Preparation of Novel Hydrolyzing Urethane Modified Thiol-Ene Networks

Abstract
: Novel tetra-functional hydrolyzing monomers were prepared from the reaction of TEOS and select alkene-containing alcohols, ethylene glycol vinyl ether or 2-allyloxy ethanol, and combined with trimethylolpropane tris(3-mercaptopropionate) (tri-thiol) in a thiol-ene “click” polymerization reaction to produce clear, colorless thiol-ene networks using both radiation and thermal-cure techniques. These networks were characterized for various mechanical characteristics, and found to posses Tg's (DSC), hardness, tack, and thermal stability (TGA) consistent with their molecular structures. A new ene-modified urethane oligomer was prepared based on the aliphatic polyisocyanate Desmodur® N 3600 and added to the thiol-ene hydrolyzable network series in increasing amounts, creating a phase-segregated material having two Tg's. An increase in water absorption in the ene-modified urethane formulations leading to a simultaneous increase in the rate of hydrolysis was supported by TGA data, film hardness measurements, and an NMR study of closely related networks. This phenomenon was attributed to the additional hydrogen bonding elements and polar functionality brought to the film with the addition of the urethane segment. SEM was utilized for visual analysis of topographical changes in the film's surface upon hydrolysis and provides support for surface-driven erosion. Coatings prepared in this study are intended for use as hydrolyzing networks for marine coatings to protect against ship fouling.
1. Introduction
The area of hydrolyzing networks or self-polishing coatings has generated much excitement in the coatings industry, for applications ranging from antifouling coatings to drug delivery systems [1-3]. A number of polymer compositions have been investigated for these applications including polylactic acids, poly ortho esters, and organo silicates. These self-polishing coatings introduce unique characteristics to films through the incorporation of hydrolytically unstable bonds, which react with water to cleave covalent bonds and result in the degradation of the local network. If designed to erode at the coating-water interface rather than in the bulk material, films presenting a continuously new, regenerated surface with time are possible [4]. Therefore, controlled rates of hydrolysis is an extremely desirable property for these coatings; however, in general the reaction kinetics and mechanisms have not been widely explored [2].
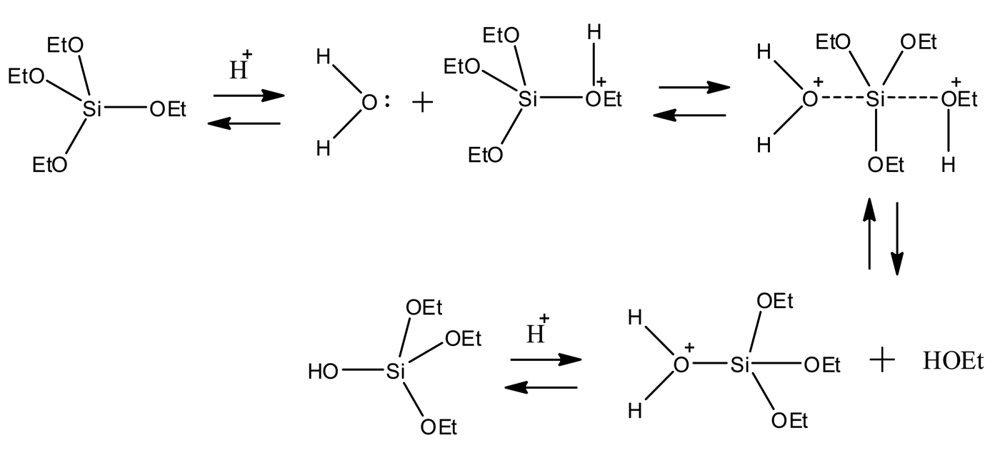
The hydrolysis of metaloxides, such as organo silicates, is discussed primarily in the context of sol-gel techniques. In this approach, metal oxides undergo various stages of hydrolysis and sequential polycondensation reactions, ultimately producing gels suitable for applications in polymeric materials [5-10], optics [7], biomaterials, and glassy or ceramic like coatings [11,12]. Tetraethyl orthosilicate (TEOS) has been widely explored using sol-gel techniques since the 1880s [8]. Scheme 1 describes an example substitution reaction of TEOS, resulting in the evolution of ethanol byproduct and the formation of a newly substituted organo-silicate [13]. This methodology can be used in the synthesis of organo-silicates having polymerizable functional groups, such as ene-modified monomers containing a C=C, while maintaining the hydrolyzing properties of the silicate center. These ene-terminated organosilicates (C=C) could be used in chemical reactions with thiols (SH) to form novel thiol-ene networks while maintaining hydrolyzing bonds.
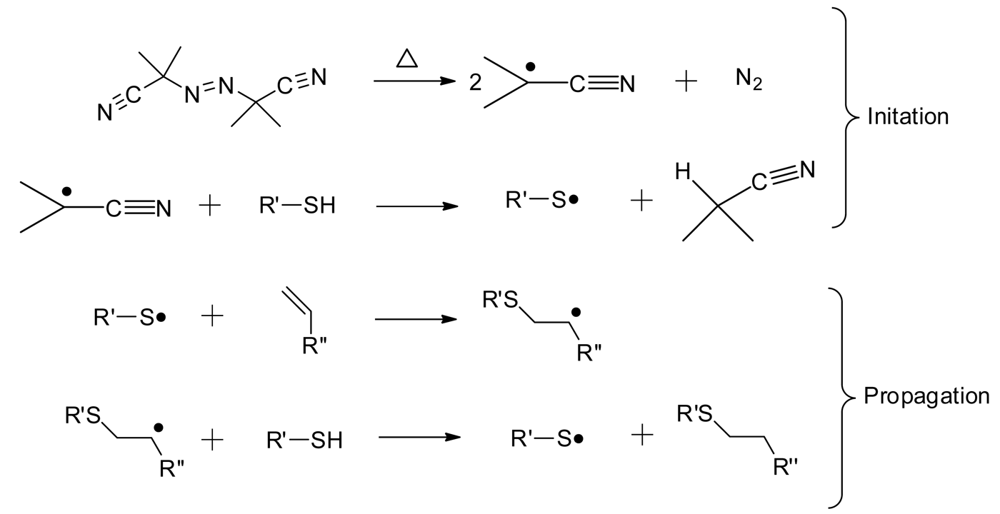
Thiol-ene “click” chemistry, as a means to form polymer networks, has been valued for its numerous advantages. Thiol-ene reactions form homogenous networks through a radical step-growth process shown generically in Scheme 2, using the thermal initiator AIBN [14,15]. The thiol-ene reaction can occur between almost any thiol and any ene, over a wide range of molecular structures and degree of functionality [16]. Since a variety of commercial thiols and enes exist, the molecular architecture is readily tailored for numerous applications to produce thoughtfully designed coatings quickly and efficiently [17]. The technical approach is amenable to either thermal or photochemical curing conditions, allowing for ready adaptation to common research and commercial polymerization methods. Other benefits of thiol-ene chemistry are a delayed gelation, insensitivity to molecular oxygen, the production of a uniform crosslink density, and high conversions of monomers [14].
Urethane reactions, as another network forming polymerization chemistry, produces materials with excellent mechanical properties for use in industrial applications such as foams, coatings, and medical devices [18]. Analogous to the thiol-ene system, a wide array of commercially available monomers exist to allow for tailoring of the resulting polymer properties [19,20]. Urethane bonds form quickly through the reaction of isocyanate (NCO) and alcohol (OH) functional groups to produce relatively inexpensive and durable coatings, having strong mechanical properties, good abrasion resistance, and thermal stability [21]. Recent efforts of the Hoyle research group [22-24] demonstrate that the modification of thiol-ene networks with urethane segments may result in the improvement of selected polymer network properties specifically imparting impact adsorption, elastic properties and fracture toughness [22,24]. Polyisocyanates can be end-capped by reacting with ene containing alcohols, resulting in urethane bridged oligomers that may be efficiently polymerized with thiols. Each monomer can be specifically chosen to modify the coating for the desired mechanical properties such as glass transition temperature (Tg) and film hardness [23]. Using these techniques, the benefits of thiol-ene click reactions and mechanical toughness of urethane bonds can be combined into one continuous film.
Our interests lie in the preparation and characterization of thiol-ene hydrolyzing networks for potential use as marine coatings to protect against ship fouling. In this effort, the new hydrolyzing moiety is a polymerizable ene-functionalized organosilicate monomer, which is readily incorporated into the polymer matrix and the hydrolysis is based on the known reactions of tetraethylorthosilicate. Ene-terminated urethane segments are prepared and added to the custom thiol-ene, hydrolyzing networks, and their effects on coating mechanical properties and rates of hydrolysis are characterized using a variety of common analytical techniques. Preliminary relative rates of hydrolysis are investigated for the prepared network series using established theoretical and kinetic models. This work is part of our ongoing effort to expand and enhance the utility of thiol-ene systems for high performance and specialized coating applications.
2. Results and Discussion
Novel tetra-ene hydrolyzing monomers are prepared as shown in Scheme 3, from the reaction of TEOS and select alkene-containing alcohols, ethylene glycol vinyl ether or 2-allyloxy ethanol. These custom tetra-ene monomers are combined with trimethylolpropane tris(3-mercaptopropionate) (tri-thiol) in a 1:1 molar equivalent of thiol to ene functional group and polymerized into crosslinked thiol-ene films. The polymerization methodology is readily amendable to a UV radiation or thermal cure process using either the radial photoinitiator Irgacure 651 or thermal initiator 2,2′-azobisiso butlyronitrile (AIBN) respectively at 1.0 wt %. Representative films and the molecular structure of the tri-thiol are illustrated in Figure 1. For this study, thin films are prepared using photopolymerization techniques, and thick films are prepared using the thermal process. Regardless of the polymerization technique employed, continuous, clear and colorless films are produced. FT-IR has been used in the past to analyze the extent of the thiol-ene radical step growth polymerization revealing percent conversion of functional groups between 85–96% [14]. Polymerization conditions are optimized using FTIR analysis, by monitoring the disappearance of the thiol peak at 2,600 cm−1 and ene peak at 1,600 cm−1, for both thermal and UV-cure to ensure the reaction has gone to completion. Prior work on thiol-ene network-forming reactions has defined the electronic requirements of the ene, and in general, ene reactivity decreases with decreasing electron density in the C=C [14]. In comparable systems using the identical thiol, the vinyl monomer is expected to polymerize at a faster rate than the allyl monomer. In this study, no differences in rates are observed in the network forming reactions. Both systems, allyl and vinyl, produce fast-curing films having high monomer conversions.
The introduction of urethane oligomer segments into the thiol-ene matrix is enabled by ene-functionalizing the commercial aliphatic polyisocyanante Desmodur 3600 with an allyl and vinyl alcohols respectively, as shown in Scheme 4. A series of compositions are prepared, as described in Table 1, by varying the relative mol % of the tetra-ene hydrolyzable monomer and the urethane segment used in the polymerization process, always keeping an overall 1:1 molar equivalent of ene to thiol. By convention the samples are described according to the nature of the alcohol employed, either vinyl or allyl, followed by the mol % of the hydrolyzable component used. For example, vinyloxy-100 originates from the hydrolyzable monomer prepared from the vinyl alcohol and includes no (0 mol %) of the urethane modifier.
Surface properties of each formulation produced in Table 1 are characterized—on a 0.1 mm thick UV-cured thin film—through film hardness and tack measurements, using a TA XTplus Texture Analyzer. The hardness and tack measurements, summarized in Table 2, are related in that the hardness measurement characterizes the force required to penetrate a 1″ probe tip 10% into a film thickness at a controlled rate of decent and the tack measurement describes the force required to remove the same probe tip at the same rate from the surface after resting. The values obtained are listed in grams applied to the system to perform the desired action on the probe tip. All tack and hardness values represent averages of five individual measurements, and the error was calculated from the largest deviation from the average calculated. Networks formed from both vinyl and allyl hydrolyzable monomers demonstrated comparable hardness and tack values, and the incorporation of the corresponding ene-modified urethane had little effect on the overall hardness and tack of the films. All formulations had higher than typical hardness values for thiol-ene polymer films due to the contribution from the organosilicate moiety and are described as having low tack, which is typical of other thiol-ene films characterized recently by our lab [17] For example, the same thiol used in this study when combined in a 1:1 functional group equivalent with pentaerythritol allyl ether (tri-ene) possesses an average hardness of 72 ± 2 g and tack of 18 ± 2 g, respectively.
Physical properties of the thiol-ene network were characterized using a 5 mg thermal cured sample. The glass transition temperatures (Tg) of the hydrolyzable networks prepared in Table 1 are obtained using differential scanning calorimetry (DSC) and are also reported in Table 2. All formulations are characterized by a Tg at approximately 22–23 °C, resulting from the thiol-ene matrix constituents. The addition of the urethane to the network gave a second Tg around 26–31 °C, representative of the contribution of the isolated urethane segments. To support the theory that the second Tg was due to the addition of the urethane segment, Desmodur N 3600, when capped with methanol and characterized separately, yields a corresponding Tg of 29 °C.
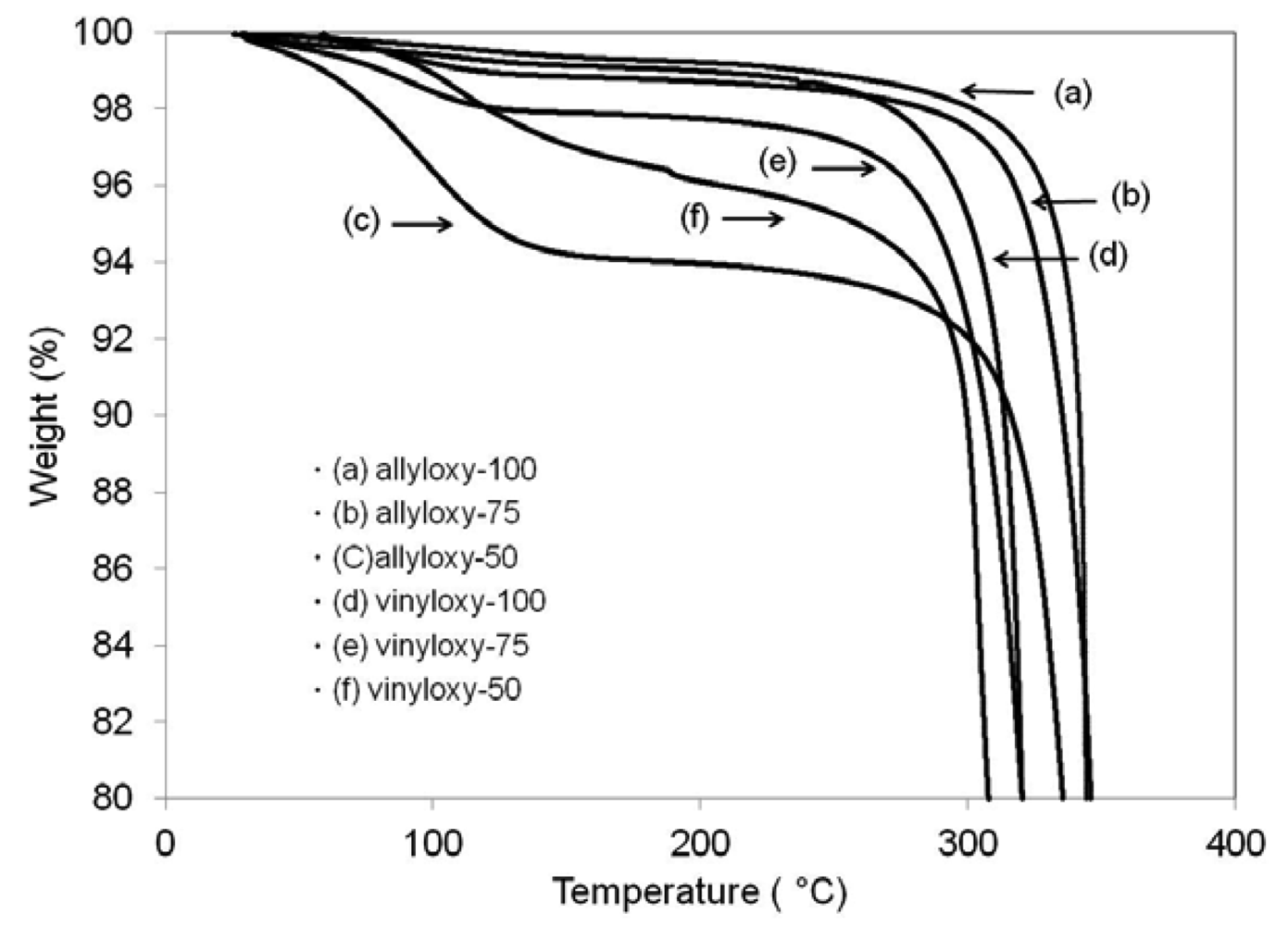
Thermal stability was analyzed by monitoring weight loss with increasing temperature using a TA instruments Q5000 Thermal Gravimetric Analysis (TGA). A representative plot of each hydrolyzable sample is shown in Figure 2. Due to the hygroscopic nature of the films, all samples were normalized for absorbed water from the atmosphere by subtracting detected mass loss from room temperature to 100 °C. Onset values were calculated as a 10% mass loss from 100 °C and recorded in Table 2. In general, allyloxy systems have higher degradation onset temperatures by approximately ∼20 °C than their vinyloxy counterparts, and are therefore modestly more thermally stable. However, both systems exhibited high overall thermal stability exhibiting onset values from 311 to 341 °C. The addition of urethane oligomer to both allyl and vinyl systems did not negatively impact calculated thermal degradation onset temperatures. An interesting trend in the hygroscopic properties of the films is suggested by the TGA data. As the amount of urethane in the network is increased, the amount of water absorbed by the film under controlled conditions increased. The incorporation of the more polar urethane segments, which are capable of hydrogen bonding, increases the water uptake of the films over the same time duration.
Ultimately for a hydrolyzable film to be successfully employed as a commercial coating, rates of hydrolysis leading to a determination of both the coating's lifetime and defining application parameters, such as applied film thickness, are required. Although this might initially seem simple, obtaining rates (or relative rates) of hydrolysis for the films produced during this investigation has proven to be a significant undertaking. The remaining sections of the discussion section review the various approaches our group has used to investigate the constituent-dependent relative rates of hydrolysis for our film series. As detailed in Scheme 1, water is a necessary reagent for the hydrolysis process to occur and, as suggested by the initial TGA experiments, chemical composition of the networks will influence the rate in which water is absorbed into the film. This work represents the preliminary characterization of the hydrolysis of our organosilicate-based thiol-ene networks.
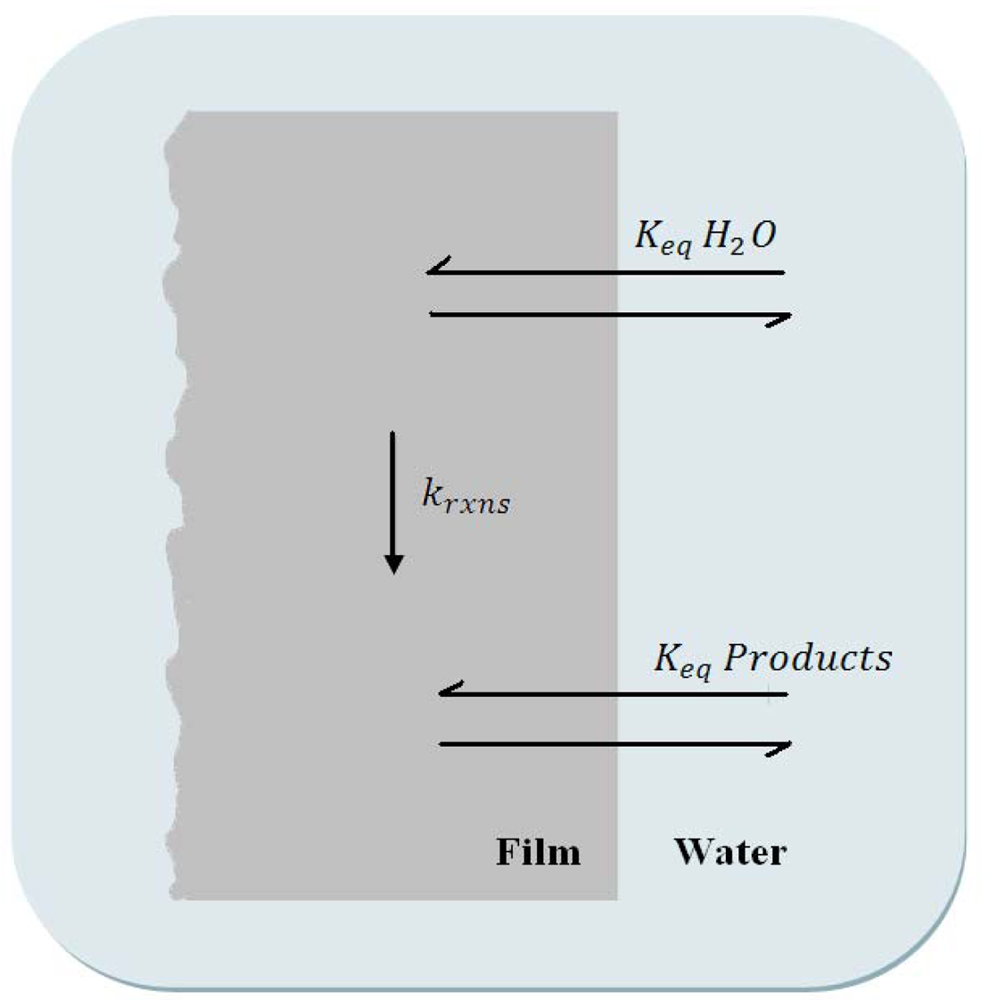
A careful balance must be made between the various rate constants involved in a hydrolytic process. In a simplified diagram, Figure 3, a general description of the important rate constants and equilibria are described. At the coating surface, water must diffuse into the coating; Keq H2O defines the mass transport properties of water at the coating surface. One or more reactions between water molecules and coating network bonds must occur within the coating, krxns. And finally the products of these reactions must be released from the coating surface, Keq Products. For a successful hydrolyzing coating, the rate of hydrolysis (krxns) should be rapid compared to the rate of water diffusion into the film, thereby water is essentially used as it enters the polymer. This ensures that film erosion takes place at the surface, resulting in a self-polishing polymer.
Visual inspection of images collected using the scanning electron microscopy (SEM) technique suggests that there are significant changes in topography of the films at short exposure times to water which in part supports surface erosion. An example coating, allyloxy-100 was photopolymerized on a mica substrate and exposed to artificial salt water. SEM images were taken before and after 48 h of water exposure and are provided in Figure 4. Initially, one would think that the easiest way to monitor film erosion is to monitor film thickness as a function of water exposure time. However, since the first step of the hydrolytic process involves water uptake of the films, the films are not initially dimensionally stable and show a slight increase in film thickness simultaneous with the changes in topography observed. Moreover, the initial uptake of water into the film is expected to alter the surface properties, and a brief investigation into the time-dependent surface property changes when exposed to a humid environment ensued.
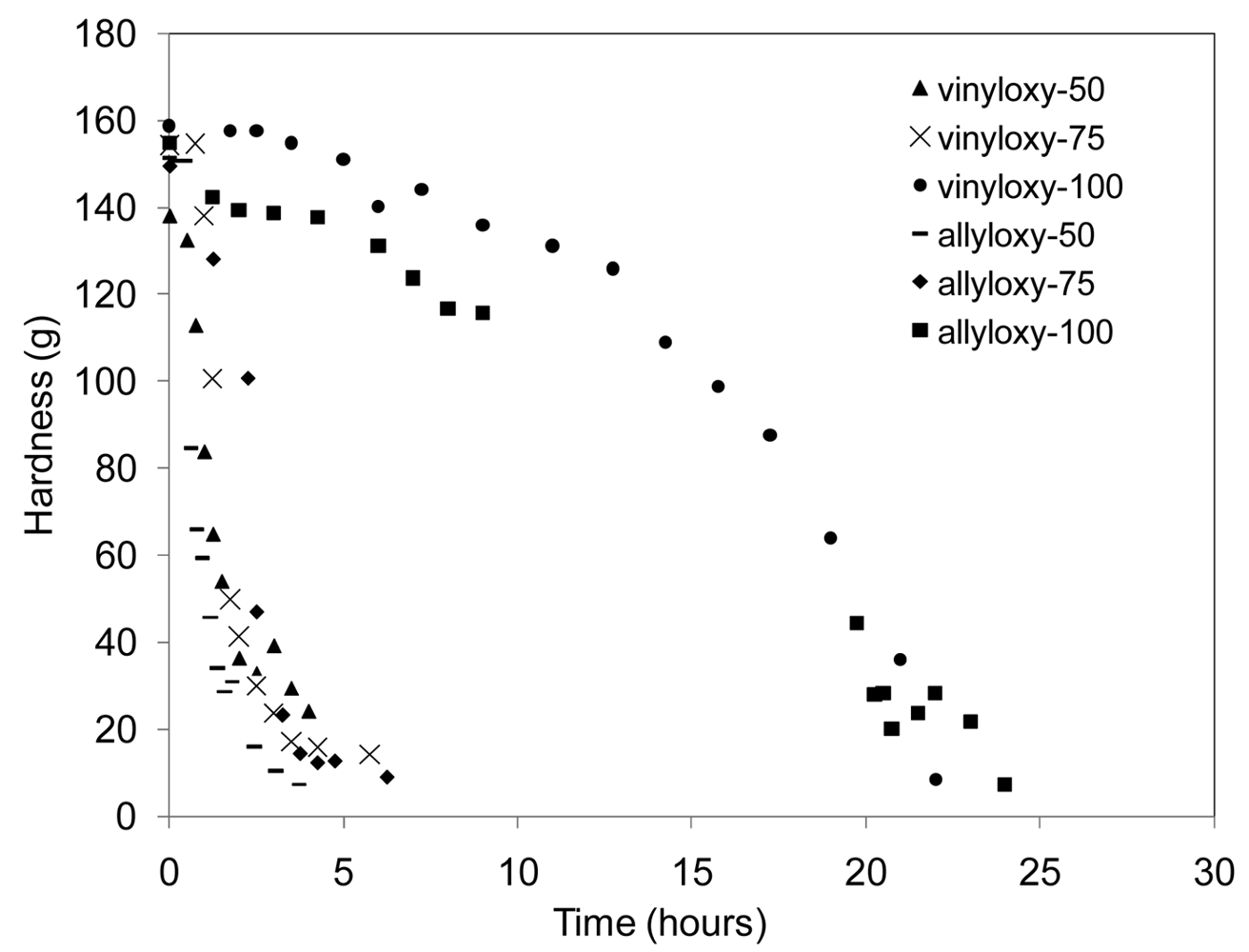
Initial changes in film hardness (changes in surface characteristics) were monitored for all six network compositions, Table 1, while samples were exposed to a constant 50% relative humidity (RH) environment. Results are reported in Figure 5. Since the chemistry of the hydrolysis reaction is the same in all samples, the change in film hardness is proportional to the rate of water uptake of the film and impacts the resulting rate of film hydrolysis by increasing the local concentration of water available, not specifying bulk or surface erosion. Using this indirect assessment of the relative rates of hydrolysis among the series, there were no significant changes between the allyl and vinyl-based compositions. However, the addition of the urethane greatly impacted the rate of water uptake by the sample, leading to drastic changes in film hardness with time. This analysis reinforces the TGA conclusions that the addition of the urethane segments creates a more hydrophilic network through hydrogen bonding among water and polar functional groups within the polymer. However, increasing the amount of urethane in the network from 25–50 mol % had no additional effect on the rate of hydrolysis. The dramatic differences in water uptake demonstrated by this series of films suggest the ability to tailor Keq H2O through the incorporation of polar segments for desired applications.
Although the indirect assessment offered by the film hardness study suggests that the rate of water uptake, Keq H2O, is tunable in the thiol-ene hydrolyzing networks by changing the molecular functionality of the constituents, no direct rate of hydrolysis is measured. A well-known kinetic model exists for the quantitative comparison among polymer networks used for the controlled-release/drug-delivery applications of pharmaceuticals [25]. The theoretical model, referred to as the Higuchi model, accounts for the relative water permeability of the networks and the concentrations, solubility, and diffusion of agents in the network. The common representation of the time-dependent model is provided as Equation (1), where Q is defined as the amount of compound released and KH is the Higuchi dissolution constant.
As another means to explore the hydrolysis rates of our thiol-ene networks, the group prepared a new series of networks containing a modified fluorinated hydrolyzable monomer, which could be detected by 19F NMR. Table 3 describes the series, which consisted of two distinct model systems: fluorinated networks and fluorinated blends. In the blends, the fluorinated alcohol 2,2,3,3-tetrafluoro-1-propanol was added to the existing thiol-ene networks as an additive; in these samples there exists no chemical bond between the fluorinated additive and the thiol-ene network. These samples are given the identifier “blend” in the sample name, and the concentration of fluorinated additive is varied among the series to match the concentration present in the analogous network samples. In the networks, a new hydrolyzable monomer was prepared according to Scheme 3 in which one leg of the silicate is substituted with the fluorinated oxy group. A small change in network density of these samples is expected upon polymerization, as the functionality of the hydrolyzable monomer is altered from F4 to F3. Since no detectable difference in polymerization rate is observed between the vinyl and allyl monomers, only vinyl monomers are investigated by 19F NMR and no allyl systems were explored.
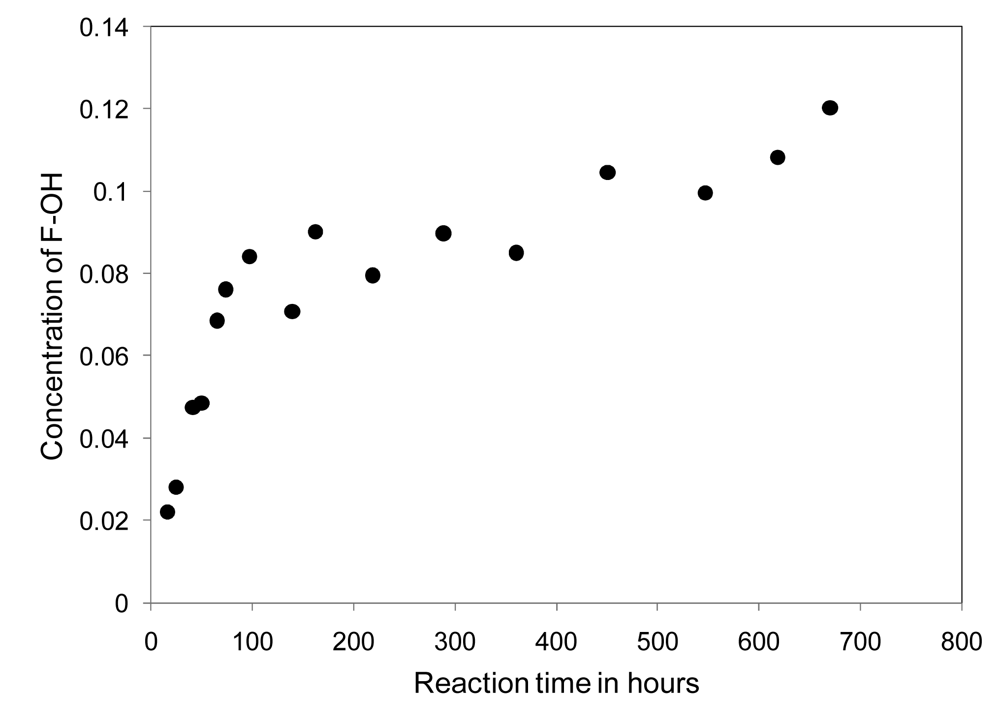
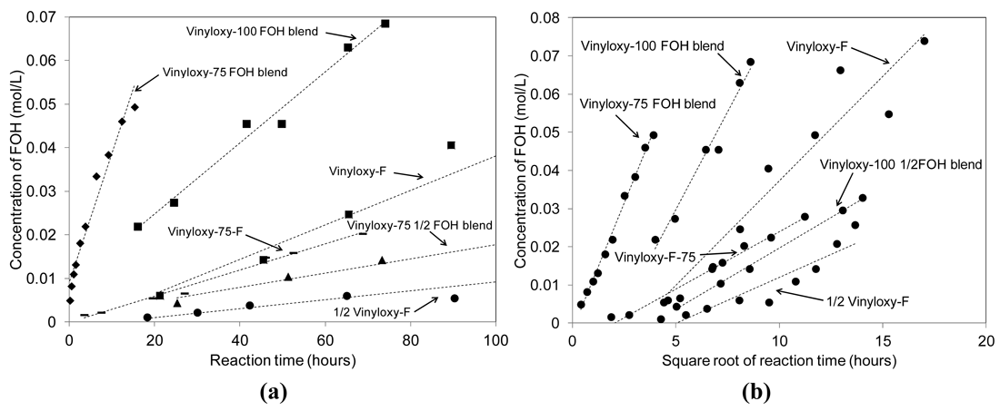
Networks are prepared as previously described and uniform 12 g thiol-ene network molded puck samples, containing various amounts of fluorinated material as listed in Table 3, are submersed in deuterated water for analysis by NMR. The increasing concentration of the fluorinated alcohol present in the solvent is monitored versus time, using integrated NMR peak areas of the downfield F signal at −126 δ against an internal standard, converted to concentrations using a calibration curve, and plotted against reaction time. A sample concentration profile is provided in Figure 6.
Figure 7(a,b) illustrates a comparison among the initial rates obtained from the concentration versus time plots for each of the six fluorine-modified systems as well as presents the plots for the Higuchi treatment on the same. Plots of the concentration data to zero and first order release models are provided in supplementary information. Appearance of 2,2,3,3-tetrafluoro-1-propanol is faster in blended samples [Table 3, entries (4–6)] and the observed reaction kinetics are found to fit the Higuchi model with excellent correlation coefficients, as expected since the system is described well by a molecule trapped within a polymer matrix that must diffuse out. The Higuchi model does not account for any chemical reactions that must occur in the film before a species may diffuse out of the matrix, and therefore the fluorinated network samples [Table 3, entries (1–3)] are expect to deviate from this model according to the rates of the chemical reactions that must occur during hydrolysis. In further kinetic analysis using a combination of theoretical models, it may be possible to extract the absolute rate constants for the film-based hydrolysis reactions, which is the focus of ongoing research and the subject of a follow-on publication.
3. Experimental Section
3.1. Materials
Unless otherwise noted, materials were used as received. Ethylene glycol vinyl ether (97%), 2-allyloxy ethanol (97%), tetraethyl orthosilicate (TEOS, 98%), trimethylolpropane tris(3-mercaptopropionate) (tri-thiol, 96–99%), 2-2′-azobis(2-methylpropoionitrate) (98%), 2,2,3,3-tetrafluoro-1-propanol (98%), 2,2′-azobisiso butlyronitrile (ABIN 98%), and dibutyltin diaularate (95%) were obtained from Aldrich. Irgacure 651 and Desmodur® 3600 were obtained from Ciba Specialty Chemicals and Bayer Material Science, respectively. Chloroform and toluene were obtained from Fischer and distilled just prior to use. 1H and 13C NMR were taken at 400 MHz in CDCl3 with an internal standard of tetramethylsilane (TMS). F NMR was taken at 400 MHz in D2O obtained from Cambridge Isotope Laboratory using an internal standard of trifluroacetic acid-D (99.5%) from Aldrich. All chemical shifts are reported in ppm as a down field shift from TMS. FT-IR analysis was done on a Nicolet Nexus 470 FT-IR from Thermo Electro Corporation. Elemental analysis was provided by Atlanta Microlab, INC. GC-MS was performed on a Perkin Elmer Clarus 600 Gas Chromatography using a Perkin Elmer ClarusT Mass Spectrometer, with the following parameters: 250 °C injection port, He carrier gas, 30 m × 0.25 mm PDMS capillary column.
3.2. Synthesis of Vinyl, Allyl, and Fluorine-Modified Ene-Terminated Hydrolyzable Monomers
Ethylene glycol vinyl ether or 2-allyloxy ethanol was combined with tetraethyl orthosilicate in a 4:1 stochiometric ratio in a round bottom equipped with a short path distillation apparatus. The solution was heated to 105 °C and held for 24 h, while the ethanol product was removed under a nitrogen stream and collected in a receiving flask. The reaction was monitored by NMR observing the disappearance of the TEOS CH3 groups at 1.2 δ. The remaining TEOS was used to determine percent yield. The resulting clear, colorless viscous liquids were obtained in 92 and 97% yield for tetrakis(2-(vinyloxy)ethyl) orthosilicate and tetrakis(2-(allyloxy)ethyl) orthosilicate, respectively. Samples were concentrated and used without further purification.
Tetrakis(2-(vinyloxy)ethyl) orthosilicate
(FTIR): 3,075, 2,935, 2,859, 1,647, 1,075 cm−1; 1H-NMR (CDCl3): 6.50 (m, 4H), 4.18 (m, 4H), 4.02 (m, 8H), 4.01 (m, 4H), 3.8 (m, 8H) δ; 13C NMR (CDCl3): 151.77, 86.65, 68.68, 62.26 δ; EA: (experimental) C 50.25, H 7.72, (theoretical) C 51.04, H 7.50; GC MS parent ion corresponding to trifunctional monomer decomposition product at 289 m/z.
Tetrakis(2-(allyloxy)ethyl) orthosilicate
(FTIR): 3,050, 2,940, 2,884, 1,616, 1,076 cm−1; 1H-NMR (CDCL3): 5.90 (m, 4H), 5.30 (m, 4H), 5.15 (m, 4H), 4.05 (m, 8H), 3.90 (m, 8H), 3.55 (m, 8H) δ; 13C NMR (CDCL3): 134.91, 116.79, 72.06, 62.86 δ; EA: (experimental) C 55.31, H 8.56, (theoretical) C 55.50, H 8.30; GC MS parent ion corresponding to trifunctional monomer decomposition product at 331 m/z.
A slightly modified procedure was used to produce a fluorinated hydrolyzable monomer, 2,2,3,3,-tetrafluro propyl tris(2-vinyloxy) ethyl orthosilicate. Ethylene glycol vinyl ether was combined with tetraethyl orthosilicate and 2,2,3,3-tetrafluoro-1-propanol in a 3:1:1 stochiometric ratio in a round bottom equipped with a short path distillation apparatus. Dried and distilled toluene was added to the solution in an equal volume ratio. The solution was heated to 85 °C and held for 48 h, while the toluene/ethanol azeotrope was removed under a nitrogen stream and collected in a receiving flask. Remaining toluene was removed under reduced pressure. Product 2,2,3,3,-tetrafluro propyl tris(2-vinyloxy) ethyl orthosilicate was obtained as a viscous liquid in 88% yield determined by NMR and used without further purification.
2,2,3,3,-Tetrafluro propyl tris(2-vinyloxy) ethyl orthosilicate
(FTIR): 2,939, 2,890, 1,618, 1,070 cm−1; 1H-NMR (CDCL3): 6.50 (m, 3H), 6.0 (m, 1H) 4.25 (m, 6H), 4.05 (m, 6H), 4.01 (m, 3H), 3.8 (m, 3H) δ; (CDCL3): δ; 13C NMR (CDCL3): 151.67, 112.10, 108.20, 86.80, 68.65, 62.00134 δ; EA: (experimental) C 46.19, H 7.04, F 6.79; (theoretical) C 42.85, H 5.75; F 18.07; GC parent ion corresponding to trifunctional monomer decomposition product at 333 m/z.
To produce the ene-endcapped isocyanate oligomer a 3-neck, 125-mL, round-bottom flask was equipped with an addition funnel, water condenser, and a temperature probe. All glassware was oven dried before use, assembled, and cooled under nitrogen. Desmodur® N 3600 was titrated separately to yield an equivalent weight per NCO group of 143 eq. wt., which corrected the commercially supplied information of 183 eq. wt. A 1:1 ratio of functional groups (NCO vs. OH) was maintained. Ethylene glycol vinyl ether or 2-allyloxy ethanol was dissolved at 50 wt % solids and charged to a round bottom flask equipped with an addition funnel containing a solution of the isocyanate also in chloroform at 50 wt %. The dibutyltin dilaurate catalyst was added at 0.1 wt % of total solids to the ethylene glycol vinyl ether solution. The isocyanate solution was slowly added over 10 min, with stirring. Once all isocyanate solution was added, the reaction was slowly heated to 55 °C, and the reaction progress was monitored by IR spectroscopy for the disappearance of isocyanate NCO functional group at 2,200 cm−1. Ene-functionalized isocyanates were used as prepared without further purification.
3.3. General Procedure for Film Preparation
The hydrolyzable monomers and ene-modified urethane oligomer were combined with tri-thiol, in concentrations to maintain a 1:1 stochiometric ratio of functional groups (thiol:ene). Photoinitiator (Irgacure 651) or thermal initiator (AIBN) was added at 1 wt % solids, and the resulting mixture was combined in a high shear mixer at 2,000 rpm for 5 min. The 100% solids sample was drawn on clean aluminum coupons using an eight-path wet film applicator (Paul N. Gardner C.) on #4. Radiation curing of thin films containing Irgacure 651 was performed on a Fusion System Corporation Epiq 6000 UV system, 1.095 W/cm2, and clear colorless films approximately 0.1 mil t hick were produced. For thick thermal cured films using AIBN, 5 g of the 100% solids sample was poured into a 10 g Teflon release mold and placed in an oven at 65 °C overnight. The resulting films were smooth, continuous, and easily removed from the Teflon release mold.
3.4. Mechanical Characterization, Bulk Tack and Hardness, and Thermal Stability
Film hardness of thin-film samples was characterized using a TA XTplus Texture Analyzer (Godelming, Surrey, UK). Using compression test mode, the applied force required to penetrate 10% of the film thickness with a one inch round probe tip at a probe insertion speed of 0.1 mm/s was determined. The amount of UV exposure needed to ensure proper curing was defined as the plateau onset in bulk hardness verses radiation dose and held constant for the rest of the investigation. In tack measurements the probe tip and applied force (determined from hardness data) was held for 10 s, and then the probe tip was withdrawn at a constant rate from the film of 0.1 mm/s. Force measurements are recorded as grams per unit time, and the highest force point is recorded as the peak tack force.
Mechanical analysis was investigated using Differential Scanning Calorimetry (DSC) on a TA Instruments #2920 Modulated DSC. Glass transitions (Tg) were monitored and correlated to additive content. Cured films were removed from aluminum coupons and cut into ∼5 mg samples for DSC analysis. Samples were monitored over the temperature range −50 to 100 °C in a heat/cool/heat cycle at 10 °C/min. The Tg was taken from the second heating cycle.
Thermal stability was analyzed by Thermal Gravimetric Analysis (TGA) analyzing weight changes of the sample with increasing temperature. A Q5000 TGA was used in high resolution mode (variable rate) with an initial heating rate of 10 °C/min from room temperature to 600 °C. All samples were fully cured films and cut into ∼5 mg sample size.
3.5. SEM Visual Analysis and NMR Kinetic Study
Hydrolysis study was performed on hydrolyzing networks containing the fluorine-modified 2,2,3,3,-tetrafluro propyl tris(2-vinyloxy) ethyl orthosilicate hydrolyzable group. These 12 g molded puck samples were prepared using the thermal cure procedures as previously described, maintaining a 1:1 stioichiometric ratio of thiol:ene. The puck sample was placed into 50 mL deuterated artificial ocean water prepared using Instant Ocean artificial sea salt following commercial supplier instructions and sealed. The chamber was placed on a VWR S-500 orbital shaker set to speed 3. One milliliter aliquots were removed with time and combined with 50 μL of 0.98 M trifluoroacetic acid solution as internal standard in D2O. 19F analysis was performed on a 400 MHz NMR, and integration ratios were based on the 2,2 F peak of 2,2,3,3-tetrafluoro-1-propanol at −126 δ versus the internal standard at −75 δ. The concentration of 2,2,3,3-tetrafluoro-1-propanol in the reaction chamber was extrapolated from a calibration curve produced from varied concentrations in of 2,2,3,3-tetrafluoro-1-propanol (FOH) with a constant concentration of trifluoroacetic acid internal standard and analyzing by 19F NMR integrations ratios. This study was repeated varying fluorinated sample formulations. Total sample mass was kept constant at 12 g.
Films were prepared for scanning electron microscopy (SEM) using the allyloxy-100 formulation prepared for thermal cure with 1.0 wt % AIBN. A sample size of 60 μL was added to 5 × 5 MM silica chips from Ted pella and spin coated using a photo-resist spinner from headway research inc. at 1,000 rpm for 20 s. The resulting samples were thermally cured as previously described. All samples were sputter coated with gold and analyzed at a 30° angle using an SEM Quanta FEI series 200 FEG with a scanning electron beam. The remaining films were submerged in water and placed on a VWR S-500 orbital shaker set to speed three. Water exposure time was varied, and all samples were dried under vacuum at ambient temperature for 24 h before analyzed by SEM.
4. Conclusions
A new series of organosilicate hydrolyzable monomers were prepared and combined with a commercial thiol to produce novel thiol-ene-based hydrolyzing networks. These networks were characterized for various mechanical properties, and found to posses Tg's, hardness, tack, and thermal stability consistent with their molecular structures. A new ene-modified urethane oligomer was also prepared from an aliphatic polyisocyanate and added to the thiol-ene hydrolyzable network series in increasing amounts. The addition of the urethane segment to the network created a phase-segregated material having a second Tg that is characteristic of the urethane oligomer alone. TGA data and film hardness measurements suggested an increase in water absorption for formulations containing the oligomer. Relative rates of hydrolysis obtained in an NMR study of closely related networks, further supports this assumption by revealing that the inclusion of the urethane segment to the network increases the rate of hydrolysis as monitored by 19F NMR. This phenomenon contributed to the additional hydrogen bonding elements and polar functionality brought to the film with the addition of the urethane segment. SEM was utilized for visual analysis of topographical changes in the film's surface upon hydrolysis, and films showed dramatic changes in surface structure after short times of water exposure, providing support for surface erosion.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition ID | mol % | Hydrolyzable-ene (tetra-ene) | mol % | Urethane-ene |
|---|---|---|---|---|
| vinyloxy-100 | 100 | tetrakis(2-(vinyloxy)ethyl) orthosilicate | 0 | 2-(vinyloxy)ethyl-terminated polyisocyanate |
| vinyloxy-75 | 75 | tetrakis(2-(vinyloxy)ethyl) orthosilicate | 25 | 2-(vinyloxy)ethyl-terminated polyisocyanate |
| vinyloxy-50 | 50 | tetrakis(2-(vinyloxy)ethyl) orthosilicate | 50 | 2-(vinyloxy)ethyl-terminated polyisocyanate |
| allyloxy-100 | 100 | tetrakis(2-(allyloxy)ethyl) orthosilicate | 0 | 2-(allyloxy)ethyl-terminated polyisocyanate |
| allyloxy-75 | 75 | tetrakis(2-(allyloxy)ethyl) orthosilicate | 25 | 2-(allyloxy)ethyl-terminated polyisocyanate |
| allyloxy-50 | 50 | tetrakis(2-(allyloxy)ethyl) orthosilicate | 50 | 2-(allyloxy)ethyl-terminated polyisocyanate |
| Compositions Prepared | DSC (Tg) a | TGA (onset) b | Hardness Initial c | Tack Initial d |
|---|---|---|---|---|
| vinyloxy-100 | 23 | 313 | 89 ± 5 | 4 ± 1 |
| vinyloxy-75 | 22/31 | 311 | 147 ± 7 | 4 ± 2 |
| vinyloxy-50 | 22/28 | 312 | 133 ± 4 | 4 ± 0 |
| allyloxy-100 | 23 | 341 | 127 ± 4 | 25 ± 7 |
| allyloxy-75 | 22/26 | 337 | 145 ± 7 | 5 ± 0 |
| allyloxy-50 | 23/30 | 330 | 147 ± 3 | 5 ± 1 |
a- TA Instruments 2920 DSC parameters, −50 to 150 °C at 10 °C/min heat/cool/heat, Tg taken from second heat;b- TA Instruments Q5000 TGA parameters, high resolution temperature profile at 10 °C/min, onset = temperature corresponding to 10% mass loss after water loss;c- Hardness determination, load required to penetrate 10% of film thickness with a 1″ steel probe tip;d- Tack determination, load required to remove the same 1″ probe tip after holding in contact with the film at 10% film depth for 10 s.
| Sample name a | Zero order | First order | Higuchi | Quantity FOH in sample (g) b | |||
|---|---|---|---|---|---|---|---|
| k | r2 | k | r2 | KH | r2 | ||
| (1) Vinyloxy-F | 0.0004 | 0.98 | 0.015 | 0.8 | 0.007 | 0.97 | 2.09 |
| (2) ½ Vinyloxy-F | 0.0001 | 0.92 | 0.018 | 0.92 | 0.002 | 0.86 | 1.05 |
| (3) Vinyloxy-F-75 | 0.0003 | 0.99 | 0.039 | 0.93 | 0.003 | 0.94 | 1.19 |
| (4) Vinyloxy-100FOH blend | 0.0008 | 0.99 | 0.020 | 0.97 | 0.010 | 0.97 | 2.10 |
| (5) Vinyloxy-100½FOH blend | 0.0002 | 0.99 | 0.010 | 0.89 | 0.003 | 0.99 | 1.05 |
| (6) Vinyloxy-75 FOH blend | 0.0030 | 0.97 | 0.160 | 0.82 | 0.010 | 0.99 | 2.10 |
a- First 3 entries (1–3) are fluorinated-modified networks, vinyloxy-F denotes the hydrolyzable monomer modified to contain 1 fluorinated hydrolyzable group and 3 ene groups for network formation, an F3 monomer; the remaining 2 network samples contain mixtures of ene-monomers, Vinyloxy-F was added to tetrafunctional vinyloxy-100 to create (2) ½ vinyloxy-F, allowing the sample to contain half the amount of fluorinated material, and finally (3) ene-modified urethane segment was added at 25 wt %; last 3 entries are blends (4–6), 2,2,3,3-tetrafluoro-1-propanol is added to vinyloxy-100 vinyloxy-75 formulations to create uncrosslinked blend compositions for comparison;b- formulations were varied to produce several concentrations of fluorinated samples, so comparisons could be drawn. The amount of fluorinated material in the total 12 g molded puck sample is provided in g.
Acknowledgments
The authors wish to thank the Office of Naval Research and the University Laboratory Initiative Program for their financial support. A special thank you would like to be extended to the late Charles Hoyle for his inspiration and education in the area of click chemistry.
References
- Ray, F.; Linossier, I.; Peron, J.J.; Langlois, V.; Vallee-Rehel, K. Antifouling activity of marine paints: Study of erosion. Prog. Org. Coat. 2007, 60, 194–206. [Google Scholar]
- Chang-Sik, H.; Gardella, J.A. Surface chemistry of biodegradable polymers for drug delivery systems. Chem. Rev. 2005, 105, 4205–4232. [Google Scholar]
- Yebra, D.M.; Kiil, S.; Dam-Johansesn, K. Antifouling technology—Past, present, and future steps towards efficient, and environmentally friendly antifouling coatings. Prog. Org. Coat. 2004, 50, 75–104. [Google Scholar]
- Siepmann, J.; Gopferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliver. Rev. 2001, 48, 229–247. [Google Scholar]
- Martin, J.; Hosticka, B.; Lattimer, C.; Norris, P.M. Mechanical and acoustical properties as a function of PEG concentration in macroporous silica gels. J. Non-Cryst. Solids 2001, 285, 222–229. [Google Scholar]
- Chang, H.Y.; Thangamuthu, R.; Lin, C.W. Structure-property relationships in PEG/SiO2 based proton conducting hybrid membranes—A 29Si CP/MAS solid-state NMR study. J. Membr. Sci. 2004, 228, 217–226. [Google Scholar]
- Fujihara, S.; Kitta, S. Broadband visible emissions from sol-gel-derived silica/poly(vinyl pyrrolidone) hybrids. Chem. Phys. Lett. 2004, 397, 479–483. [Google Scholar]
- Tanaka, K.; Kozuka, H. Sol-gel preparation and mechanical properties of machinable cellulose/silica and polyvinylpyrrolidone/silica composites. J. Sol-Gel Sci. Technol. 2004, 32, 73–77. [Google Scholar]
- Tamaki, R.; Chujo, Y. A synthesis of poly(vinyl alcohol)/silica gel polymer hybrids by in-situ hydrolysis method. Appl. Organomet. Chem. 1998, 12, 755–762. [Google Scholar]
- Kros, A.; Gerritsen, M.; Sprakel, V.S.I.; Sommerdijk, N.A.J.M.; Jansen, J.A.; Nolte, R.J.M. Silica-based hybrid materials as biocompatible coatings for glucose sensors. Sens. Actuat. B: Chem. 2001, 81, 68–75. [Google Scholar]
- Brus, J.; Špírková, M.; Hlavatá, D.; Strachota, A. Self-organization, structure, dynamic properties, and surface morphology of silica/epoxy films as seen by solid-state NMR, SAXS, and AFM. Macromolecules 2004, 37, 1346–1357. [Google Scholar]
- Innocenzi, P.; Kidchob, T.; Yoko, T. Hybrid organic-inorganic sol-gel materials based on epoxy-amine systems. J. Sol-Gel Sci. Technol. 2005, 35, 225–235. [Google Scholar]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley Inerscience: New York, NY, USA, 1979. [Google Scholar]
- Hoyle, C.E.; Lee, T.Y.; Roper, T. Thiol-enes: Chemistry of the past with promise for the future. J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 5301–5338. [Google Scholar]
- Yu, B.; Chan, J.W.; Hoyle, C.E.; Lowe, A.B. Sequential thiol-ene/thiol-ene and thiol-ene/thiol-yne reactions as a route to well-defined mono and bis end-functionalized poly(N-isopropylacrylamide). J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 3544–3557. [Google Scholar]
- Phillips, J.P.; Mackey, N.M.; Confait, B.S.; Heaps, D.T.; Deng, X.; Todd, M.L.; Stevenson, S.; Zhou, H.; Hoyle, C.E. Dispersion of gold nanoparticles in UV-cured, thiol-ene films by precomplexation of gold-thiol. Chem. Mater. 2008, 20, 5240–5245. [Google Scholar]
- Shin, J.; Nazarenko, S.; Phillips, J.P.; Hoyle, C.E. Physical and chemical modifications of thiol-ene networks to control activation energy of enthalpy relaxation. Polymer 2009, 50, 6281–6286. [Google Scholar]
- Krol, P. Synthesis methods, chemical structures and phase structures of linear polyurethanes. Properties and applications of linear polyurethanes in polyurethane elastomers, copolymers and ionomers. Prog. Mater. Sci. 2007, 52, 915–1015. [Google Scholar]
- Bruchmann, B. Dendritic polymers based on urethane chemistry—Syntheses and applications. Macromol. Mater. Eng. 2007, 292, 981–992. [Google Scholar]
- Prabu, A.A.; Alagar, M. Thermal and morphological properties of silicone-polyurethane-epoxy intercrosslinked matrix materials. J. Macromol. Sci. Pure 2005, A42, 175–188. [Google Scholar]
- Wynne, J.H.; Fulmer, P.A.; McCluskey, M.D.; Mackey, N.M.; Buchanan, J.P. Synthesis and development of a multifunctional self-decontaminating polyurethane coating. ACS Appl. Mater. Intfaces 2011. [Google Scholar] [CrossRef]
- Senyurt, A.F.; Hoyle, C.E.; Wei, H.; Piland, S.G.; Gould, T.E. Thermal and mechanical properties of cross-linked photopolymers based on multifunctional thiol-urethane ene monomers. Macromolecules 2007, 40, 3174–3182. [Google Scholar]
- Li, Q.; Zhou, H.; Wicks, D.A.; Hoyle, C.E. Thiourethane-based thiol-ene high Tg networks: Preparation, thermal, mechanical, and physical properties. J. Polym. Sci. Part A: Polym. Chem. 2007, 45, 5103–5111. [Google Scholar]
- Shin, J.; Matsushima, H.; Chan, J.W.; Hoyle, C.E. Segmented polythiourethane elastomers through sequential thiol-ene and thiol-isocyanate reactions. Macromolecules 2009, 42, 3294–3301. [Google Scholar]
- Gohel, M.C.; Panchal, M.K.; Jogani, V.V. Novel mathematical method for quantitative expression of deviation from the Higuchi model. AAPS PhramSciTech 2000, 1, E31. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mackey, N.M.; Confait, B.S.; Wynne, J.H.; Buchanan, J.P. Preparation of Novel Hydrolyzing Urethane Modified Thiol-Ene Networks. Polymers 2011, 3, 1849-1865. https://doi.org/10.3390/polym3041849
Mackey NM, Confait BS, Wynne JH, Buchanan JP. Preparation of Novel Hydrolyzing Urethane Modified Thiol-Ene Networks. Polymers. 2011; 3(4):1849-1865. https://doi.org/10.3390/polym3041849
Chicago/Turabian StyleMackey, Nicole M., Bridget S. Confait, James H. Wynne, and J. Paige Buchanan. 2011. "Preparation of Novel Hydrolyzing Urethane Modified Thiol-Ene Networks" Polymers 3, no. 4: 1849-1865. https://doi.org/10.3390/polym3041849
APA StyleMackey, N. M., Confait, B. S., Wynne, J. H., & Buchanan, J. P. (2011). Preparation of Novel Hydrolyzing Urethane Modified Thiol-Ene Networks. Polymers, 3(4), 1849-1865. https://doi.org/10.3390/polym3041849