Fine-Tuning of Polymeric Resins and their Interfaces with Amorphous Calcium Phosphate. A Strategy for Designing Effective Remineralizing Dental Composites

Abstract
:1. Introduction
2. Results and Discussion
2.1. Characteristics of ACP filler

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property/method | Findings |
|---|---|
| Long-range structure (XRD) | Two diffuse broad bands in 2θ(°) = (4 to 60) region typical of ACP |
| Short-range structure (FTIR) | Two typical phosphate absorption bands at (1200 to 900) cm−1 and (630 to 500) cm−1 |
| Morphology, size (SEM) | Heterogeneous particle sizes, predominantly agglomerates ranging from approx. 5 µm to 20 µm; some larger than 100 µm |
| Particle size (PSD analysis) | Particle sizes ranging from submicron to > 100 µm in diameter; average calculated median diameter, dm = 7.4 µm |
| Water content (TGA) | Zr-ACP filler contained a mass fraction of (16.1 ± 2.0)% water |
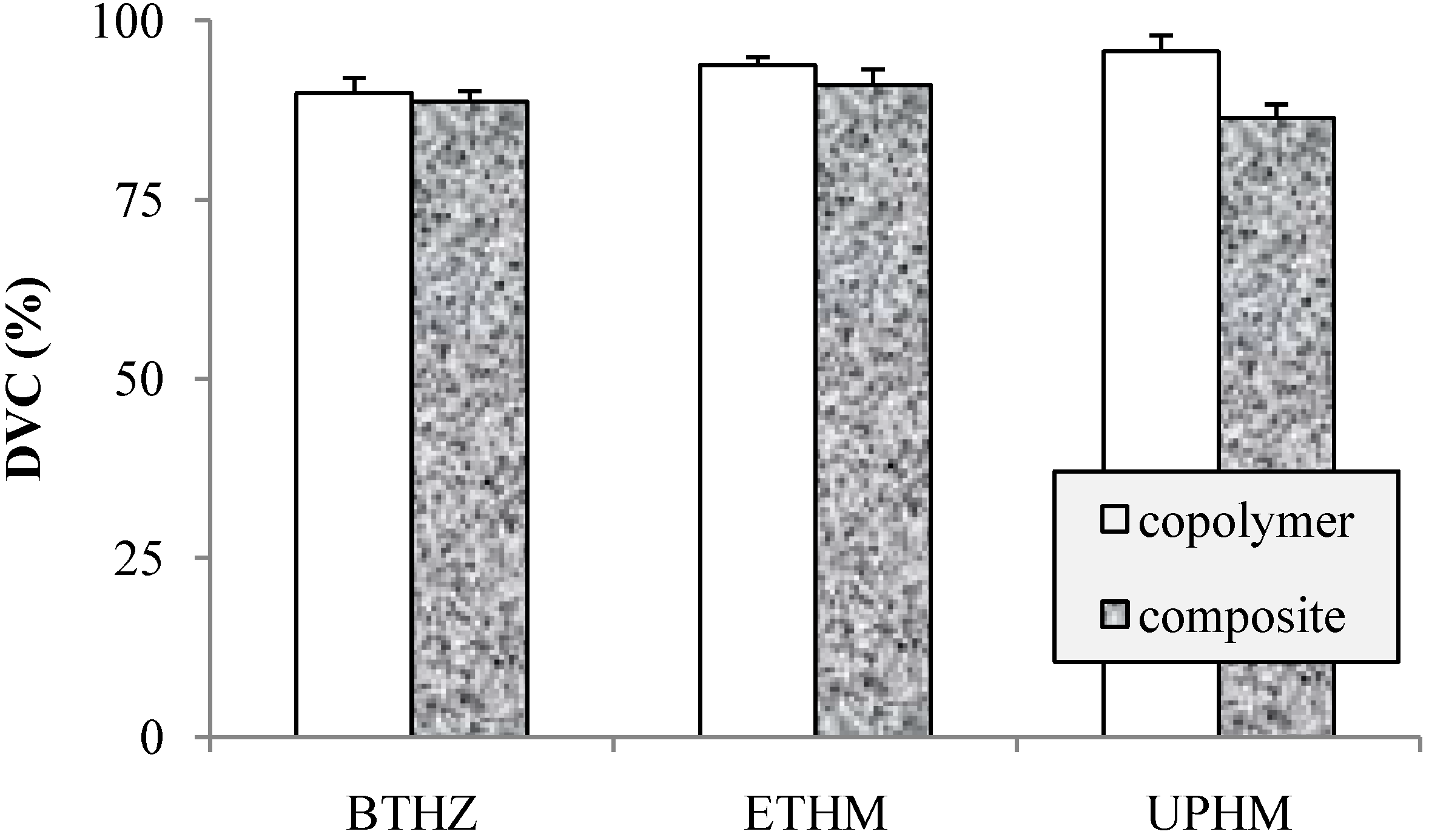
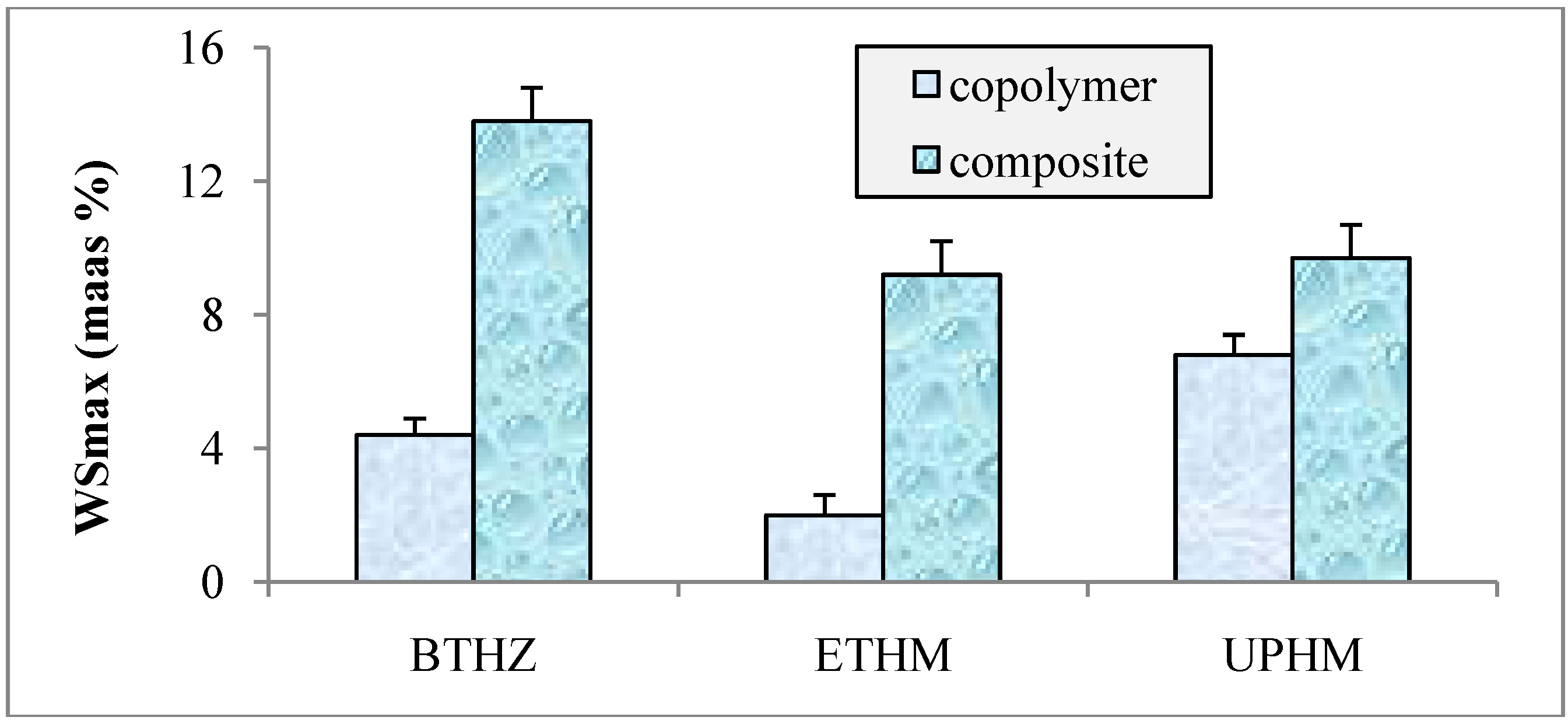
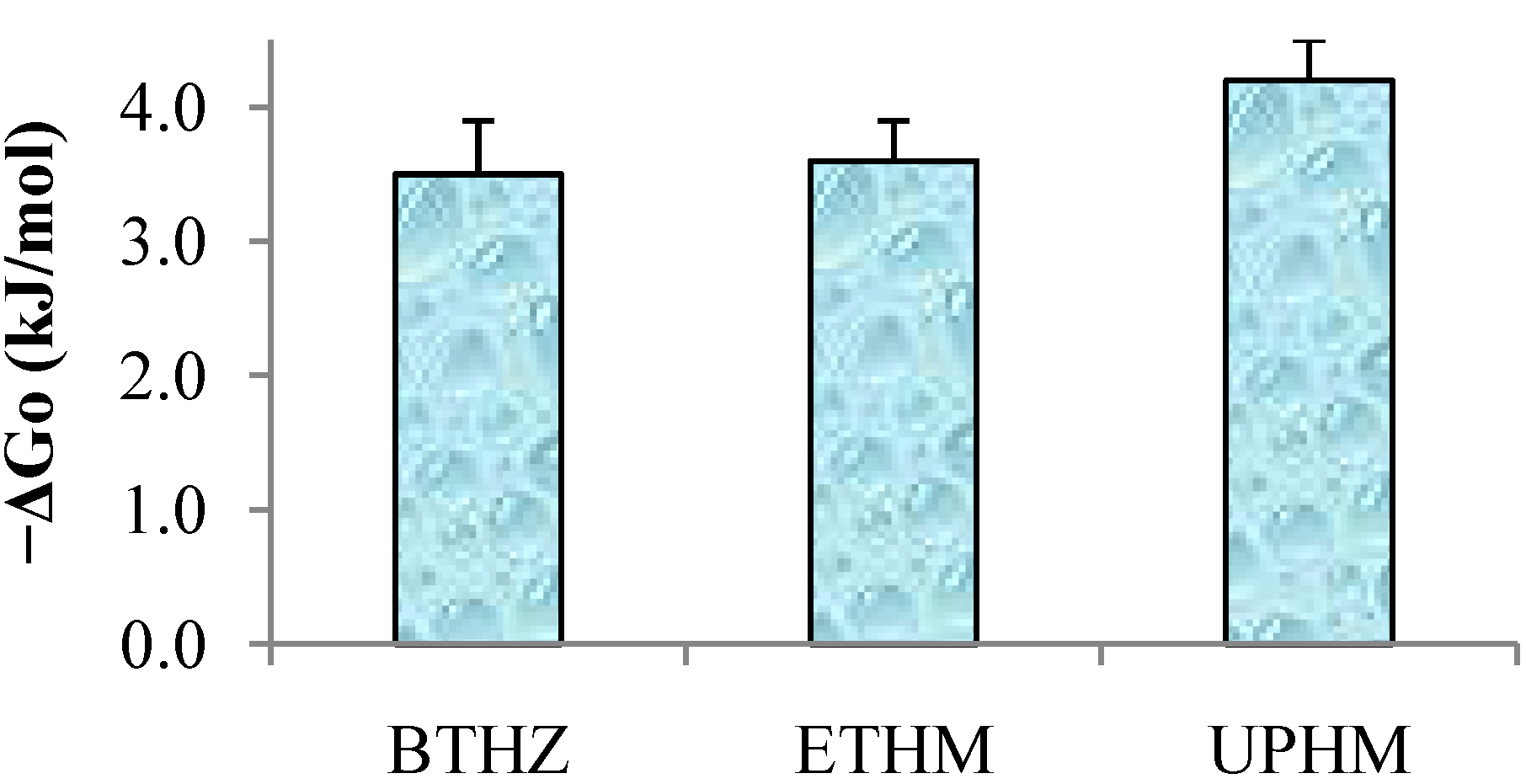
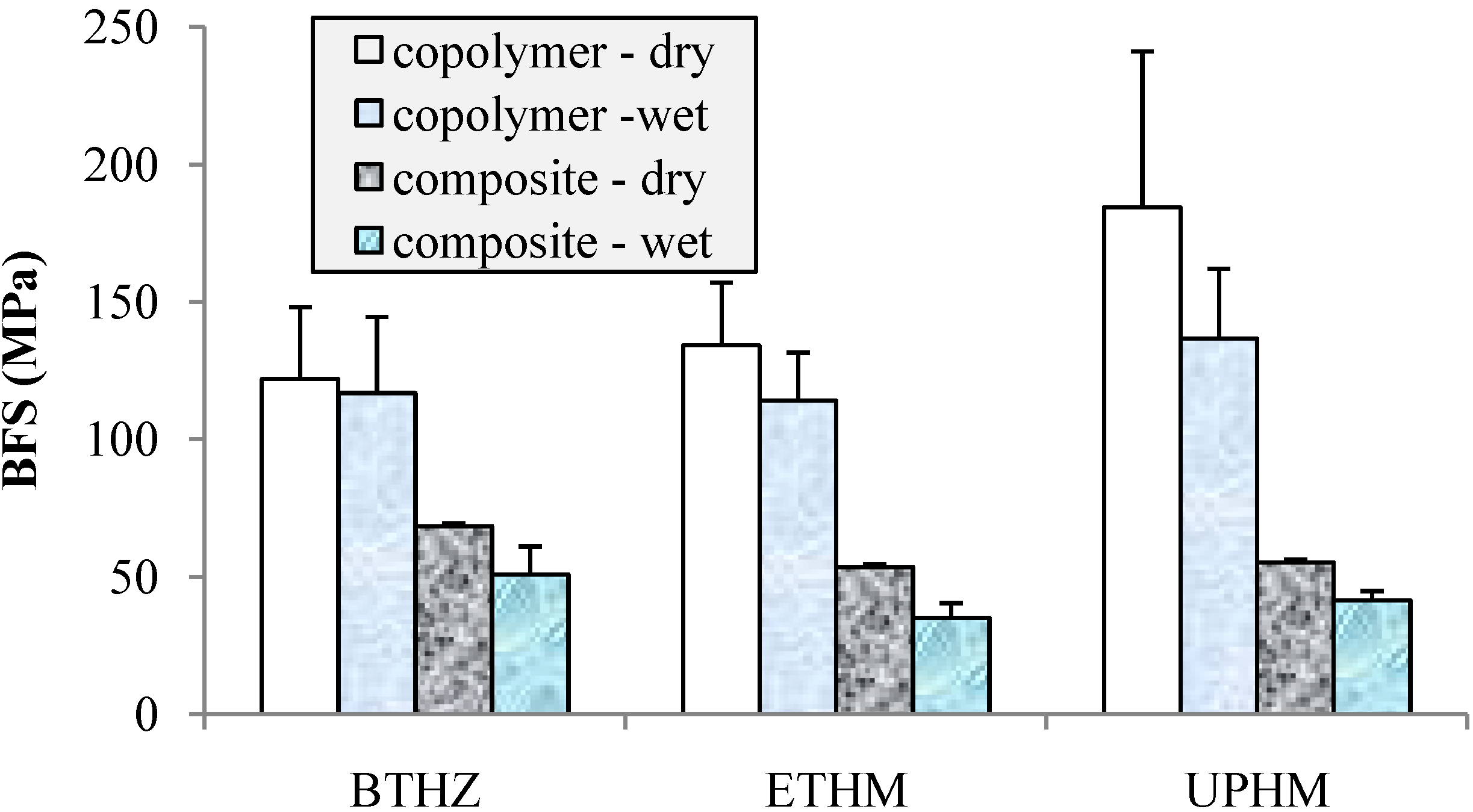
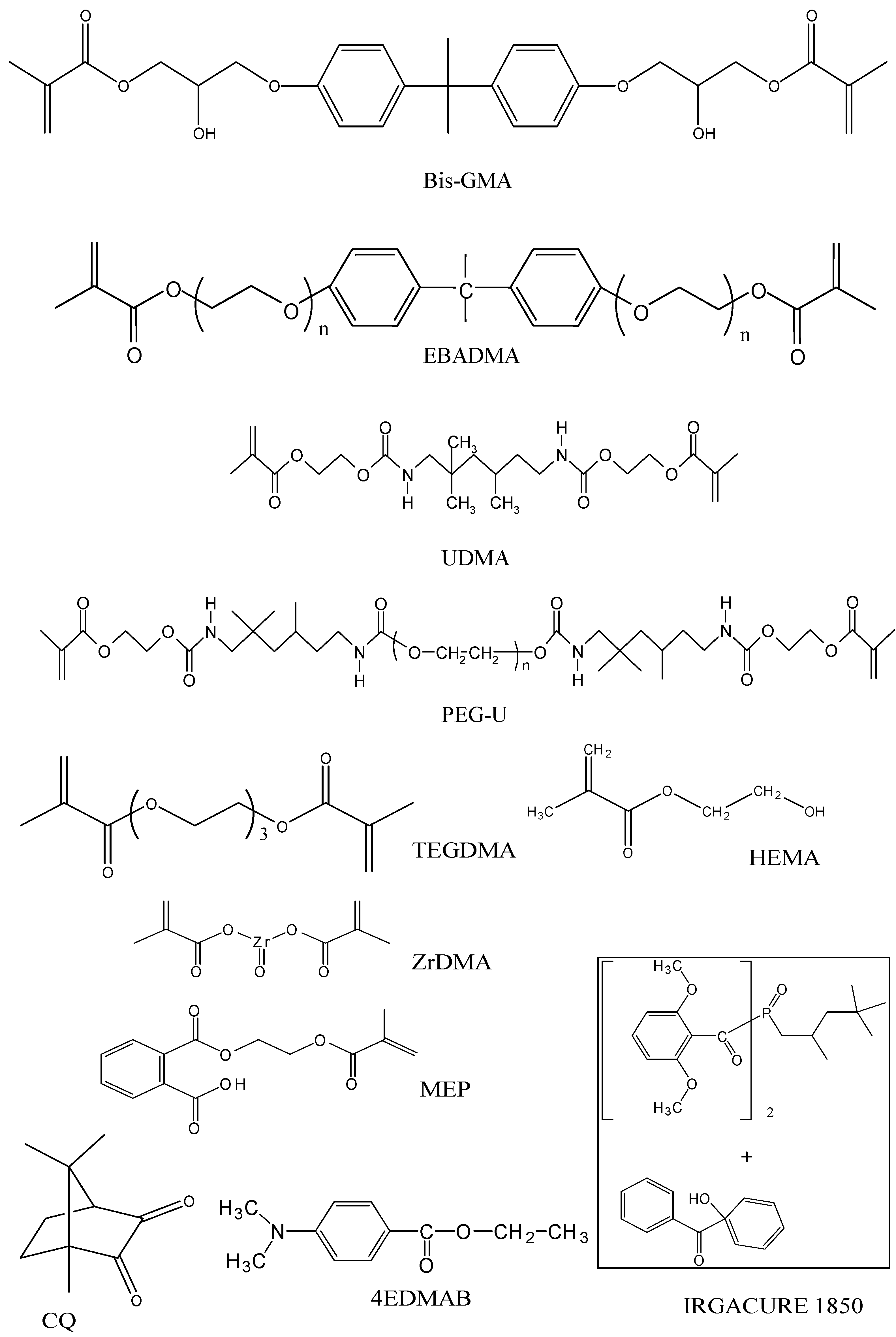
2.2. Evaluation of copolymers and composites
| Monomer/photoinitiator | BTHZ resin | ETHM resin | UPHM resin |
|---|---|---|---|
| Bis-GMA: 2,2-bis[p-(2‘-hydroxy-3-methacryloxypropoxy)phenyl]propane | 35.5 | - | - |
| EBPADMA: ethoxylated bisphenol A dimethacrylate | - | 62.8 | - |
| UDMA: urethane dimethacrylate | - | - | 48.7 |
| TEGDMA: triethylene glycol dimethacrylate | 35.5 | 23.2 | - |
| HEMA: 2-hydroxyethyl methacrylate | 27.0 | 10.4 | 17.3 |
| PEG-U: poly(ethylene glycol)-extended UDMA | - | - | 30.0 |
| ZrDMA: zirconyl dimethacrylate | 1.0 | - | - |
| MEP: methacryloyloxyethyl phthalate | - | 2.6 | 3.0 |
| CQ: camphorquinone | 0.2 | 0.2 | - |
| 4EDMAB: ethyl-4-N,N-,dimethylaminobenzoate | 0.8 | 0.8 | - |
| IRGACURE 1850: bis(2,6-dimethoxybenzoyl)-2,4,4-triethylpentyl phosphine oxide & 1-hydroxycyclohexyl phenyl ketone | - | - | 1.0 |




3. Experimental Section
3.1. Resin formulation, copolymer and composite specimen preparation

3.2. ACP filler synthesis and characterization
3.3. Degree of vinyl conversion (DVC)
3.4. Water sorption (WS)
3.5. Release of mineral ions from composites
3.6. Mechanical testing of the copolymers and composites
3.7. Statistical analysis
4. Conclusions
Abbreviations
| ACP | Amorphous calcium phosphate |
| ADAF | American Dental Association Foundation |
| ANOVA | Analysis of variance |
| BFS | Biaxial flexural strength |
| Bis-GMA | 2,2-Bis[p-(2‘-hydroxy-3‘-methacryloxypropoxy)phenyl]propane |
| BTHZ | Bis-GMA/TEGDMA/HEMA/ZrDMA resin |
| CQ | Camphorquinone |
| dm | Median diameter |
| DVC | Degree of vinyl conversion |
| EBPADMA | Ethoxylated bisphenol A dimethacrylate |
| ETHM | EBPADMA/TEGDMA/HEMA/MEP resin |
| 4EDMAB | Ethyl-4-N,N-dimethylaminobenzoate |
| FTIR | Fourier transform infrared spectroscopy |
| ΔG0 | Gibbs free energy |
| HAP | Hydroxyapatite |
| HEMA | 2-Hydroxyethyl methacrylate |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| IAP | Ion activity product |
| IRGACURE 1850 | Bis(2,6-dimethoxybenzoyl)-2,4,4-triethylpentyl phosphine oxide & 1-Hydroxycyclohexyl phenyl ketone |
| MEP | Methacryloyloxyethyl phthalate |
| NIDCR | National Institute of Dental and Craniofacial Research |
| NIR | Near infrared |
| NIST | National Institute of Standards and Technology |
| PEG-U | Poly(ethylene glycol) extended UDMA |
| PRC | Paffenbarger Research Center |
| PSD | Particle size distribution |
| SEM | Scanning electron microscopy |
| SD | Standard deviation |
| TEGDMA | Triethylene glycol dimethacrylate |
| TGA | Thermogravimetric analysis |
| UDMA | Urethane dimethacrylate |
| UPHM | UDMA/PEG-U/HEMA/MEP resin |
| WS | Water sorption |
| XRD | X-ray diffraction |
| ZrDMA | Zirconyl dimethacrylate |
Acknowledgements
Disclaimer
References and Notes
- Dorozhkin, S.V. Calcium Orthophosphates in Nature, Biology and Medicine. Materials 2009, 2, 399–498. [Google Scholar]
- Dorozhkin, S.V. Calcium Orthophosphate-based Biocomposites and Hybrid Materials. J. Mater. Sci. 2009, 44, 2343–2387. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Skrtic, D.; Hailer, A.W.; Eanes, E.D. Bioactive Polymeric Composites Based on Hybrid Amorphous Calcium Phosphate. In Polymeric Drugs & Drug Delivery Systems; Ottenbrite, R.M., Kim, S.W., Eds.; Technomics Publ. Co., Inc.: Lancaster, PA, USA, 2000; pp. 301–310. [Google Scholar]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Eichmiller, F.C.; Schumacher, G.E. Physicochemical Evaluation of Bioactive Polymeric Composites Based on Hybrid Amorphous Calcium Phosphates. J. Biomed. Mat. Res. B Appl. Biomater. 2000, 53, 381–391. [Google Scholar] [CrossRef]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous Calcium Phosphate-based Bioactive Polymeric Composites for Mineralized Tissue Regeneration. J. Res. Natl. Inst. Stands. Technol. 2003, 108, 167–182. [Google Scholar] [CrossRef]
- Langhorst, S.E.; O’Donnell, J.N.R.; Skrtic, D. In vitro Remineralization Effectiveness of Polymeric ACP Composites: Quantitative Micro-radiographic Study. Dent. Mater. 2009, 25, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.E.; Chow, L.C. Combination of Sparingly Soluble Calcium Phosphates in Slurries and Paste as Mineralizers and Cements. U.S. Patent 4,612,053, 16 September 1986. [Google Scholar]
- Chen, W.C.; Ju, P.C.; Wang, J.C.; Hung, C.C.; Lin, J.H.C. Brittle and Ductile Adjustable Cement Derived from Calcium Phosphate Cement/Polyacrylic Acid Composites. Dent. Mater. 2008, 24, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Ko, C.L.; Hung, C.C.; Tyan, Y.C.; Lai, C.H.; Chen, W.C.; Wang, C.K. Deriving Fast Setting Properties of Tetracalcium Phosphate/Dicalcium Phosphate Anhydrous Bone Cement with Nanocrystallites on the Reactant Surfaces. J. Dent. 2010, 38, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Eidelman, N. Dental Composites Based on Hybrid and Surface-modified Amorphous Calcium Phosphates. Biomaterials 2004, 25, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.N.R.; Antonucci, J.M.; Skrtic, D. Amorphous Calcium Phosphate Composites with Improved Mechanical Properties. J. Bioact. Compat. Polym. 2006, 21, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Regnault, W.F.; Antonucci, J.M.; Skrtic, D. Effect of Particle Size of an Amorphous Calcium Phosphate Filler on the Mechanical Strength and Ion Release of Polymeric Composites. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 80B, 11–17. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Liu, D.W.; Skrtic, D. Amorphous Calcium Phosphate Based Composites: Effect of Surfactants and Poly(ethylene oxide) on Filler and Composite Properties. J. Disp. Sci. Technol. 2007, 28, 819–824. [Google Scholar]
- Antonucci, J.M.; Skrtic, D. Matrix Resin Effects on Selected Physicochemical Properties of Amorphous Calcium Phosphate Composites. J. Bioact. Compat. Polym. 2005, 20, 29–49. [Google Scholar] [CrossRef]
- Skrtic, D.; Antonucci, J.M.; Liu, D.W. Ethoxylated Bisphenol A Methacrylate-based Amorphous Calcium Phosphate Composites. Acta Biomater. 2006, 2, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M. Effect of Composition of the Resin Phase on Vinyl Conversion of Amorphous Calcium Phosphate-based Composites. Polym. Int. 2007, 56, 497–505. [Google Scholar]
- O’Donnell, J.N.R.; Skrtic, D. Degree of Vinyl Conversion, Polymerization Shrinkage and Stress Development in Experimental Endodontic Composites. J. Biomim. Biomater. Tissue Eng. 2009, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stansbury, J.W.; Dickens, S.H. Network Formation and Compositional Drift During Photo-Initiated Copolymerization of Dimethacrylate Monomers. Polymer 2001, 42, 6363–6369. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Stansbury, J.W. Molecularly Designed Dental Polymers. In Desk Reference of Functional Polymers Syntheses and Applications; Arshady, R., Ed.; ACS: Washington DC, USA, 1997; pp. 719–738. [Google Scholar]
- Antonucci, J.M.; Stansbury, J.W.; Venz, S. Synthesis and Properties of Polyfluorinated Prepolymer Multifunctional Urethane Methacrylate. In Progress in Biomedical Polymers; Gebelein, C.G., Dunn, R.L., Eds.; Plenum Press: New York, NY, USA, 1990; pp. 121–131. [Google Scholar]
- Johns, J.I.; O’Donnell, J.N.R.; Skrtic, D. Selected Physicochemical Properties of the Experimental Endodontic Sealer. J. Mater Sci. Mater. Med. 2010, 21, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M. Dental Composites Based on Amorphous Calcium Phosphate––Resin Composition/Physicochemical Properties Study. J. Biomater. Appl. 2007, 21, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Lowell, L.G.; Stansbury, J.W.; Syrpes, D.C.; Bowman, C.N. Effects of Composition and Reactivity on the Reaction Kinetics of Dimethacrylate/Dimethacrylate Copolymerizations. Macromolecules 1999, 32, 3913–3921. [Google Scholar] [CrossRef]
- Simon, C.G.; Antonucci, J.M.; Liu, D.W.; Skrtic, D. In vitro Cytotoxicity of Amorphous Calcium Phosphate Composites. J. Bioact. Compat. Polym. 2005, 20, 279–295. [Google Scholar] [CrossRef]
- Momoi, Y.; McCabe, J.F. Hygroscopic Expansion of Resin Based Composites during 6 Months of Water Storage. Br. Dent. J. 1994, 176, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Tay, F.R.; Cheung, G.S.P.; Kei, L.H.; Wei, S.H.Y.; Pashley, D.H. Hygroscopic Expansion of a Compomer and a Composite on Artificial Gap Reduction. J. Dent. 2002, 30, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Anttila, E.J.; Krintilä, O.H.; Laurila, T.K.; Lassila, L.V.J.; Vallitu, P.K.; Hernberg, R.G.R. Evaluation of Polymerization Shrinkage and Hygroscopic Expansion of Fiber-reinforced Biocomposites Using Optical Fiber Bragg Grating Sensors. Dent. Mater. 2008, 24, 1720–1727. [Google Scholar] [CrossRef]
- Skrtic, D.; Hailer, A.W.; Takagi, S.; Antonucci, J.M.; Eanes, E.D. Quantitative Assessement of the Efficacy of Amorphous Calcium Phosphate/Methacrylate Composites in Remineralizing Caries-like Lesions Artificially Produced in Bovine Enamel. J. Dent. Res. 1996, 75, 1679–1686. [Google Scholar] [PubMed]
- Garcia-Fiero, J.L.; Aleman, J.V. Sorption of Water by Epoxide Prepolymers. Macromolecules 1982, 15, 1145–1149. [Google Scholar] [CrossRef]
- Arima, T.; Hamada, T.; McCabe, J.F. The Effects of Cross-linking Agents on Some Properties of HEMA-based Resins. J. Dent. Res. 1995, 74, 1597–1601. [Google Scholar] [PubMed]
- Skrtic, D.; Antonucci, J.M. Effect of Bifunctional Co-monomers on Mechanical Strength and Water Sorption of Amorphous Calcium Phosphate- and Silanized Glass-filled Bis-GMA-based Composites. Biomaterials 2003, 24, 2881–2888. [Google Scholar] [CrossRef] [PubMed]
- Stansbury, J.W.; Dickens, S.H. Determination of Double Bond Conversion in Dental Resins by Near Infrared Spectroscopy. Dent. Mater. 2001, 17, 71–79. [Google Scholar] [CrossRef] [PubMed]
- American Society for Testing and Materials. Philadelphia. Standard Test Method for Biaxial Strength (Modulus of Rupture) of Ceramic Substrates (re-approved 1991). ASTM F394-78, 1991.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antonucci, J.M.; Skrtic, D. Fine-Tuning of Polymeric Resins and their Interfaces with Amorphous Calcium Phosphate. A Strategy for Designing Effective Remineralizing Dental Composites. Polymers 2010, 2, 378-392. https://doi.org/10.3390/polym2040378
Antonucci JM, Skrtic D. Fine-Tuning of Polymeric Resins and their Interfaces with Amorphous Calcium Phosphate. A Strategy for Designing Effective Remineralizing Dental Composites. Polymers. 2010; 2(4):378-392. https://doi.org/10.3390/polym2040378
Chicago/Turabian StyleAntonucci, Joseph M., and Drago Skrtic. 2010. "Fine-Tuning of Polymeric Resins and their Interfaces with Amorphous Calcium Phosphate. A Strategy for Designing Effective Remineralizing Dental Composites" Polymers 2, no. 4: 378-392. https://doi.org/10.3390/polym2040378
APA StyleAntonucci, J. M., & Skrtic, D. (2010). Fine-Tuning of Polymeric Resins and their Interfaces with Amorphous Calcium Phosphate. A Strategy for Designing Effective Remineralizing Dental Composites. Polymers, 2(4), 378-392. https://doi.org/10.3390/polym2040378