
MD-DFT Calculations on Dissociative Absorption Configurations of FOX-7 on (001)- and (101)-Oriented Crystalline Parylene Protective Membranes


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Surface Structures of the (001)- and (101)-Oriented Crystalline Parylene Protective Membranes
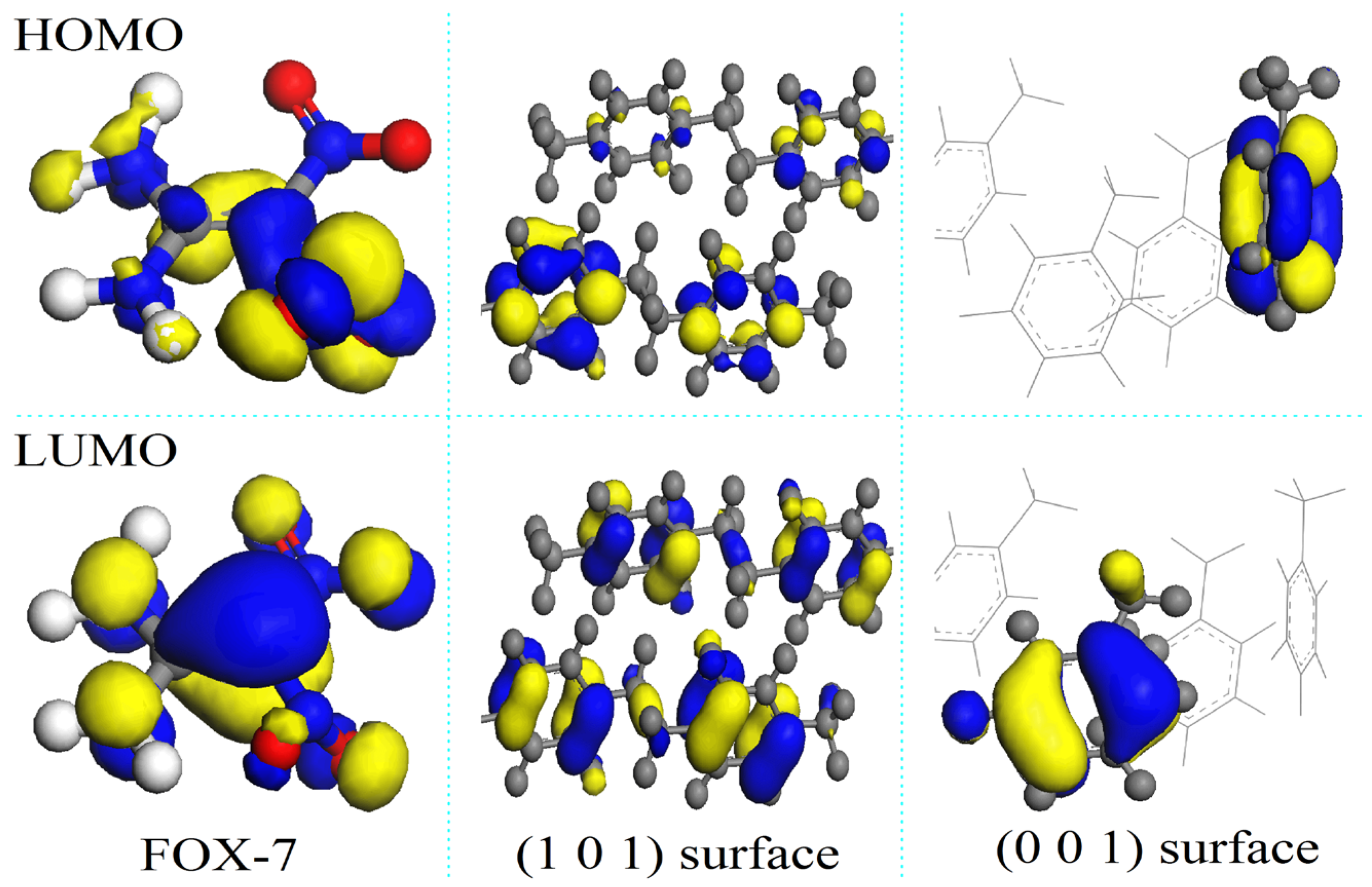
3.2. Adsorption Configurations of FOX-7 on the (001)- and (101)-Oriented Crystalline Parylene Protective Membranes
3.3. Nitro-to-Nitrite Rearrangement of the Decomposition Products of FOX-7 on the Parylene Membranes
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rose, A.; Zhu, Z.; Madigan, C.F.; Swager, T.M.; Bulovi, V. Sensitivity gains in chemosensing by lasing action in organic polymers. Nature 2005, 434, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Anniyappan, M.; Talawar, M.B.; Venugopalan, S.B.; Gandhe, R. Synthesis Method for preparation of fine TATB (2–5 μm) and its evaluation in plastic bonded explosive (PBX) formulations. J. Hazard. Mater. 2006, 137, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Song, J.; Zhao, F.; Ma, H.; Gao, H.; Chang, C.; Ren, Y.; Hu, R. Thermal behavior, specific heat capacity and adiabatic time-to-explosion of G(FOX-7). J. Hazard. Mater. 2008, 158, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Evers, J.; Klapötke, T.M.; Mayer, P.; Oehlinger, G.; Welch, J. alpha and beta-FOX-7, polymorphs of a high energy density material, studied by X-ray single crystal and powder investigations in the temperature range from 200 to 423 K. Inorg. Chem. 2006, 45, 4996–5007. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, H. High-pressure behavior of crystalline FOX-7 by density functional theory calculations. Comput. Mater. Sci. 2008, 42, 689–703. [Google Scholar] [CrossRef]
- Vo, T.T.; Parrish, D.A.; Shreeve, J.M. 1,1-Diamino-2,2-dintroethene (FOX-7) in copper and nickel diamine complexes and copper FOX-7. Inorg. Chem. 2012, 51, 1963–1968. [Google Scholar] [CrossRef]
- Booth, R.S.; Lam, C.S.; Brynteson, M.D.; Wang, L.; Butler, L.J. Elucidating the Decomposition Mechanism of Energetic Materials with Geminal Dinitro Groups Using 2-Bromo-2-nitropropane Photodissociation. J. Chem. Phys. A 2013, 117, 9531–9547. [Google Scholar] [CrossRef]
- Bishop, M.M.; Velisavljevic, N.; Chellappa, R.; Vohra, Y.K. High Pressure–Temperature Phase Diagram of 1,1-Diamino-2,2-dinitroethylene (FOX-7). J. Phys. Chem. A 2015, 119, 9739–9747. [Google Scholar] [CrossRef]
- Yuan, X.T.; Yang, H.S.; Wang, L.L. Effects of aestivation on the energy budget of sea cucumber Apostichopus japonicus (Selenka) (Echinodermata: Holothuroidea). Acta Ecol. Sin. 2008, 27, 3155–3161. [Google Scholar] [CrossRef]
- Hu, A.; Larade, B.; Rachid, H.A.; Lussier, L.S.; Guo, H. Theoretical Prediction of Heats of Sublimation of Energetic Materials Using Pseudo-Atomic Orbital Density Functional Theory Calculations. Propellants Explos. Pyrotech. 2007, 32, 331–337. [Google Scholar] [CrossRef]
- Sorescu, D.C.; Boatz, J.A.; Thompson, D.L. First-principles calculations of the adsorption of nitromethane and 1,1-diamino-2,2-dinitroethylene (FOX-7) molecules on the Al(111) surface. J. Phys. Chem. B 2004, 107, 8953–8964. [Google Scholar] [CrossRef]
- Rashkeev, S.N.; Kuklja, M.M.; Zerilli, F.J. Electronic excitations and decomposition of 1,1-diamino-2,2-dinitroethylene. Appl. Phys. Lett. 2003, 82, 1371–1373. [Google Scholar] [CrossRef]
- Yan, A.H.; Son, S.F.; Jackson, T.L.; Venugopal, P. Validation of numerical simulations for nano-aluminum composite solid propellants. J. Propuls. Power 2011, 27, 1280–1287. [Google Scholar] [CrossRef]
- Sonmez, G.; Shen, C.K.F.; Rubin, Y.; Wudl, F. A Red, Green, and Blue (RGB) Polymeric Electrochromic Device (PECD): The Dawning of the PECD Era. Angew. Chem. J. Ger. Chem. Soc. 2005, 43, 1489–1502. [Google Scholar] [CrossRef]
- Tompa, A.S. The ultrastructure and mineralogy of the dart from Philomycus carolinianus (Pulmonata: Gastropoda) with a brief survey of the occurrence of darts in land snails. Veliger 1980, 23, 35–42. [Google Scholar]
- Castorina, T.C.; Smetana, A.F. Effect of polymer coating on ammonium nitrate substrate. J. Appl. Polym. Sci. 2010, 18, 1373–1383. [Google Scholar] [CrossRef]
- Brown, K.E.; Monroe, D.C.; Brown, G.W. Effect of pellet coatings on PETN porosity and slapper detonator efficacy. AIP Conf. Proc. 2017, 1793, 040011. [Google Scholar] [CrossRef]
- Dai, X.; Xu, J.; Wen, Y.; Li, Y.; Huang, F.; Li, M.; Zeng, Q. Delay Mechanism of β→δ Phase Transition of Cyclotetramethylene Tetranitramine in Polymer Bonded Explosive Formulations by Heat Conduction Obstacle. Propellants Explos. Pyrotech. 2016, 41, 637–640. [Google Scholar] [CrossRef]
- Smalara, K.; Giełdo, A.; Bobrowski, M.; Rybicki, J.; Czaplewski, C. Theoretical study of polymerization mechanism of p-xylylene based polymers. J. Phys. Chem. A 2010, 114, 4296–4303. [Google Scholar] [CrossRef]
- Bae, J.; Suh, M.J.; Kim, B.S. Optimal production of 7,10-dihydroxy-8(E)-hexadecenoic acid from palmitoleic acid by Pseudomonas aeruginosa PR3. New Biotechnol. 2010, 27, 352–357. [Google Scholar] [CrossRef]
- Bian, L.; Shu, Y.; Wang, X. Computational investigation on the new high energy density material of aluminum enriched 1,1-diamino-2,2-dinitroethylene. J. Mol. Model. 2013, 19, 131–138. [Google Scholar] [CrossRef]
- Kahouli, A.; Sylvestre, A.; Pairis, S. Effect of ClH aromatic substitution on structural and dielectric properties of poly(p-xylylene). Polymer 2012, 53, 3001–3007. [Google Scholar] [CrossRef]
- Mattos, E.C.; Moreira, E.D.; Diniz, M.F. Characterization of Polymer-Coated RDX and HMX Particles. Propellants Explos. Pyrotech. 2008, 33, 44–50. [Google Scholar] [CrossRef]
- Zhao, L.; Gou, Y.; Li, H. A Network Pharmacology Approach to Determine Active Compounds and Action Mechanisms of Ge-Gen-Qin-Lian Decoction for Treatment of Type 2 Diabetes. J. Evid.-Based Complement. Altern. Med. eCAM 2014, 2014, 495840. [Google Scholar] [CrossRef]
- Streltsov, D.R.; Mailyan, K.A.; Gusev, A.V. Structure and optical properties of thin poly(p-xylylene)—Silver nanocomposite films prepared by low-temperature vapor deposition polymerization. Polymer 2015, 71, 60–69. [Google Scholar] [CrossRef]
- Frac, I.; Kucinska, M.; Gawrys, P. Ambipolar organic thin film transistors prepared with a one step solution technique. Synth. Met. 2016, 220, 197–201. [Google Scholar] [CrossRef]
- Sroka-Bartnicka, A.; Olejniczak, S.; Ciesielski, W. Solid State NMR Study and Density Functional Theory (DFT) Calculations of Structure and Dynamics of Poly(p-xylylenes). J. Phys. Chem. B 2009, 113, 5464–5472. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Zeman, S.; Sánchez Jiménez, P.E. The effect of polymer matrices on the thermal hazard properties of RDX-based PBXs by using model-free and combined kinetic analysis. J. Phys. Chem. C 2014, 271, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Shu, Y.; Wang, X. A molecular dynamics study on permeabilityof gases through parylene AF8 membranes. Polym. Adv. Technol. 2012, 23, 1429–1528. [Google Scholar] [CrossRef]
- Bian, L.; Shu, Y.; Xu, J. Computational Investigation on Adsorption and Diffusion of 4 Gas Molecules in High Pressure PPX Structure. Integr. Ferroelectr. 2011, 127, 83–90. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Bian, L.; Shu, Y.; Li, H. Sorption and permeation of gaseous molecules in amorphous and crystalline PPX C membranes: Molecular dynamics and grand canonical Monte Carlo simulation studies. Chin. Phys. B 2012, 7, 074208. [Google Scholar] [CrossRef]
- Sorescu, D.C.; Boatz, J.A.; Thompson, D.L. Classical and Quantum-Mechanical Studies of Crystalline FOX-7 (1,1-Diamino-2,2-dinitroethylene). J. Phys. Chem. A 2001, 105, 5010–5021. [Google Scholar] [CrossRef]
- Taylor, D.E.; Rob, F.; Rice, B.M. A molecular dynamics study of 1,1-diamino-2,2-dinitroethylene (FOX-7) crystal using a symmetry adapted perturbation theory-based intermolecular force field. Phys. Chem. Chem. Phys. 2011, 13, 16629–16636. [Google Scholar] [CrossRef]
- Isoda, S.; Tsuji, M.; Ohara, M. Structural analysis of β-form poly(p-xylyene) starting from a high-resolution image. Polymers 1983, 24, 1155–1161. [Google Scholar] [CrossRef]
- Chung, Y.; Murmann, B.; Selvarasah, S. Low-voltage and short-channel pentacene field-effect transistors with top-contact geometry using parylene-C shadow masks. Appl. Phys. Lett. 2010, 96, 133306. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yadav, V.G.; Sarah, D.L. Cell and protein compatibility of parylene-C surfaces. Langmuir 2007, 23, 11718–11725. [Google Scholar] [CrossRef]
- He, X.; Zhang, F.; Wang, R. Preparation of a carbon nanotube/carbon fiber multi-scale reinforcement by grafting multi-walled carbon nanotubes onto the fibers. Carbon 2007, 45, 2559–2563. [Google Scholar] [CrossRef]
- Hanyaloglu, B.; Aydinli, A.; Oye, M. Low dielectric constant Parylene-F-like films for intermetal dielectric applications. Appl. Phys. Lett. 1999, 74, 606–608. [Google Scholar] [CrossRef]
- Diaham, S.; Bechara, M.; Locatelli, M.L. Influence of crystallization-induced amorphous phase confinement on α- and β-relaxation molecular mobility in parylene F. J. Appl. Phys. 2011, 110, 063703. [Google Scholar] [CrossRef]
- Sutcliffe, R.; Lee, W.W.; Gaynor, J.F. Characterization and aluminum metallization of a parylene AF-4 surface. Appl. Surf. Sci. 1998, 126, 43–56. [Google Scholar] [CrossRef]
- Sabri, S.S.; Levesque, P.L.; Aguirre, C.M. Graphene field effect transistors with parylene gate dielectric. Appl. Phys. Lett. 2009, 95, 242104. [Google Scholar] [CrossRef]
- Zhang, J.X.; Zhang, L.D.; Xu, W. Surface plasmon polaritons: Physics and applications. J. Phys. D Appl. Phys. 2012, 45, 113001. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parylene C [36,37,38] | Parylene F [39,40] | Parylene AF4 [41] | Parylene AF8 [21] | |
|---|---|---|---|---|
| a (nm) | 0.95 | 0.88 | 1.15 | 1.17 |
| b (nm) | 2.15 | 2.15 | 1.95 | 2.13 |
| c (nm) | 4.21 | 4.64 | 3.72 | 3.85 |
| Density (g·cm−3) | 1.29 (1.27) | 1.13 (1.11) | 1.71 (1.58) | 2.17 (1.93) |
| Tg (K) | 332 (347) | 312 (289) | 840 (773) | 610 (673) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, W.; Bian, L.; Dong, F.; Nie, J.; Yang, J. MD-DFT Calculations on Dissociative Absorption Configurations of FOX-7 on (001)- and (101)-Oriented Crystalline Parylene Protective Membranes. Polymers 2024, 16, 438. https://doi.org/10.3390/polym16030438
Luo W, Bian L, Dong F, Nie J, Yang J. MD-DFT Calculations on Dissociative Absorption Configurations of FOX-7 on (001)- and (101)-Oriented Crystalline Parylene Protective Membranes. Polymers. 2024; 16(3):438. https://doi.org/10.3390/polym16030438
Chicago/Turabian StyleLuo, Weihui, Liang Bian, Faqin Dong, Jianan Nie, and Jingjie Yang. 2024. "MD-DFT Calculations on Dissociative Absorption Configurations of FOX-7 on (001)- and (101)-Oriented Crystalline Parylene Protective Membranes" Polymers 16, no. 3: 438. https://doi.org/10.3390/polym16030438
APA StyleLuo, W., Bian, L., Dong, F., Nie, J., & Yang, J. (2024). MD-DFT Calculations on Dissociative Absorption Configurations of FOX-7 on (001)- and (101)-Oriented Crystalline Parylene Protective Membranes. Polymers, 16(3), 438. https://doi.org/10.3390/polym16030438