
Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems

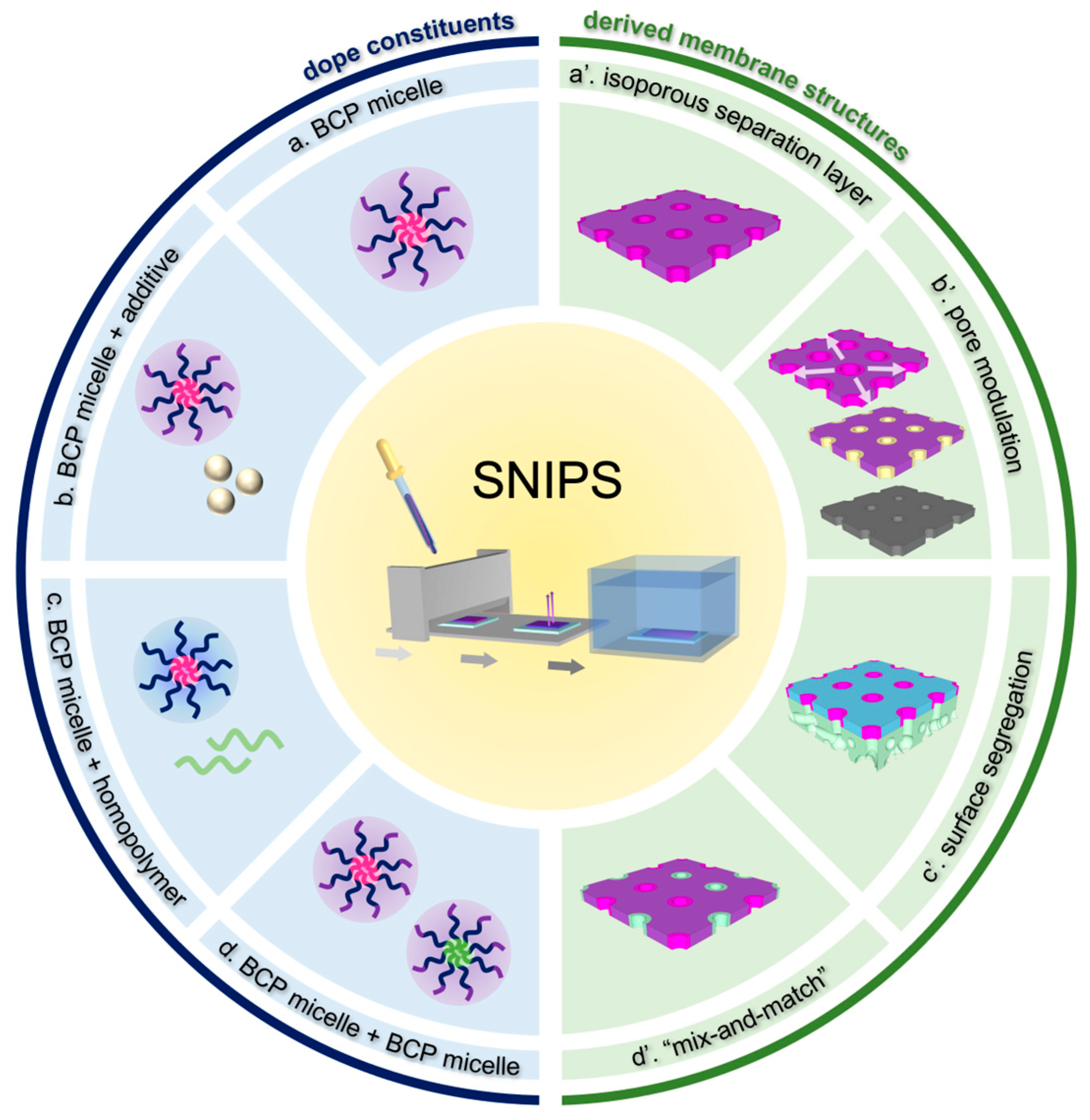{kind=link}
{kind=link}
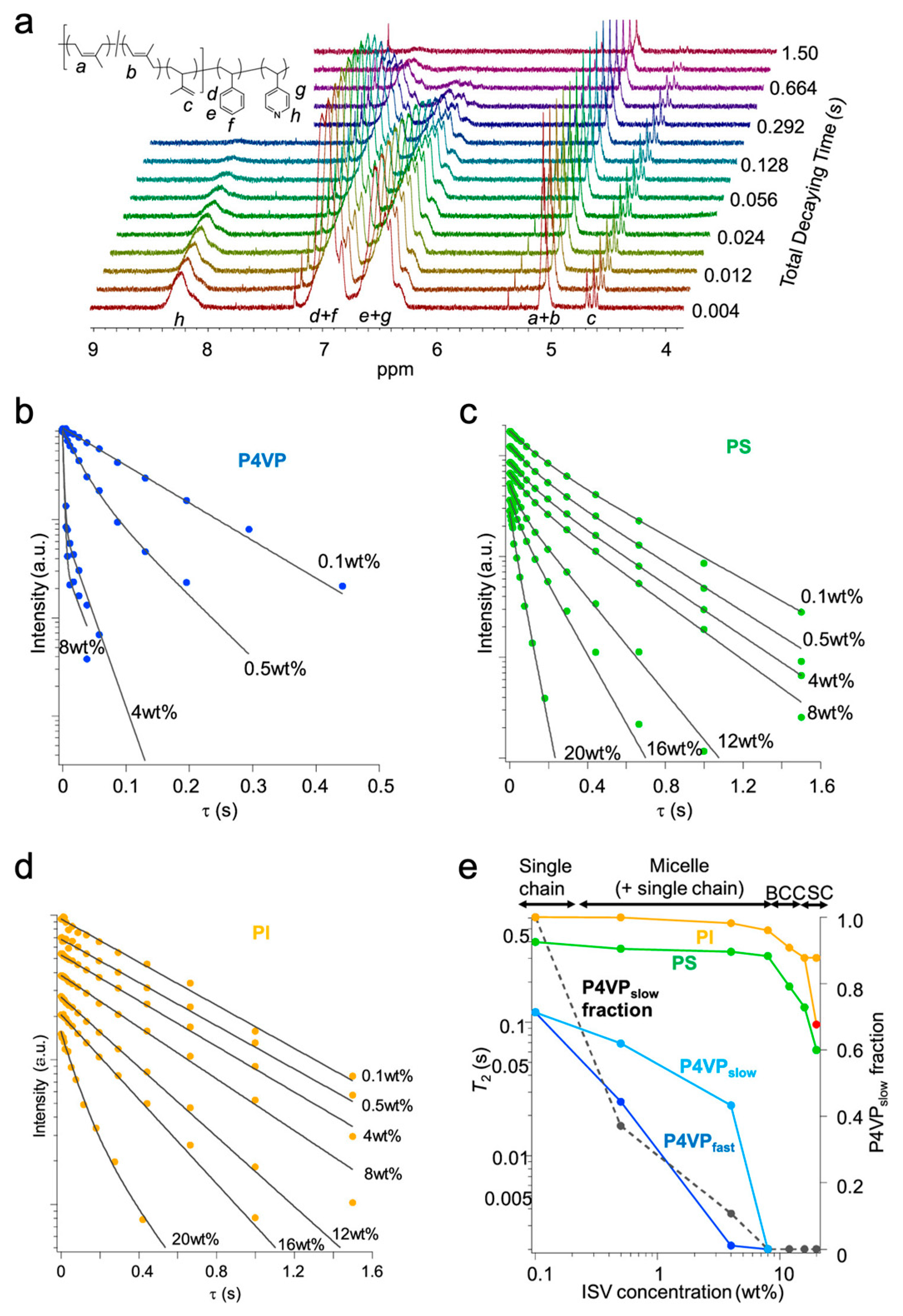{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
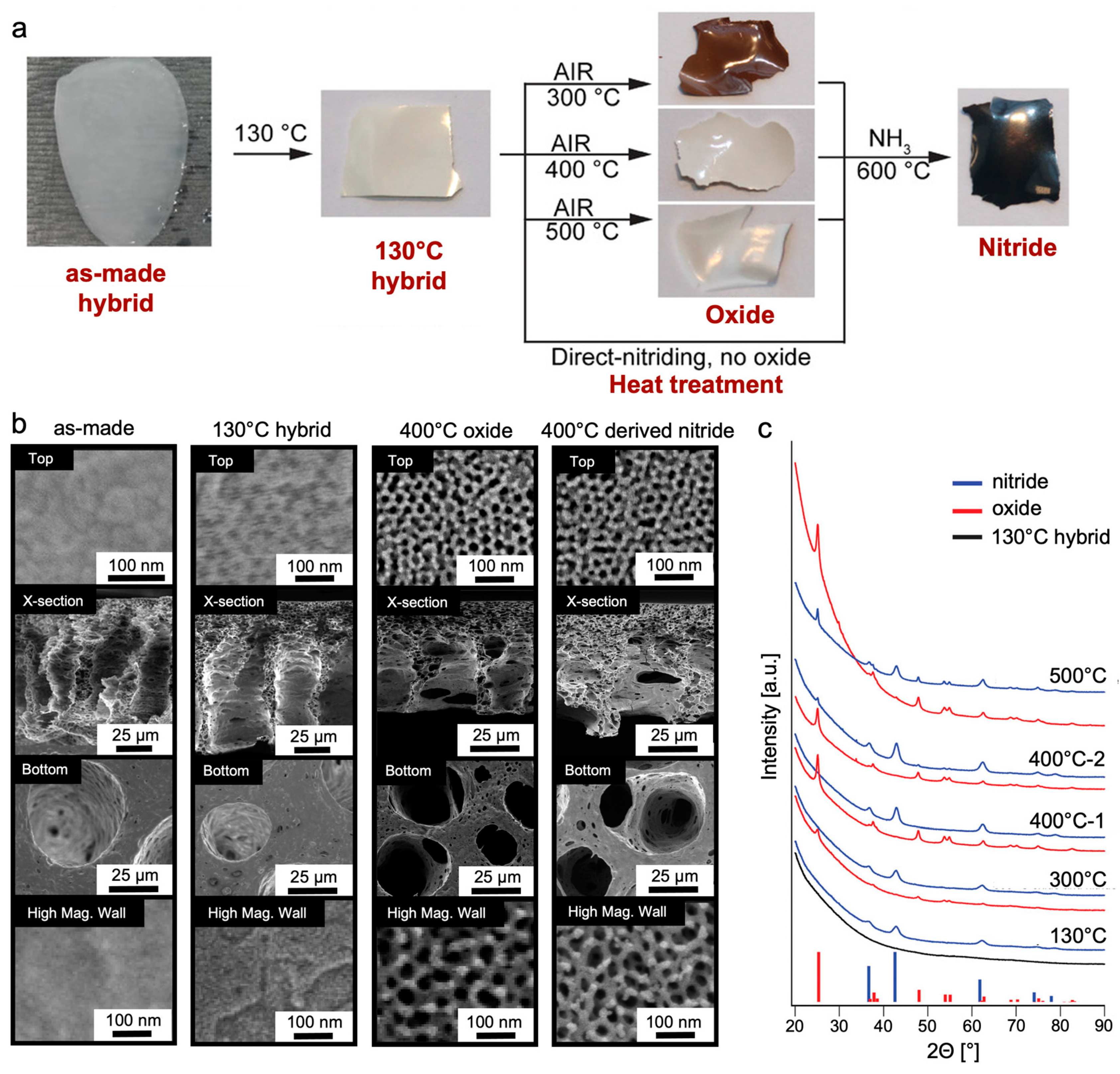{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Single-Component ISV SNIPS Membranes
2.1. General Overview of Structure and Properties
2.2. Elucidation of Structure Formation Mechanisms
2.3. Perspectives on Single-Component ISV SNIPS: The Inverted Designer Cycle and Templating Inorganic Materials
3. Multicomponent Approaches to SNIPS-Derived Asymmetric Porous Materials
3.1. CNIPS-Derived Membranes from BCP plus Additives in the Dope
3.1.1. CNIPS-Derived Membranes from Organic Additives
3.1.2. CNIPS-Derived Organic–Inorganic Hybrid Membranes from Inorganic Additives
3.1.3. CNIPS-Derived Inorganic Membranes from Inorganic Additives
CNIPS-Derived Asymmetric Carbons
CNIPS-Derived Asymmetric Nitrides
Electrical Double Layer Capacitors from CNIPS-Derived Asymmetric Inorganic Materials
3.2. Surface SNIPS (S2NIPS) Derived Membranes from BCP plus Homopolymer in the Dope
3.3. “Mix-and-Match” Derived Asymmetric Membranes from Mixtures of Chemically Distinct BCPs in the Dope
3.4. Perspectives on Multicomponent SNIPS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, Z.; Yu, Y.; Wang, L. Learning from Nature: Use Material Architecture to Break the Performance Tradeoffs. Mater. Des. 2019, 168, 107650. [Google Scholar] [CrossRef]
- Shannon, M.A.; Bohn, P.W.; Elimelech, M.; Georgiadis, J.G.; Mariñas, B.J.; Mayes, A.M. Science and Technology for Water Purification in the Coming Decades. Nature 2008, 452, 301–310. [Google Scholar] [CrossRef]
- Park, H.B.; Kamcev, J.; Robeson, L.M.; Elimelech, M.; Freeman, B.D. Maximizing the Right Stuff: The Trade-off between Membrane Permeability and Selectivity. Science 2017, 356, eaab0530. [Google Scholar] [CrossRef] [Green Version]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sakai, F.; Su, L.; Liu, Y.; Wei, K.; Chen, G.; Jiang, M. Progressive Macromolecular Self-Assembly: From Biomimetic Chemistry to Bio-Inspired Materials. Adv. Mater. 2013, 25, 5215–5256. [Google Scholar] [CrossRef]
- Meng, X. A Mini-Review on Bio-Inspired Polymer Self-Assembly: Single-Component and Interactive Polymer Systems. Emerg. Top. Life Sci. 2022, 6, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, D.; Wang, T. Hierarchical Structures of Bone and Bioinspired Bone Tissue Engineering. Small 2016, 12, 4611–4632. [Google Scholar] [CrossRef] [PubMed]
- Dorin, R.M.; Marques, D.S.; Sai, H.; Vainio, U.; Phillip, W.A.; Peinemann, K.-V.; Nunes, S.P.; Wiesner, U. Solution Small-Angle X-ray Scattering as a Screening and Predictive Tool in the Fabrication of Asymmetric Block Copolymer Membranes. ACS Macro Lett. 2012, 1, 614–617. [Google Scholar] [CrossRef]
- LOEB, S.; SOURIRAJAN, S. Sea Water Demineralization by Means of an Osmotic Membrane. In Saline Water Conversion—II; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1963; Volume 38, pp. 117–132. ISBN 978-0-8412-0039-5. [Google Scholar]
- Strathmann, H.; Kock, K. The Formation Mechanism of Phase Inversion Membranes. Desalination 1977, 21, 241–255. [Google Scholar] [CrossRef]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock Polymers: Panacea or Pandora’s Box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef]
- Peinemann, K.-V.; Abetz, V.; Simon, P.F.W. Asymmetric Superstructure Formed in a Block Copolymer via Phase Separation. Nat. Mater 2007, 6, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Rangou, S.; Abetz, C.; Filiz, V.; Abetz, V. Structure Formation of Integral Asymmetric Composite Membranes of Polystyrene-Block-Poly(2-Vinylpyridine) on a Nonwoven. Macromol. Mater. Eng. 2012, 297, 790–798. [Google Scholar] [CrossRef]
- Hahn, J.; Filiz, V.; Rangou, S.; Clodt, J.; Jung, A.; Buhr, K.; Abetz, C.; Abetz, V. Structure Formation of Integral-Asymmetric Membranes of Polystyrene-Block-Poly(Ethylene Oxide). J. Polym. Sci. Part B Polym. Phys. 2013, 51, 281–290. [Google Scholar] [CrossRef]
- Karunakaran, M.; Shevate, R.; Peinemann, K.-V. Nanostructured Double Hydrophobic Poly(Styrene-b-Methyl Methacrylate) Block Copolymer Membrane Manufactured via a Phase Inversion Technique. RSC Adv. 2016, 6, 29064–29071. [Google Scholar] [CrossRef] [Green Version]
- Schöttner, S.; Schaffrath, H.-J.; Gallei, M. Poly(2-Hydroxyethyl Methacrylate)-Based Amphiphilic Block Copolymers for High Water Flux Membranes and Ceramic Templates. Macromolecules 2016, 49, 7286–7295. [Google Scholar] [CrossRef]
- Höhme, C.; Hahn, J.; Lademann, B.; Meyer, A.; Bajer, B.; Abetz, C.; Filiz, V.; Abetz, V. Formation of High Thermally Stable Isoporous Integral Asymmetric Block Copolymer Membranes. Eur. Polym. J. 2016, 85, 72–81. [Google Scholar] [CrossRef]
- Phillip, W.A.; Dorin, R.M.; Werner, J.; Hoek, E.M.V.; Wiesner, U.; Elimelech, M. Tuning Structure and Properties of Graded Triblock Terpolymer-Based Mesoporous and Hybrid Films. Nano Lett. 2011, 11, 2892–2900. [Google Scholar] [CrossRef]
- Jung, A.; Filiz, V.; Rangou, S.; Buhr, K.; Merten, P.; Hahn, J.; Clodt, J.; Abetz, C.; Abetz, V. Formation of Integral Asymmetric Membranes of AB Diblock and ABC Triblock Copolymers by Phase Inversion. Macromol. Rapid Commun. 2013, 34, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Weidman, J.L.; Mulvenna, R.A.; Boudouris, B.W.; Phillip, W.A. Unusually Stable Hysteresis in the PH-Response of Poly(Acrylic Acid) Brushes Confined within Nanoporous Block Polymer Thin Films. J. Am. Chem. Soc. 2016, 138, 7030–7039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gu, Y.; Li, Y.M.; Beaucage, P.A.; Kao, T.; Wiesner, U. Dynamically Responsive Multifunctional Asymmetric Triblock Terpolymer Membranes with Intrinsic Binding Sites for Covalent Molecule Attachment. Chem. Mater. 2016, 28, 3870–3876. [Google Scholar] [CrossRef]
- Saleem, S.; Rangou, S.; Abetz, C.; Filiz, V.; Abetz, V. Isoporous Membranes from Novel Polystyrene-b-Poly(4-Vinylpyridine)-b-Poly(Solketal Methacrylate) (PS-b-P4VP-b-PSMA) Triblock Terpolymers and Their Post-Modification. Polymers 2020, 12, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Abetz, V. Nonequilibrium Processes in Polymer Membrane Formation: Theory and Experiment. Chem. Rev. 2021, 121, 14189–14231. [Google Scholar] [CrossRef] [PubMed]
- Dorin, R.M.; Sai, H.; Wiesner, U. Hierarchically Porous Materials from Block Copolymers. Chem. Mater. 2014, 26, 339–347. [Google Scholar] [CrossRef]
- Werber, J.R.; Osuji, C.O.; Elimelech, M. Materials for Next-Generation Desalination and Water Purification Membranes. Nat. Rev. Mater. 2016, 1, 16018. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.M.; Gu, Y.; Dorin, R.M.; Wiesner, U. Tuning Substructure and Properties of Supported Asymmetric Triblock Terpolymer Membranes. Polymer 2016, 107, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Dorin, R.M.; Phillip, W.A.; Sai, H.; Werner, J.; Elimelech, M.; Wiesner, U. Designing Block Copolymer Architectures for Targeted Membrane Performance. Polymer 2014, 55, 347–353. [Google Scholar] [CrossRef]
- Hibi, Y.; Hesse, S.A.; Yu, F.; Thedford, R.P.; Wiesner, U. Structural Evolution of Ternary Amphiphilic Block Copolymer Solvent Systems for Phase Inversion Membrane Formation. Macromolecules 2020, 53, 4889–4900. [Google Scholar] [CrossRef]
- Mika, A.M.; Childs, R.F. Acid/Base Properties of Poly(4-Vinylpyridine) Anchored within Microporous Membranes. J. Membr. Sci. 1999, 152, 129–140. [Google Scholar] [CrossRef]
- Qiu, X.; Yu, H.; Karunakaran, M.; Pradeep, N.; Nunes, S.P.; Peinemann, K.-V. Selective Separation of Similarly Sized Proteins with Tunable Nanoporous Block Copolymer Membranes. ACS Nano 2013, 7, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Rahman, M.M.; Abetz, C.; Höhme, A.-L.; Sperling, E.; Abetz, V. Chemically Tailored Multifunctional Asymmetric Isoporous Triblock Terpolymer Membranes for Selective Transport. Adv. Mater. 2020, 32, 1907014. [Google Scholar] [CrossRef] [Green Version]
- Poole, J.L.; Donahue, S.; Wilson, D.; Li, Y.M.; Zhang, Q.; Gu, Y.; Ferebee, R.; Lu, Z.; Dorin, R.M.; Hancock, L.F.; et al. Biocatalytic Stimuli-Responsive Asymmetric Triblock Terpolymer Membranes for Localized Permeability Gating. Macromol. Rapid Commun. 2017, 38, 1700364. [Google Scholar] [CrossRef] [PubMed]
- Riasi, M.S.; Tsaur, L.; Li, Y.M.; Zhang, Q.; Wiesner, U.; Yeghiazarian, L. Stochastic Microstructure Delineation and Flow Simulation in Asymmetric Block Copolymer Ultrafiltration Membranes. J. Membr. Sci. 2023, 668, 121163. [Google Scholar] [CrossRef]
- Pendergast, M.M.; Mika Dorin, R.; Phillip, W.A.; Wiesner, U.; Hoek, E.M.V. Understanding the Structure and Performance of Self-Assembled Triblock Terpolymer Membranes. J. Membr. Sci. 2013, 444, 461–468. [Google Scholar] [CrossRef]
- Li, Y.M.; Zhang, Q.; Álvarez-Palacio, J.R.; Hakem, I.F.; Gu, Y.; Bockstaller, M.R.; Wiesner, U. Effect of Humidity on Surface Structure and Permeation of Triblock Terpolymer Derived SNIPS Membranes. Polymer 2017, 126, 368–375. [Google Scholar] [CrossRef]
- Nunes, S.P.; Sougrat, R.; Hooghan, B.; Anjum, D.H.; Behzad, A.R.; Zhao, L.; Pradeep, N.; Pinnau, I.; Vainio, U.; Peinemann, K.-V. Ultraporous Films with Uniform Nanochannels by Block Copolymer Micelles Assembly. Macromolecules 2010, 43, 8079–8085. [Google Scholar] [CrossRef]
- Nunes, S.P.; Karunakaran, M.; Pradeep, N.; Behzad, A.R.; Hooghan, B.; Sougrat, R.; He, H.; Peinemann, K.-V. From Micelle Supramolecular Assemblies in Selective Solvents to Isoporous Membranes. Langmuir 2011, 27, 10184–10190. [Google Scholar] [CrossRef] [PubMed]
- Abetz, V. Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2015, 36, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Oss-Ronen, L.; Schmidt, J.; Abetz, V.; Radulescu, A.; Cohen, Y.; Talmon, Y. Characterization of Block Copolymer Self-Assembly: From Solution to Nanoporous Membranes. Macromolecules 2012, 45, 9631–9642. [Google Scholar] [CrossRef]
- Stegelmeier, C.; Filiz, V.; Abetz, V.; Perlich, J.; Fery, A.; Ruckdeschel, P.; Rosenfeldt, S.; Förster, S. Topological Paths and Transient Morphologies during Formation of Mesoporous Block Copolymer Membranes. Macromolecules 2014, 47, 5566–5577. [Google Scholar] [CrossRef]
- Marques, D.S.; Vainio, U.; Chaparro, N.M.; Calo, V.M.; Bezahd, A.R.; Pitera, J.W.; Peinemann, K.-V.; Nunes, S.P. Self-Assembly in Casting Solutions of Block Copolymer Membranes. Soft Matter 2013, 9, 5557–5564. [Google Scholar] [CrossRef]
- Gu, Y.; Dorin, R.M.; Tan, K.W.; Smilgies, D.-M.; Wiesner, U. In Situ Study of Evaporation-Induced Surface Structure Evolution in Asymmetric Triblock Terpolymer Membranes. Macromolecules 2016, 49, 4195–4201. [Google Scholar] [CrossRef]
- Grzetic, D.J.; Cooper, A.J.; Delaney, K.T.; Fredrickson, G.H. Modeling Microstructure Formation in Block Copolymer Membranes Using Dynamical Self-Consistent Field Theory. ACS Macro Lett. 2023, 12, 8–13. [Google Scholar] [CrossRef]
- Hampu, N.; Werber, J.R.; Chan, W.Y.; Feinberg, E.C.; Hillmyer, M.A. Next-Generation Ultrafiltration Membranes Enabled by Block Polymers. ACS Nano 2020, 14, 16446–16471. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthi, G.; Hadwiger, M.; Ben-Romdhane, M.; Behzad, A.R.; Madhavan, P.; Nunes, S.P. 3D Membrane Imaging and Porosity Visualization. Ind. Eng. Chem. Res. 2016, 55, 3689–3695. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.; Zhang, Z.; Abetz, C.; Abetz, V.; Segal-Peretz, T. Atomic Layer Deposition Enables Multi-Modal Three-Dimensional Electron Microscopy of Isoporous Membranes. Nanoscale 2023, 15, 3219–3229. [Google Scholar] [CrossRef]
- Gao, H.; Zhong, S.; Dangayach, R.; Chen, Y. Understanding and Designing a High-Performance Ultrafiltration Membrane Using Machine Learning. Environ. Sci. Technol. 2023. [Google Scholar] [CrossRef]
- Gu, Y.; Werner, G.J.; Dorin, R.M.; Robbins, S.W.; Wiesner, U. Graded Porous Inorganic Materials Derived from Self-Assembled Block Copolymer Templates. Nanoscale 2015, 7, 5826–5834. [Google Scholar] [CrossRef]
- Hu, X.-H.; Xiong, S. Fabrication of Nanodevices Through Block Copolymer Self-Assembly. Front. Nanotechnol. 2022, 4, 762996. [Google Scholar] [CrossRef] [Green Version]
- Shevate, R.; Karunakaran, M.; Kumar, M.; Peinemann, K.-V. Polyanionic PH-Responsive Polystyrene-b-Poly(4-Vinyl Pyridine-N-Oxide) Isoporous Membranes. J. Membr. Sci. 2016, 501, 161–168. [Google Scholar] [CrossRef]
- Hoffman, J.R.; Phillip, W.A. 100th Anniversary of Macromolecular Science Viewpoint: Integrated Membrane Systems. ACS Macro Lett. 2020, 9, 1267–1279. [Google Scholar] [CrossRef]
- Zhang, Z.; Rahman, M.M.; Bajer, B.; Scharnagl, N.; Abetz, V. Highly Selective Isoporous Block Copolymer Membranes with Tunable Polyelectrolyte Brushes in Soft Nanochannels. J. Membr. Sci. 2022, 646, 120266. [Google Scholar] [CrossRef]
- Matsen, M.W. Phase Behavior of Block Copolymer/Homopolymer Blends. Macromolecules 1995, 28, 5765–5773. [Google Scholar] [CrossRef]
- Koning, C.; Van Duin, M.; Pagnoulle, C.; Jerome, R. Strategies for Compatibilization of Polymer Blends. Prog. Polym. Sci. 1998, 23, 707–757. [Google Scholar] [CrossRef]
- Hester, J.F.; Mayes, A.M. Design and Performance of Foul-Resistant Poly(Vinylidene Fluoride) Membranes Prepared in a Single-Step by Surface Segregation. J. Membr. Sci. 2002, 202, 119–135. [Google Scholar] [CrossRef]
- Epps, T.H.; Chatterjee, J.; Bates, F.S. Phase Transformations Involving Network Phases in ISO Triblock Copolymer−Homopolymer Blends. Macromolecules 2005, 38, 8775–8784. [Google Scholar] [CrossRef]
- Tada, A.; Geng, Y.; Wei, Q.; Hashimoto, K.; Tajima, K. Tailoring Organic Heterojunction Interfaces in Bilayer Polymer Photovoltaic Devices. Nat. Mater. 2011, 10, 450–455. [Google Scholar] [CrossRef]
- Sai, H.; Tan, K.W.; Hur, K.; Asenath-Smith, E.; Hovden, R.; Jiang, Y.; Riccio, M.; Muller, D.A.; Elser, V.; Estroff, L.A.; et al. Hierarchical Porous Polymer Scaffolds from Block Copolymers. Science 2013, 341, 530–534. [Google Scholar] [CrossRef]
- Lindsay, A.P.; Lewis, R.M.I.; Lee, B.; Peterson, A.J.; Lodge, T.P.; Bates, F.S. A15, σ, and a Quasicrystal: Access to Complex Particle Packings via Bidisperse Diblock Copolymer Blends. ACS Macro Lett. 2020, 9, 197–203. [Google Scholar] [CrossRef]
- Gu, Y.; Wiesner, U. Tailoring Pore Size of Graded Mesoporous Block Copolymer Membranes: Moving from Ultrafiltration toward Nanofiltration. Macromolecules 2015, 48, 6153–6159. [Google Scholar] [CrossRef]
- Clodt, J.I.; Rangou, S.; Schröder, A.; Buhr, K.; Hahn, J.; Jung, A.; Filiz, V.; Abetz, V. Carbohydrates as Additives for the Formation of Isoporous PS-b-P4VP Diblock Copolymer Membranes. Macromol. Rapid Commun. 2013, 34, 190–194. [Google Scholar] [CrossRef]
- Madhavan, P.; Peinemann, K.-V.; Nunes, S.P. Complexation-Tailored Morphology of Asymmetric Block Copolymer Membranes. ACS Appl. Mater. Interfaces 2013, 5, 7152–7159. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, G.; Yi, Z.; Qiu, Y.; Liu, L.; Gao, C. Tailoring the Pore Size and Permeability of Isoporous Membranes through Blending with Poly(Ethylene Glycol): Toward the Balance of Macro- and Microphase Separation. J. Membr. Sci. 2020, 598, 117755. [Google Scholar] [CrossRef] [Green Version]
- Rangou, S.; Buhr, K.; Filiz, V.; Clodt, J.I.; Lademann, B.; Hahn, J.; Jung, A.; Abetz, V. Self-Organized Isoporous Membranes with Tailored Pore Sizes. J. Membr. Sci. 2014, 451, 266–275. [Google Scholar] [CrossRef]
- Yu, H.; Qiu, X.; Nunes, S.P.; Peinemann, K.-V. Self-Assembled Isoporous Block Copolymer Membranes with Tuned Pore Sizes. Angew. Chem. Int. Ed. 2014, 53, 10072–10076. [Google Scholar] [CrossRef] [PubMed]
- Shevate, R.; Kumar, M.; Karunakaran, M.; Canlas, C.; Peinemann, K.-V. Surprising Transformation of a Block Copolymer into a High Performance Polystyrene Ultrafiltration Membrane with a Hierarchically Organized Pore Structure. J. Mater. Chem. A 2018, 6, 4337–4345. [Google Scholar] [CrossRef]
- Zhang, Z.; Simon, A.; Abetz, C.; Held, M.; Höhme, A.-L.; Schneider, E.S.; Segal-Peretz, T.; Abetz, V. Hybrid Organic–Inorganic–Organic Isoporous Membranes with Tunable Pore Sizes and Functionalities for Molecular Separation. Adv. Mater. 2021, 33, 2105251. [Google Scholar] [CrossRef]
- Guldin, S.; Kohn, P.; Stefik, M.; Song, J.; Divitini, G.; Ecarla, F.; Ducati, C.; Wiesner, U.; Steiner, U. Self-Cleaning Antireflective Optical Coatings. Nano Lett. 2013, 13, 5329–5335. [Google Scholar] [CrossRef]
- Gu, Y.; Dorin, R.M.; Wiesner, U. Asymmetric Organic–Inorganic Hybrid Membrane Formation via Block Copolymer–Nanoparticle Co-Assembly. Nano Lett. 2013, 13, 5323–5328. [Google Scholar] [CrossRef]
- Gallei, M.; Rangou, S.; Filiz, V.; Buhr, K.; Bolmer, S.; Abetz, C.; Abetz, V. The Influence of Magnesium Acetate on the Structure Formation of Polystyrene-Block-Poly(4-Vinylpyridine)-Based Integral-Asymmetric Membranes. Macromol. Chem. Phys. 2013, 214, 1037–1046. [Google Scholar] [CrossRef]
- Shevate, R.; Kumar, M.; Cheng, H.; Hong, P.-Y.; Behzad, A.R.; Anjum, D.; Peinemann, K.-V. Rapid Size-Based Protein Discrimination inside Hybrid Isoporous Membranes. ACS Appl. Mater. Interfaces 2019, 11, 8507–8516. [Google Scholar] [CrossRef]
- Zhang, Z.; Rahman, M.M.; Abetz, C.; Abetz, V. High-Performance Asymmetric Isoporous Nanocomposite Membranes with Chemically-Tailored Amphiphilic Nanochannels. J. Mater. Chem. A 2020, 8, 9554–9566. [Google Scholar] [CrossRef]
- Hesse, S.A.; Werner, J.G.; Wiesner, U. One-Pot Synthesis of Hierarchically Macro- and Mesoporous Carbon Materials with Graded Porosity. ACS Macro Lett. 2015, 4, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Chen, I.-W.; Liu, F.; Yang, C.; Bi, H.; Xu, F.; Huang, F. Nitrogen-Doped Mesoporous Carbon of Extraordinary Capacitance for Electrochemical Energy Storage. Science 2015, 350, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-W.; Li, F.; Liu, M.; Lu, G.Q.; Cheng, H.-M. 3D Aperiodic Hierarchical Porous Graphitic Carbon Material for High-Rate Electrochemical Capacitive Energy Storage. Angew. Chem. Int. Ed. 2008, 47, 373–376. [Google Scholar] [CrossRef]
- Hesse, S.A.; Beaucage, P.A.; Smilgies, D.-M.; Wiesner, U. Structurally Asymmetric Porous Carbon Materials with Ordered Top Surface Layers from Nonequilibrium Block Copolymer Self-Assembly. Macromolecules 2021, 54, 2979–2991. [Google Scholar] [CrossRef]
- Hesse, S.A.; Fritz, K.E.; Beaucage, P.A.; Susca, E.M.; Suntivich, J.; Wiesner, U. Oxides and Nitrides with Asymmetric Pore Structure from Block Copolymer Co-Assembly and Non-Solvent Induced Phase Separation. Macromol. Chem. Phys. 2023, 224, 2200304. [Google Scholar] [CrossRef]
- Fritz, K.E.; Beaucage, P.A.; Matsuoka, F.; Wiesner, U.; Suntivich, J. Mesoporous Titanium and Niobium Nitrides as Conductive and Stable Electrocatalyst Supports in Acid Environments. Chem. Commun. 2017, 53, 7250–7253. [Google Scholar] [CrossRef]
- Aricò, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; van Schalkwijk, W. Nanostructured Materials for Advanced Energy Conversion and Storage Devices. Nat. Mater. 2005, 4, 366–377. [Google Scholar] [CrossRef]
- Rolison, D.R.; Long, J.W.; Lytle, J.C.; Fischer, A.E.; Rhodes, C.P.; McEvoy, T.M.; Bourg, M.E.; Lubers, A.M. Multifunctional 3D Nanoarchitectures for Energy Storage and Conversion. Chem. Soc. Rev. 2008, 38, 226–252. [Google Scholar] [CrossRef]
- Hesse, S.A.; Fritz, K.E.; Beaucage, P.A.; Thedford, R.P.; Yu, F.; Di Salvo, F.J.; Suntivich, J.; Wiesner, U. Materials Combining Asymmetric Pore Structures with Well-Defined Mesoporosity for Energy Storage and Conversion. ACS Nano 2020, 14, 16897–16906. [Google Scholar] [CrossRef]
- Werner, J.G.; Hoheisel, T.N.; Wiesner, U. Synthesis and Characterization of Gyroidal Mesoporous Carbons and Carbon Monoliths with Tunable Ultralarge Pore Size. ACS Nano 2014, 8, 731–743. [Google Scholar] [CrossRef]
- Guillen, G.R.; Pan, Y.; Li, M.; Hoek, E.M.V. Preparation and Characterization of Membranes Formed by Nonsolvent Induced Phase Separation: A Review. Ind. Eng. Chem. Res. 2011, 50, 3798–3817. [Google Scholar] [CrossRef] [Green Version]
- Bucher, T.; Filiz, V.; Abetz, C.; Abetz, V. Formation of Thin, Isoporous Block Copolymer Membranes by an Upscalable Profile Roller Coating Process—A Promising Way to Save Block Copolymer. Membranes 2018, 8, 57. [Google Scholar] [CrossRef]
- Hampu, N.; Werber, J.R.; Hillmyer, M.A. Co-Casting Highly Selective Dual-Layer Membranes with Disordered Block Polymer Selective Layers. ACS Appl. Mater. Interfaces 2020, 12, 45351–45362. [Google Scholar] [CrossRef] [PubMed]
- Hibi, Y.; Wiesner, U. Surface Segregation and Self-Assembly of Block-Copolymer Separation Layers on Top of Homopolymer Substructures in Asymmetric Ultrafiltration Membranes from a Single Casting Step. Adv. Funct. Mater. 2021, 31, 2009387. [Google Scholar] [CrossRef]
- Li, Y.M.; Srinivasan, D.; Vaidya, P.; Gu, Y.; Wiesner, U. Asymmetric Membranes from Two Chemically Distinct Triblock Terpolymers Blended during Standard Membrane Fabrication. Macromol. Rapid Commun. 2016, 37, 1689–1693. [Google Scholar] [CrossRef]
- Choi, S.-H.; Bates, F.S.; Lodge, T.P. Molecular Exchange in Ordered Diblock Copolymer Micelles. Macromolecules 2011, 44, 3594–3604. [Google Scholar] [CrossRef]
- Radjabian, M.; Abetz, V. Tailored Pore Sizes in Integral Asymmetric Membranes Formed by Blends of Block Copolymers. Adv. Mater. 2015, 27, 352–355. [Google Scholar] [CrossRef]
- Yu, H.; Qiu, X.; Moreno, N.; Ma, Z.; Calo, V.M.; Nunes, S.P.; Peinemann, K.-V. Self-Assembled Asymmetric Block Copolymer Membranes: Bridging the Gap from Ultra- to Nanofiltration. Angew. Chem. Int. Ed. 2015, 54, 13937–13941. [Google Scholar] [CrossRef]
- Plamper, F.A.; Ruppel, M.; Schmalz, A.; Borisov, O.; Ballauff, M.; Müller, A.H.E. Tuning the Thermoresponsive Properties of Weak Polyelectrolytes: Aqueous Solutions of Star-Shaped and Linear Poly(N,N-Dimethylaminoethyl Methacrylate). Macromolecules 2007, 40, 8361–8366. [Google Scholar] [CrossRef]
- Huang, T.; Moosa, B.A.; Hoang, P.; Liu, J.; Chisca, S.; Zhang, G.; AlYami, M.; Khashab, N.M.; Nunes, S.P. Molecularly-Porous Ultrathin Membranes for Highly Selective Organic Solvent Nanofiltration. Nat. Commun. 2020, 11, 5882. [Google Scholar] [CrossRef]
- Jiang, Z.; Dong, R.; Evans, A.M.; Biere, N.; Ebrahim, M.A.; Li, S.; Anselmetti, D.; Dichtel, W.R.; Livingston, A.G. Aligned Macrocycle Pores in Ultrathin Films for Accurate Molecular Sieving. Nature 2022, 609, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cai, Y.; Zhang, C.; Wei, W.; Chen, C.; Liu, L.; Yang, K.; Ma, Y.; Wang, Y.; Tseng, C.-C.; et al. Fast Water Transport and Molecular Sieving through Ultrathin Ordered Conjugated-Polymer-Framework Membranes. Nat. Mater. 2022, 21, 1183–1190. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, L.; Deng, H.; Liu, Q.; You, X.; Yuan, J.; Jiang, Z.; Zhang, S. Ultrathin Membranes for Separations: A New Era Driven by Advanced Nanotechnology. Adv. Mater. 2022, 34, 2108457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leunissen, M.E.; Christova, C.G.; Hynninen, A.-P.; Royall, C.P.; Campbell, A.I.; Imhof, A.; Dijkstra, M.; van Roij, R.; van Blaaderen, A. Ionic Colloidal Crystals of Oppositely Charged Particles. Nature 2005, 437, 235–240. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaur, L.; Wiesner, U.B. Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems. Polymers 2023, 15, 2020. https://doi.org/10.3390/polym15092020
Tsaur L, Wiesner UB. Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems. Polymers. 2023; 15(9):2020. https://doi.org/10.3390/polym15092020
Chicago/Turabian StyleTsaur, Lieihn, and Ulrich B. Wiesner. 2023. "Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems" Polymers 15, no. 9: 2020. https://doi.org/10.3390/polym15092020
APA StyleTsaur, L., & Wiesner, U. B. (2023). Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems. Polymers, 15(9), 2020. https://doi.org/10.3390/polym15092020