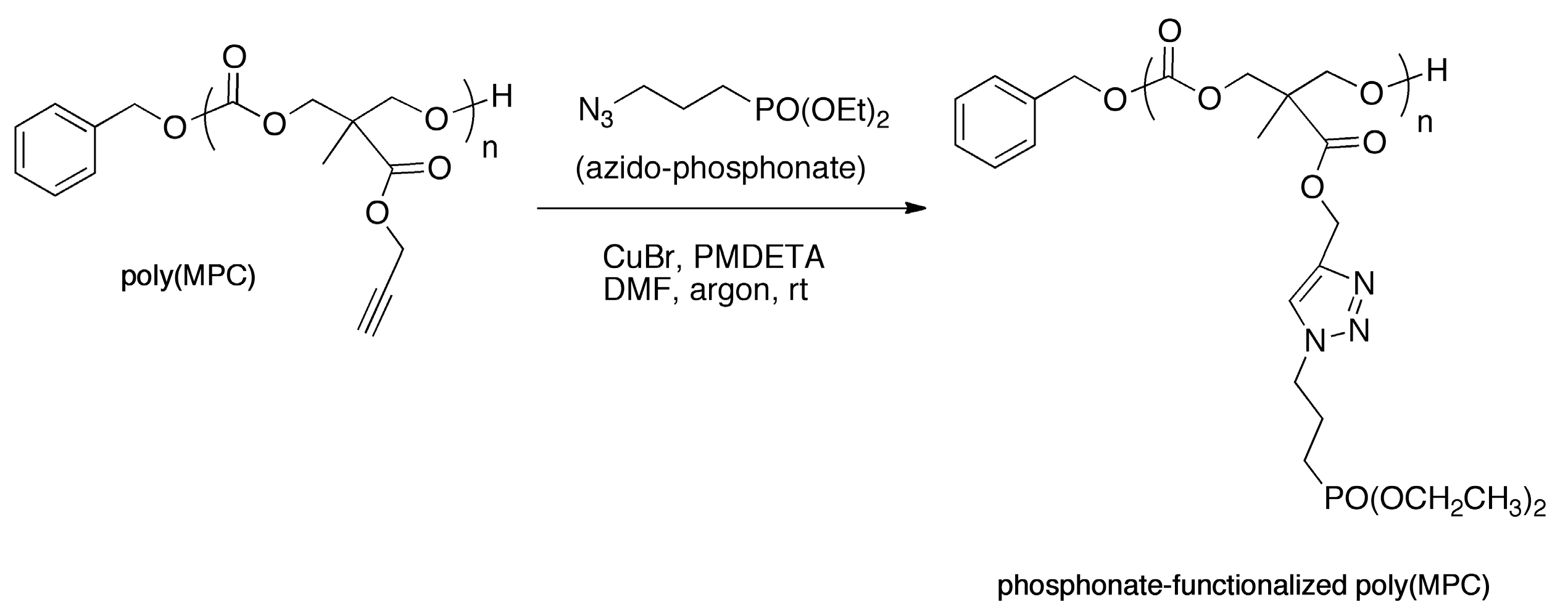
2.11. Click Synthesis of Phosphonated-Functionalized Polycarbonate
3. Results and Discussion
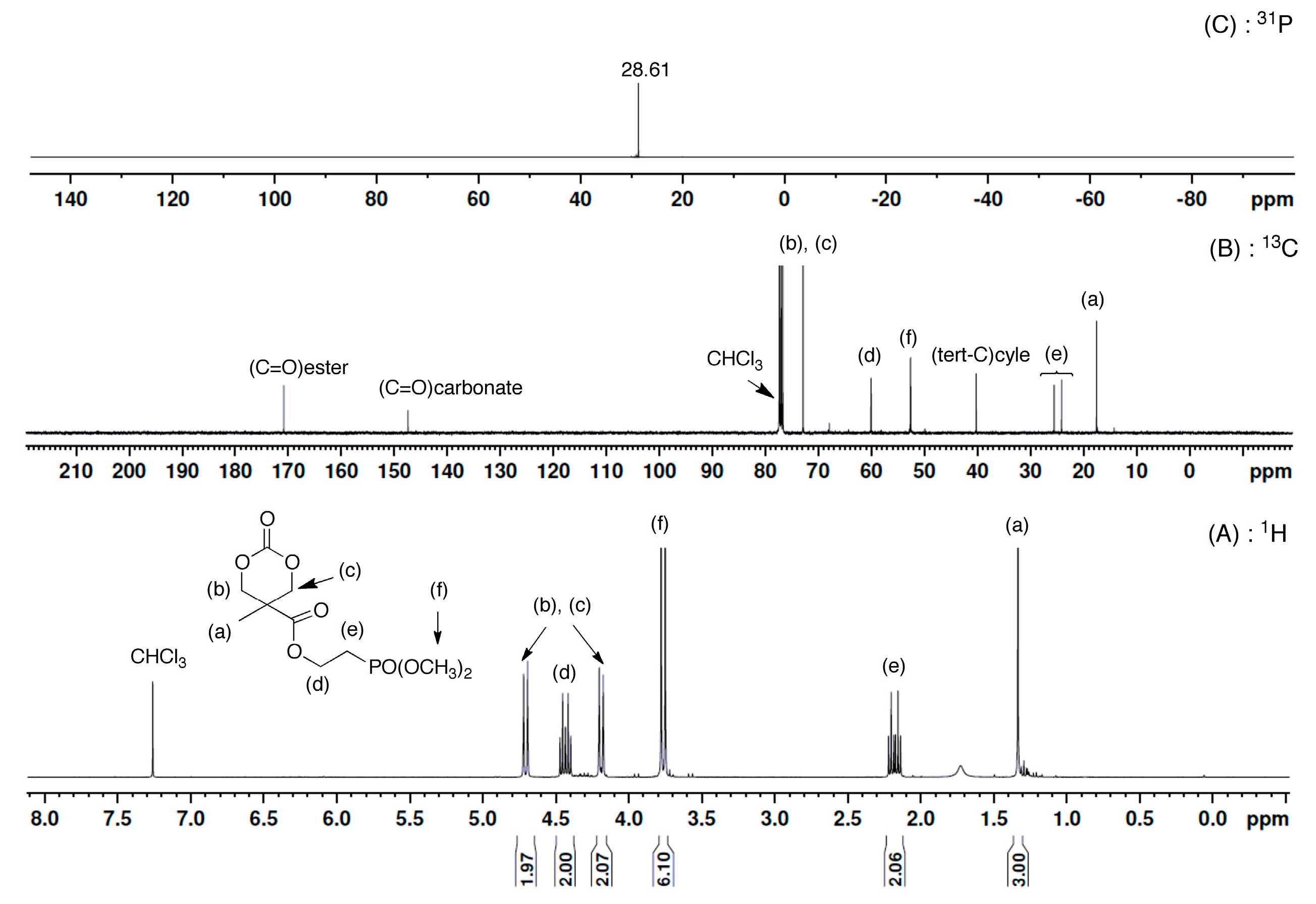
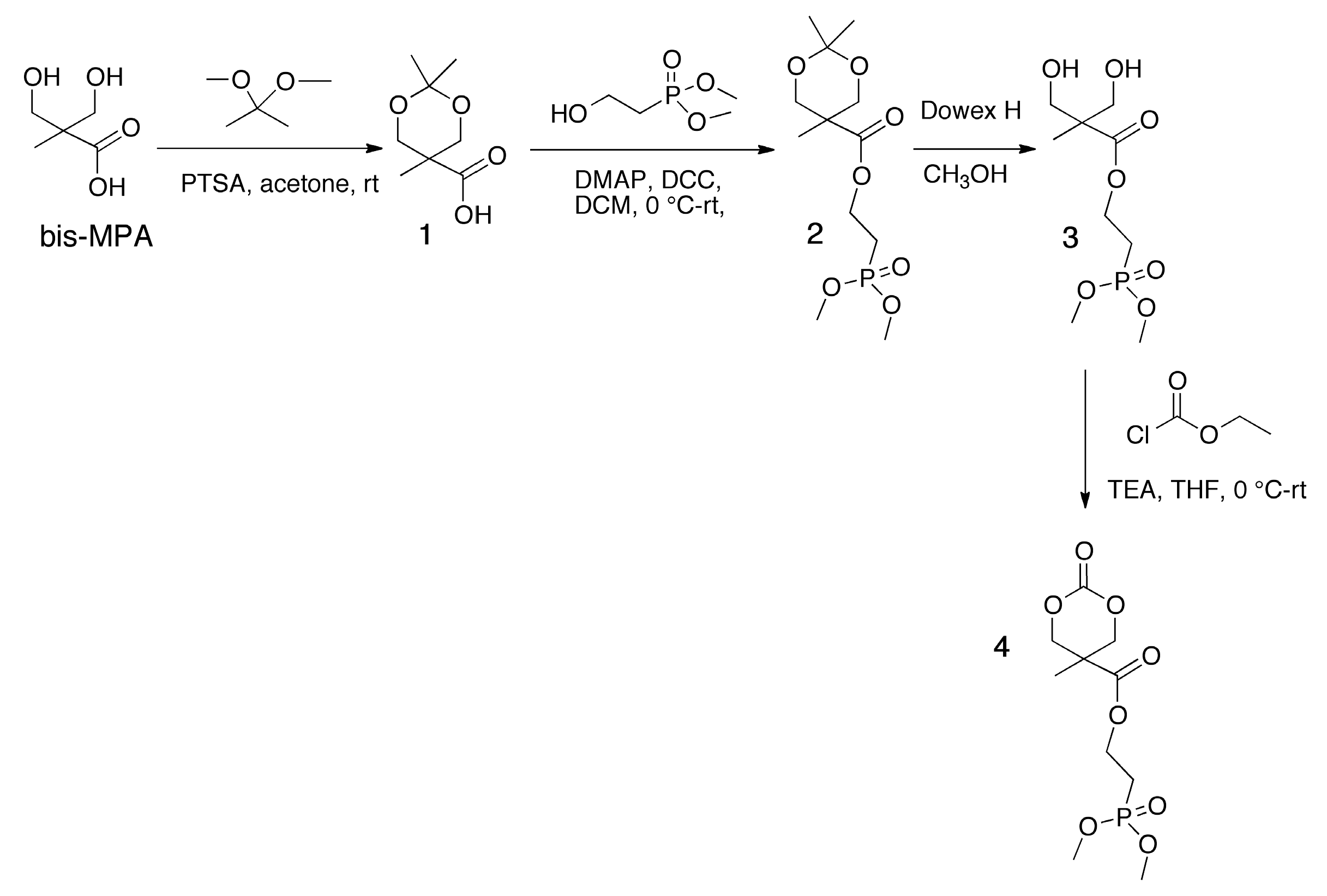
3.1. Synthesis of Phosphonate-Functionalized Cyclic Carbonate Monomer
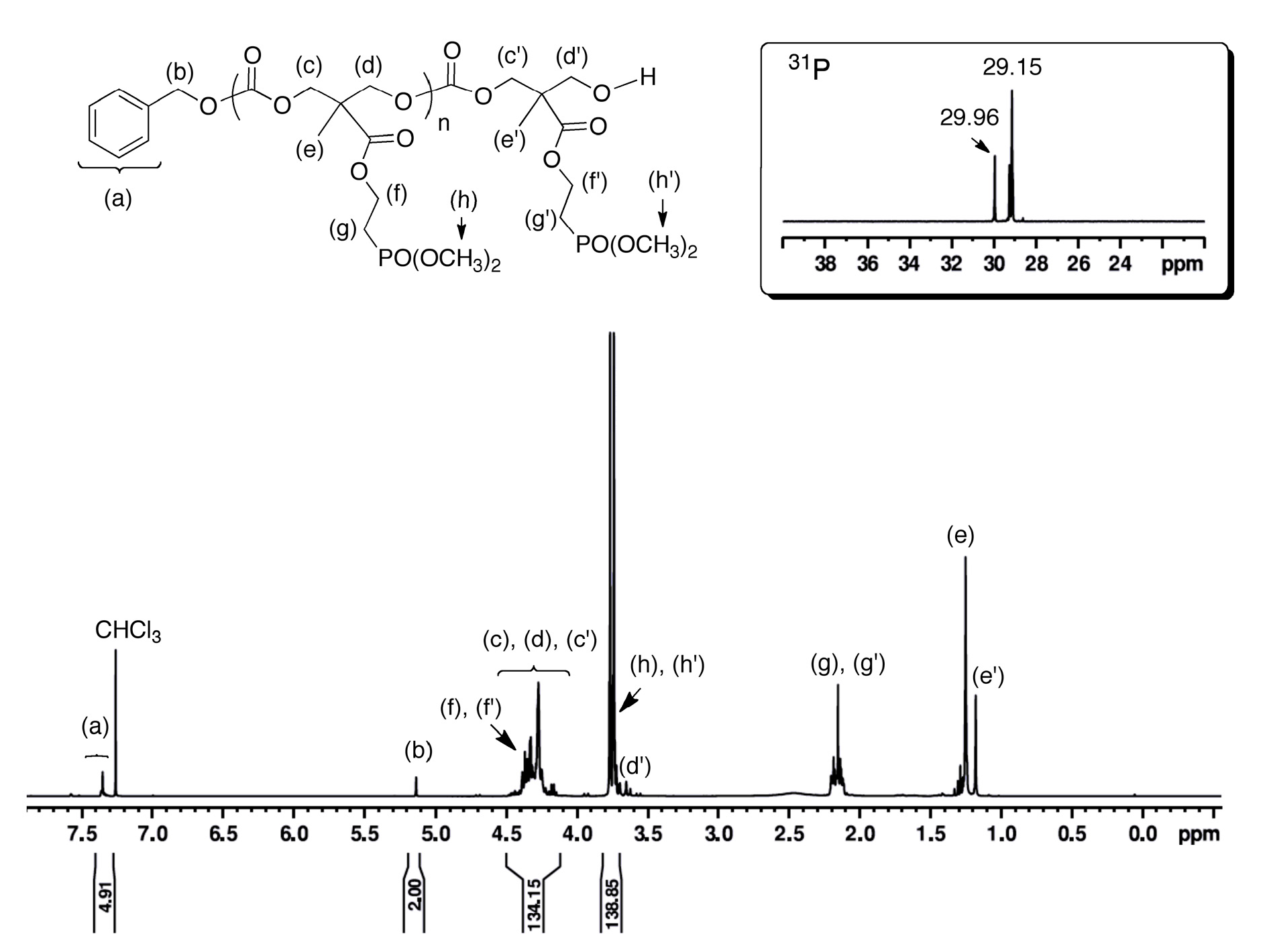
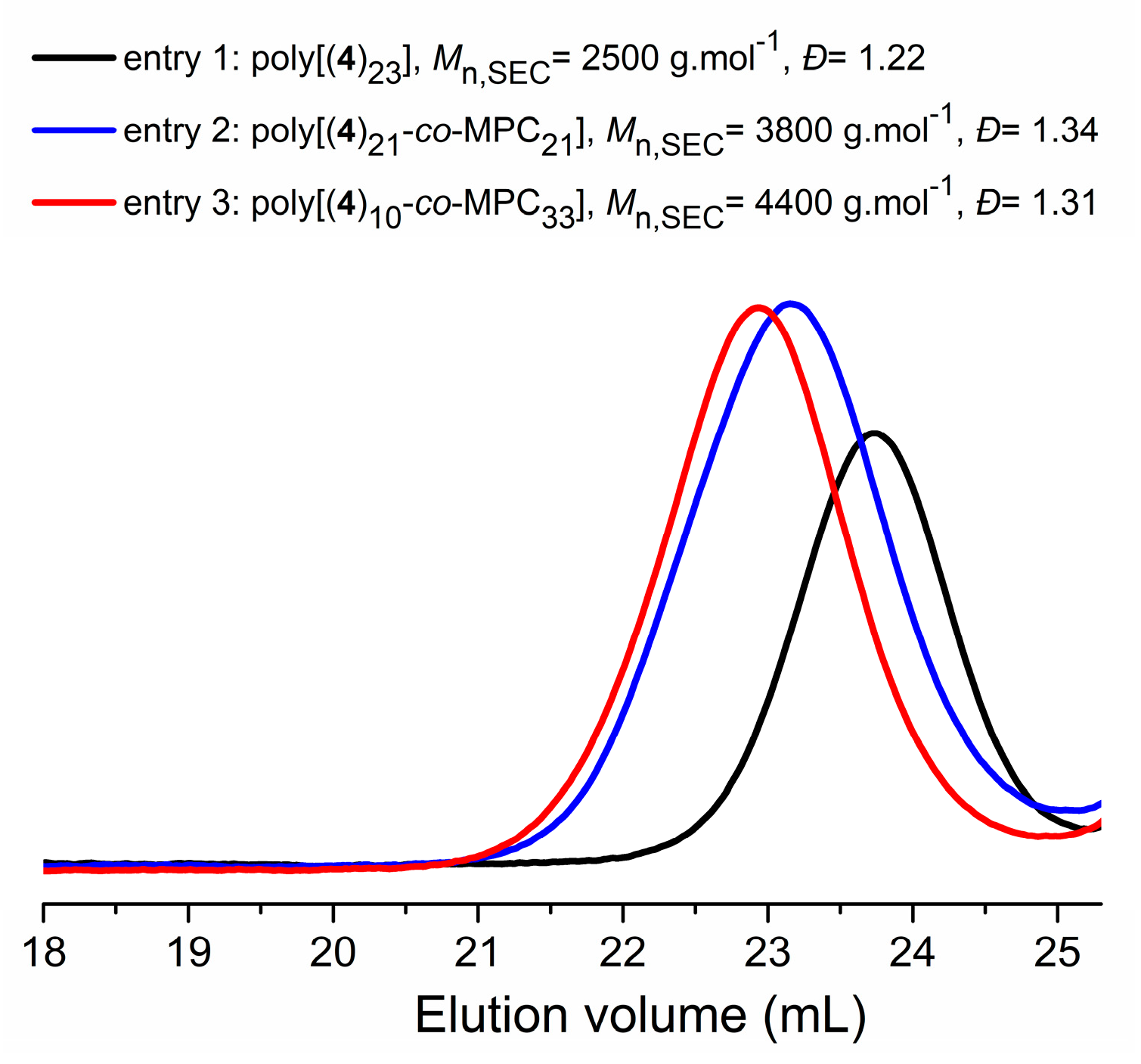
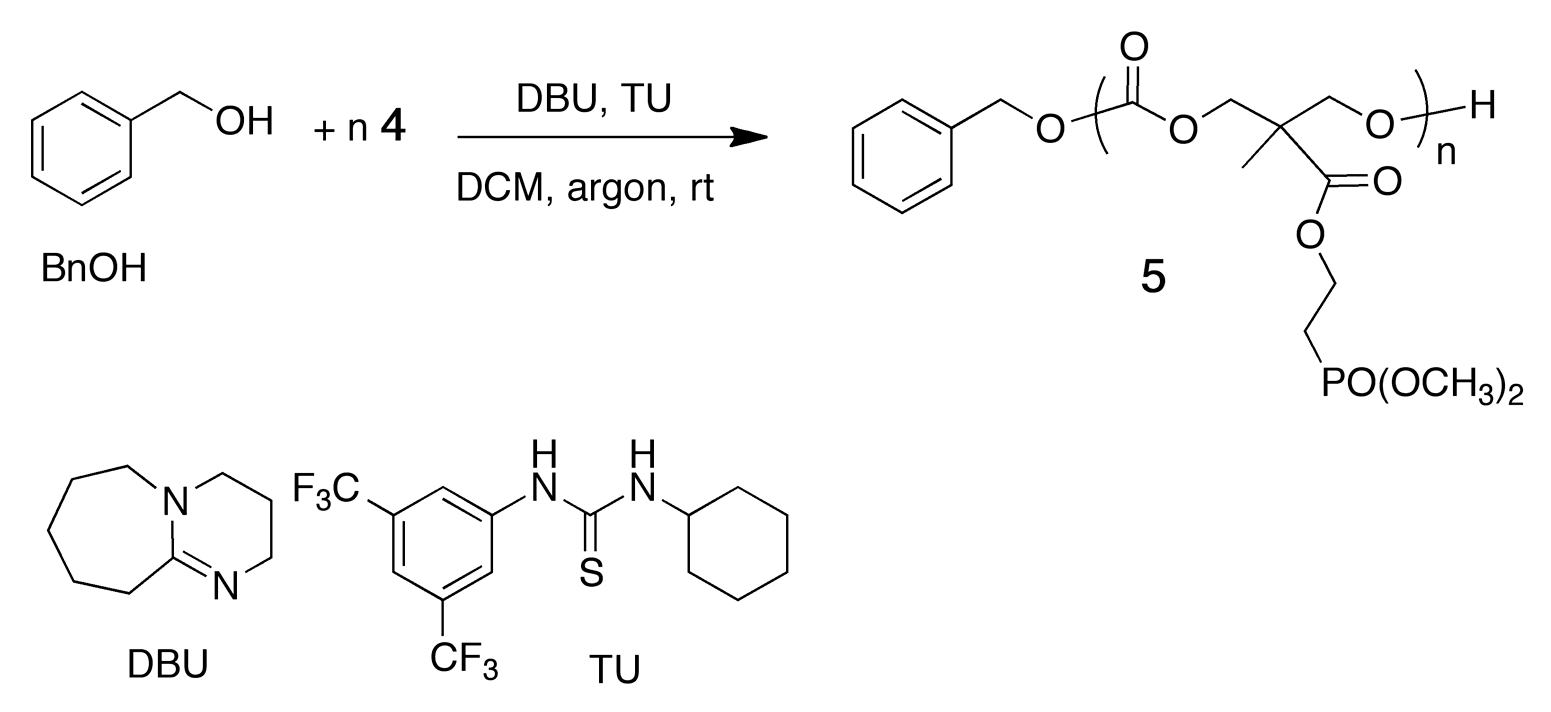
3.2. ROP Synthesis of Well-Defined Phosphonate-Functionalized (Co)polycarbonates
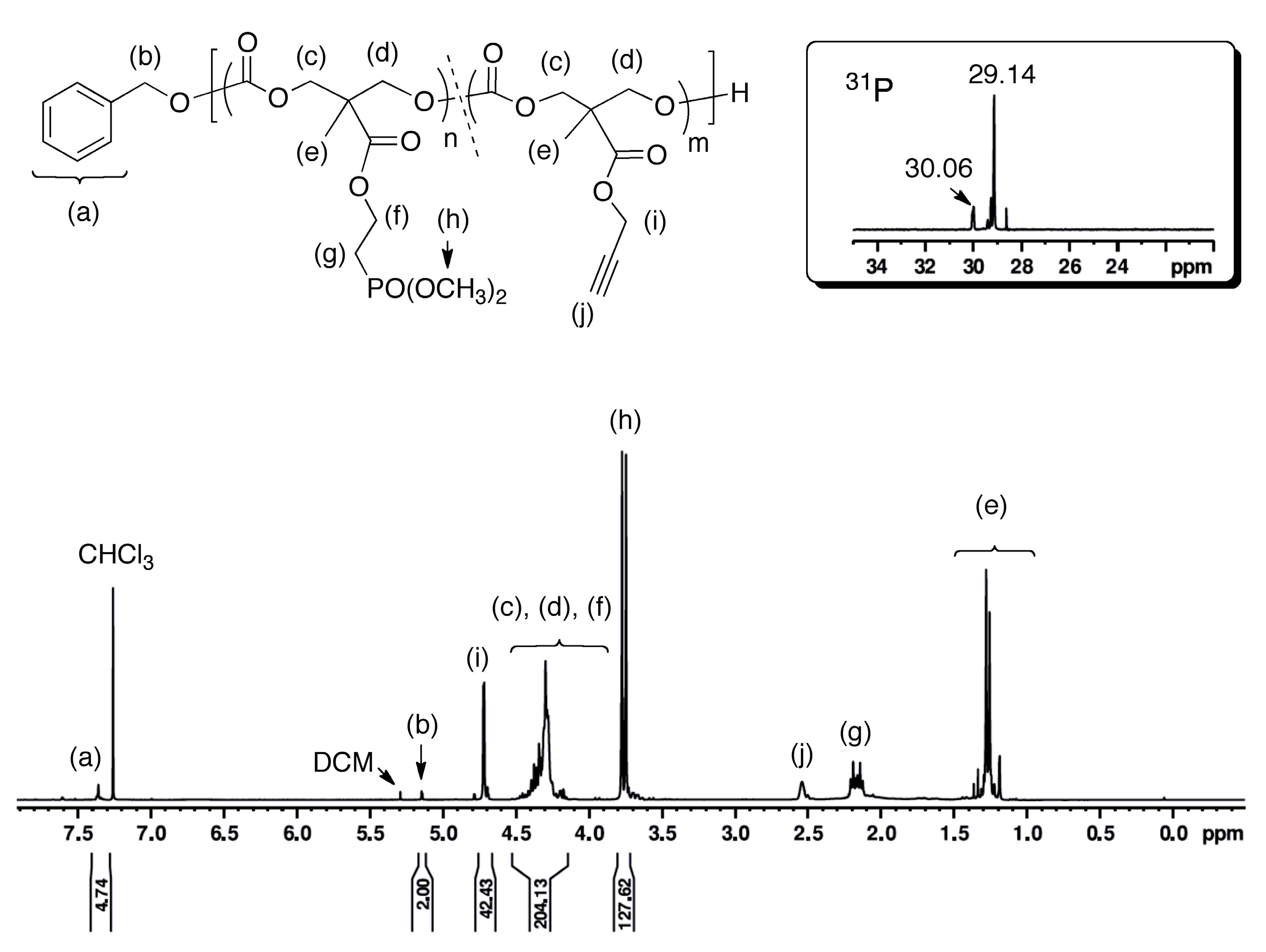
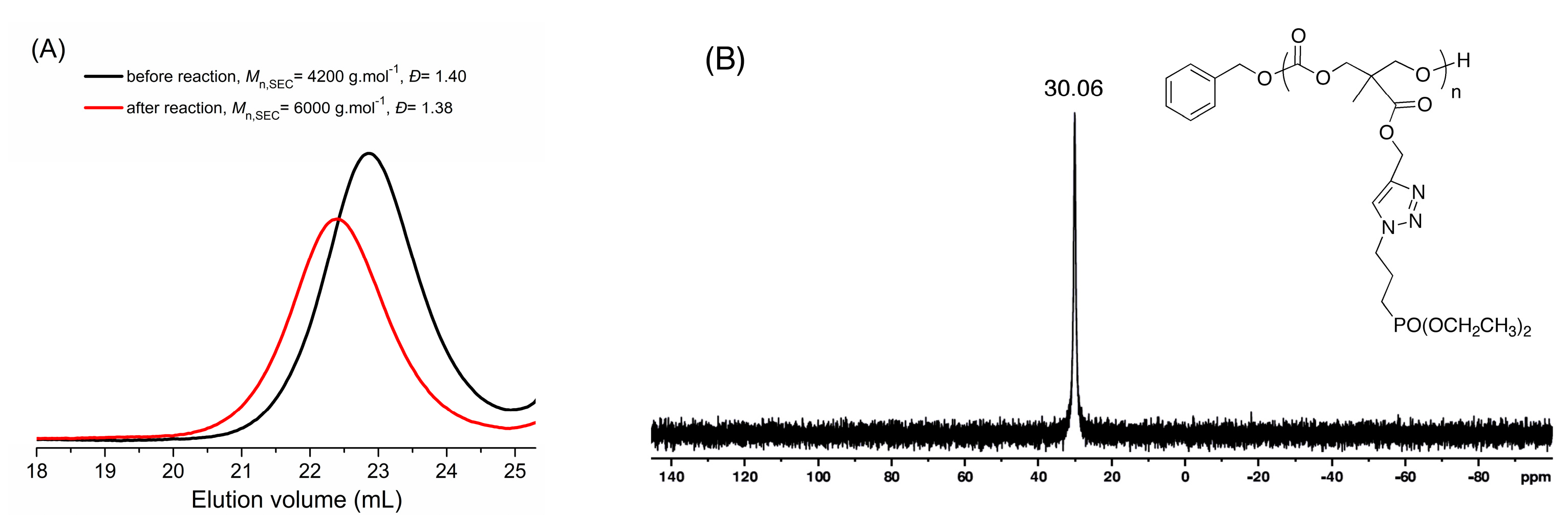
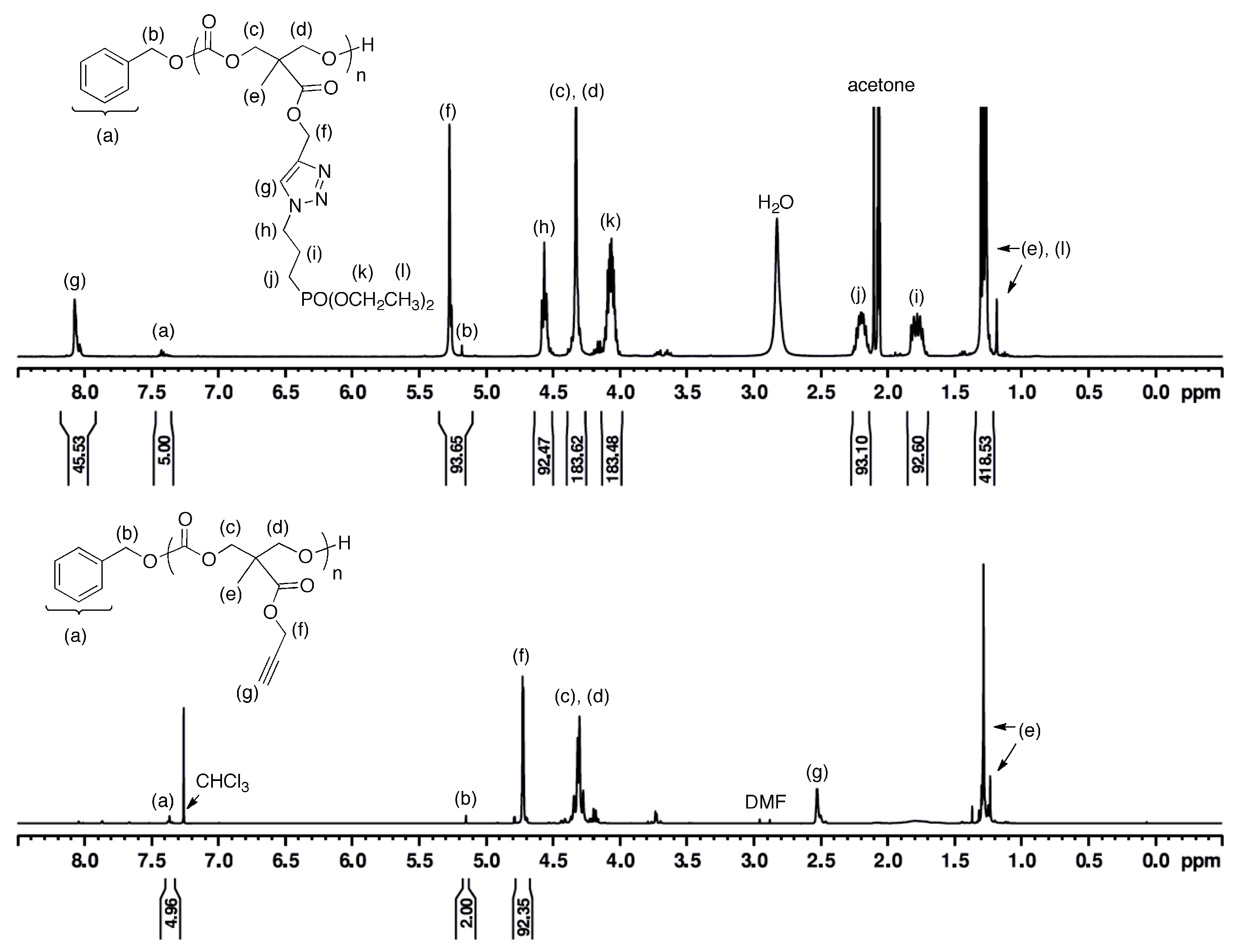
3.3. Post-Polymerization Synthesis of Phosphonate-Functionalized Polycarbonate
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.J. Phosphorus-Containing Polymers: A Great Opportunity for the Biomedical Field. Biomacromolecules 2011, 12, 1973–1982. [Google Scholar] [CrossRef]
- MacArie, L.; Ilia, G. Poly(vinylphosphonic Acid) and Its Derivatives. Prog. Polym. Sci. 2010, 35, 1078–1092. [Google Scholar] [CrossRef]
- Wehbi, M.; Mehdi, A.; Negrell, C.; David, G.; Alaaeddine, A.; Améduri, B. Phosphorus-Containing Fluoropolymers: State of the Art and Applications. ACS Appl. Mater. Interfaces 2020, 12, 38–59. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P. Review of Phosphorus-Based Polymers for Mineral Scale and Corrosion Control in Oilfield. Polymers 2022, 14, 2673. [Google Scholar] [CrossRef]
- Price, D.; Cunliffe, L.K.; Bullett, K.J.; Hull, T.R.; Milnes, G.J.; Ebdon, J.R.; Hunt, B.J.; Joseph, P. Thermal Behaviour of Covalently Bonded Phosphate and Phosphonate Flame Retardant Polystyrene Systems. Polym. Degrad. Stab. 2007, 92, 1101–1114. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, L.; Cheng, Z.; Zhu, X. A Novel Methacrylate with a Bisphosphonate Group: RAFT Polymerization and Flame Retardant Property of the Resultant Polymers. Polym. Chem. 2015, 6, 2283–2289. [Google Scholar] [CrossRef]
- Vahabi, H.; Ferry, L.; Longuet, C.; Sonnier, R.; Negrell-Guirao, C.; David, G.; Lopez-Cuesta, J.-M. Theoretical and Empirical Approaches to Understanding the Effect of Phosphonate Groups on the Thermal Degradation for Two Chemically Modified PMMA. Eur. Polym. J. 2012, 48, 604–612. [Google Scholar] [CrossRef]
- Beaugeard, V.; Muller, J.; Graillot, A.; Ding, X.; Robin, J.-J.; Monge, S. Acidic Polymeric Sorbents for the Removal of Metallic Pollution in Water: A Review. React. Funct. Polym. 2020, 152, 104599. [Google Scholar] [CrossRef]
- Ho, H.T.; Coupris, J.; Pascual, S.; Fontaine, L.; Lequeux, T.; Pham, T.N. Synthesis and Characterization of Innovative Well-Defined Difluorophosphonylated-(Co)polymers by RAFT Polymerization. Polym. Chem. 2015, 6, 4597–4604. [Google Scholar]
- Solimando, X.; Catel, Y.; Moszner, N.; Robin, J.J.; Monge, S. Smart Functionalized Phosphonic Acid Based Copolymers: New Structures for Old Purposes. Polym. Chem. 2020, 11, 3237–3250. [Google Scholar] [CrossRef]
- David, G.; Negrell-Guirao, C.; Iftene, F.; Boutevin, B.; Chougrani, K. Recent Progress on Phosphonate Vinyl Monomers and Polymers Therefore Obtained by Radical (Co)polymerization. Polym. Chem. 2012, 3, 265–274. [Google Scholar] [CrossRef]
- Ho, H.T.; Pascual, S.; Livi, S.; Mebold, E.; Fontaine, L.; Lequeux, T.; Pham, T.N. A Straightforward Synthesis of Well-Defined Difluorophosphonylated Terminated Poly(ε-Caprolactone) for Grafting onto Iron Oxide Magnetic Nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2453–2458. [Google Scholar] [CrossRef]
- Liu, N.; Yao, C.; Lin, F.; Liu, B.; Cui, D. An Intensification and Integration Process of Preparing Thermal Stable Polylactide End-Capped by Phosphate Ester. Polymer 2015, 80, 104–108. [Google Scholar] [CrossRef]
- Pu, L.; Xu, J.; Sun, Y.; Fang, Z.; Chan-Park, M.B.; Duan, H. Cationic Polycarbonate-Grafted Superparamagnetic Nanoparticles with Synergistic Dual-Modality Antimicrobial Activity. Biomater. Sci. 2016, 4, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K. Poly(trimethylene Carbonate)-Based Polymers Engineered for Biodegradable Functional Biomaterials. Biomater. Sci. 2016, 4, 9–24. [Google Scholar] [CrossRef]
- Tempelaar, S.; Mespouille, L.; Coulembier, O.; Dubois, P.; Dove, A.P. Synthesis and Post-Polymerisation Modifications of Aliphatic Poly(Carbonate)s Prepared by Ring-Opening Polymerisation. Chem. Soc. Rev. 2013, 42, 1312–1336. [Google Scholar] [CrossRef] [Green Version]
- Becker, G.; Wurm, F.R. Functional Biodegradable Polymers: Via Ring-Opening Polymerization of Monomers without Protective Groups. Chem. Soc. Rev. 2018, 47, 7739–7782. [Google Scholar] [CrossRef] [Green Version]
- Mady, M.F.; Charoensumran, P.; Ajiro, H.; Kelland, M.A. Synthesis and Characterization of Modified Aliphatic Polycarbonates as Environmentally Friendly Oilfield Scale Inhibitors. Energy Fuels 2018, 32, 6746–6755. [Google Scholar] [CrossRef]
- Pratt, R.C.; Lohmeijer, B.G.G.; Long, D.A.; Lundberg, P.N.P.; Dove, A.P.; Li, H.; Wade, C.G.; Waymouth, R.M.; Hedrick, J.L. Exploration, Optimization, and Application of Supramolecular Thiourea-Amine Catalysts for the Synthesis of Lactide (Co)polymers. Macromolecules 2006, 39, 7863–7871. [Google Scholar] [CrossRef]
- Lu, C.; Shi, Q.; Chen, X.; Lu, T.; Xie, Z.; Hu, X.; Ma, J.; Jing, X. Sugars-Grafted Aliphatic Biodegradable Poly(L-lactide-co-carbonate)s by Click Reaction and Their Specific Interaction with Lectin Molecules. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 3204–3217. [Google Scholar] [CrossRef]
- Truong, V.; Blakey, I.; Whittaker, A.K. Hydrophilic and Amphiphilic Polyethylene Glycol-Based Hydrogels with Tunable Degradability Prepared by “Click” Chemistry. Biomacromolecules 2012, 13, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Nederberg, F.; Lohmeijer, B.G.G.; Leibfarth, F.; Pratt, R.C.; Choi, J.; Dove, A.P.; Waymouth, R.M.; Hedrick, J.L. Organocatalytic Ring Opening Polymerization of Trimethylene Carbonate. Biomacromolecules 2007, 8, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.W.; Zhang, X.; Brennan, M.K.; Sardon, H.; Engler, A.C.; Fox, C.H.; Frank, C.W.; Waymouth, R.M.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization of Trimethylene Carbonate to Yield a Biodegradable Polycarbonate. J. Chem. Educ. 2015, 92, 708–713. [Google Scholar] [CrossRef]
- Pratt, R.C.; Nederberg, F.; Waymouth, R.M.; Hedrick, J.L. Tagging Alcohols with Cyclic Carbonate: A Versatile Equivalent of (Meth)acrylate for Ring-Opening Polymerization. Chem. Commun. 2008, 114–116. [Google Scholar] [CrossRef]
- Clément, B.; Grignard, B.; Koole, L.; Jérôme, C.; Lecomte, P. Metal-Free Strategies for the Synthesis of Functional and Well-Defined Polyphosphoesters. Macromolecules 2012, 45, 4476–4486. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Controlled Ring-Opening Polymerisation of Cyclic Phosphates, Phosphonates and Phosphoramidates Catalysed by Heteroleptic BHT-Alkoxy Magnesium Complexes. Polym. Chem. 2017, 8, 6806–6816. [Google Scholar] [CrossRef]
- Steinbach, T.; Ritz, S.; Wurm, F.R. Water-Soluble Poly(phosphonate)s via Living Ring-Opening Polymerization. ACS Macro Lett. 2014, 3, 244–248. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. “Click” Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Golas, P.L.; Matyjaszewski, K. Marrying Click Chemistry with Polymerization: Expanding the Scope of Polymeric Materials. Chem. Soc. Rev. 2010, 39, 1338–1354. [Google Scholar] [CrossRef]
- Ho, H.T.; Tintaru, A.; Rollet, M.; Gigmes, D.; Phan, T.N.T. A Post-Polymerization Functionalization Strategy for the Synthesis of Sulfonyl (Trifluoromethanesulfonyl)imide Functionalized (Co)polymers. Polym. Chem. 2017, 8, 5660–5665. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [M]0:[BnOH]0:[TU]0:[DBU]0 | 4/MPC a | Time (min) | Conv. b (%) | Mn,thc (g·mol−1) | Mn,NMRd (g·mol−1) | Mn,SECe (g·mol−1) | Ðe |
|---|---|---|---|---|---|---|---|---|
| 1 | 30:1:1.27:1.20 | 100/0 | 100 | 90 | 8100 | 6920 | 2500 | 1.22 |
| 2 | 40:1:1.23:1.22 | 50/50 | 45 | 100 | 9990 | 10480 | 3800 | 1.34 |
| 3 | 41:1:1.23:1.22 | 22/78 | 45 | 100 | 9110 | 9600 | 4400 | 1.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, H.T.; Nguyen, N.H.; Rollet, M.; Phan, T.N.T.; Gigmes, D. Phosphonate-Functionalized Polycarbonates Synthesis through Ring-Opening Polymerization and Alternative Approaches. Polymers 2023, 15, 955. https://doi.org/10.3390/polym15040955
Ho HT, Nguyen NH, Rollet M, Phan TNT, Gigmes D. Phosphonate-Functionalized Polycarbonates Synthesis through Ring-Opening Polymerization and Alternative Approaches. Polymers. 2023; 15(4):955. https://doi.org/10.3390/polym15040955
Chicago/Turabian StyleHo, Hien The, Nam Hoai Nguyen, Marion Rollet, Trang N. T. Phan, and Didier Gigmes. 2023. "Phosphonate-Functionalized Polycarbonates Synthesis through Ring-Opening Polymerization and Alternative Approaches" Polymers 15, no. 4: 955. https://doi.org/10.3390/polym15040955
APA StyleHo, H. T., Nguyen, N. H., Rollet, M., Phan, T. N. T., & Gigmes, D. (2023). Phosphonate-Functionalized Polycarbonates Synthesis through Ring-Opening Polymerization and Alternative Approaches. Polymers, 15(4), 955. https://doi.org/10.3390/polym15040955