Thermal Properties and Thermal Degradation of Cellulose Tri-Stearate (CTs)

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of CTs
2.3. Characterizations of CTs
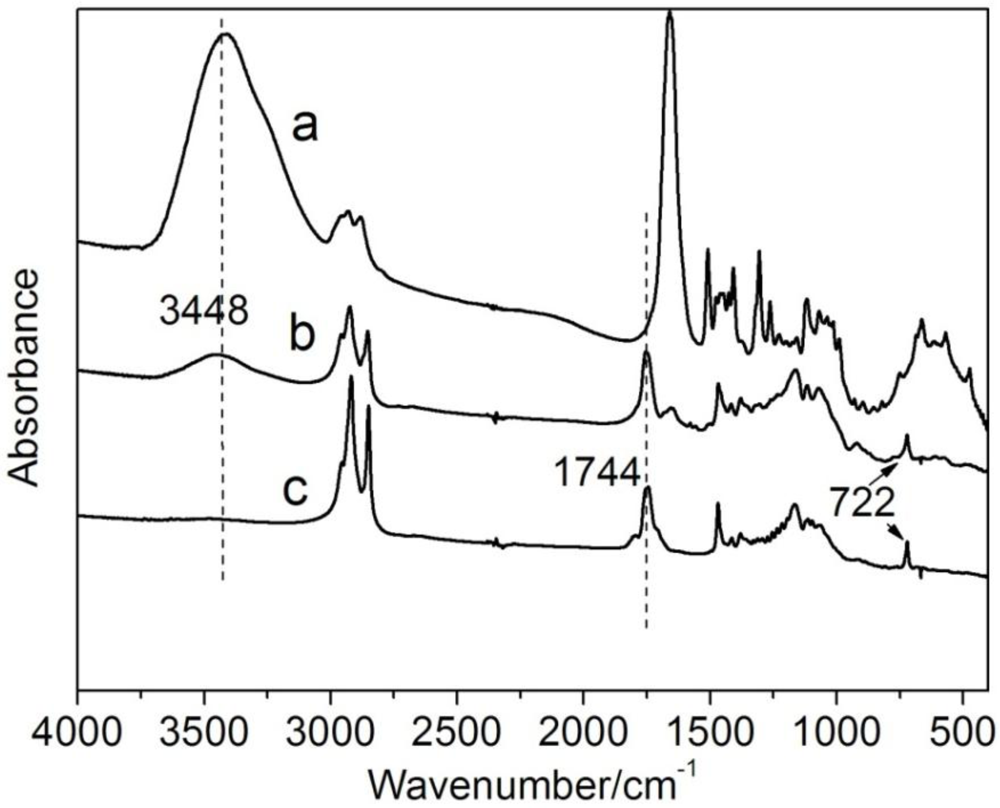
2.3.1. FTIR
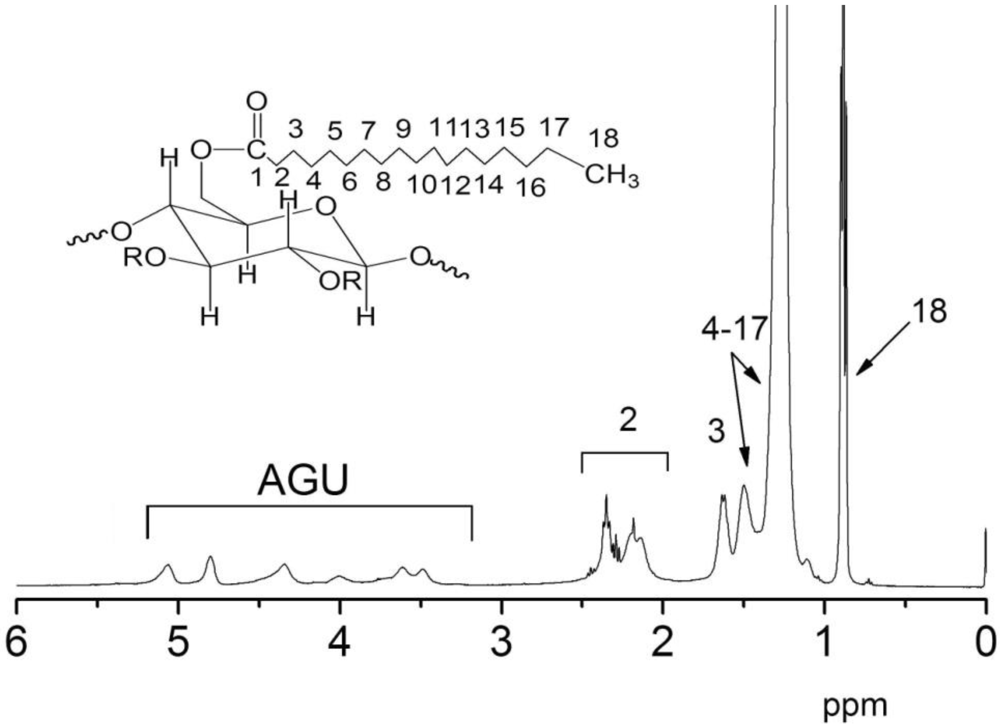
2.3.2. 1H-NMR
2.3.3. Degree of Substitution (DS)
2.3.4. Thermal Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of CTs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Molar ratio of AGU: TFAA: SA a | Reaction temperature | Reaction Time | Solvent added | WI c | Yield | DS |
|---|---|---|---|---|---|---|---|
| °C | h | % | % | ||||
| 1 | 1:3.3:3.63 | 50 | 5 | / | 256 | 89 | 2.80 |
| 2 | 1:3.3:3.63 | 50 | 4 | Chloroform/50 mL | 261 | 88 | 2.85 |
| 3 | 1:3.3:3.63 | 50 | 5 | Chloroform/50 mL | 313 | 97 | 2.95 |
| 2.99 d | |||||||
| 4 | 1:3.3:3.63 | 50 | 5 | Toluene/50 mL | 276 | 90 | 2.90 |
| 5 b | 1:10:11 | 50 | 5 | / | 308 | 92 | 2.97 |
| 6 | 1:2.6:2.9 | 50 | 5 | Chloroform/100 mL | 209 | 95 | 2.45 |


3.2. Thermal Properties of CTs


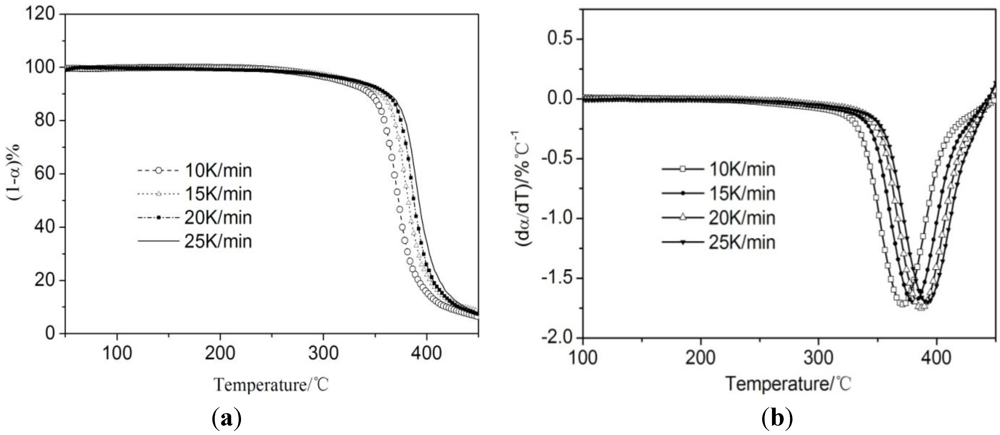
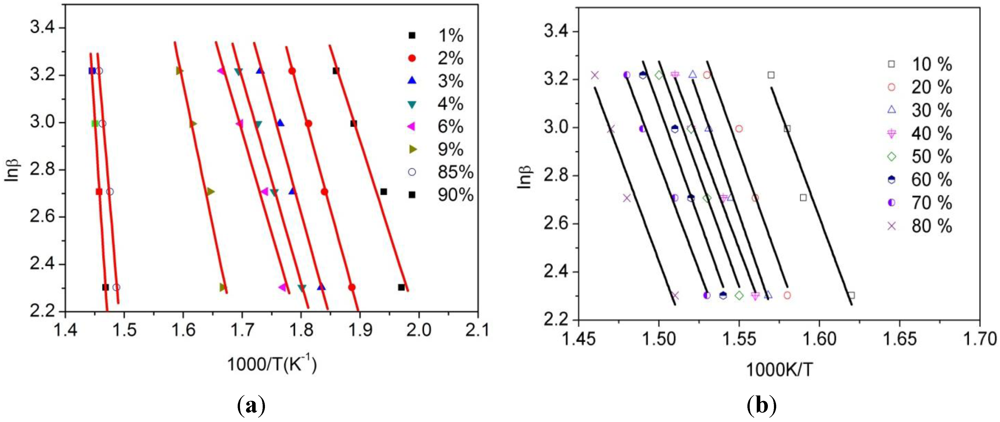
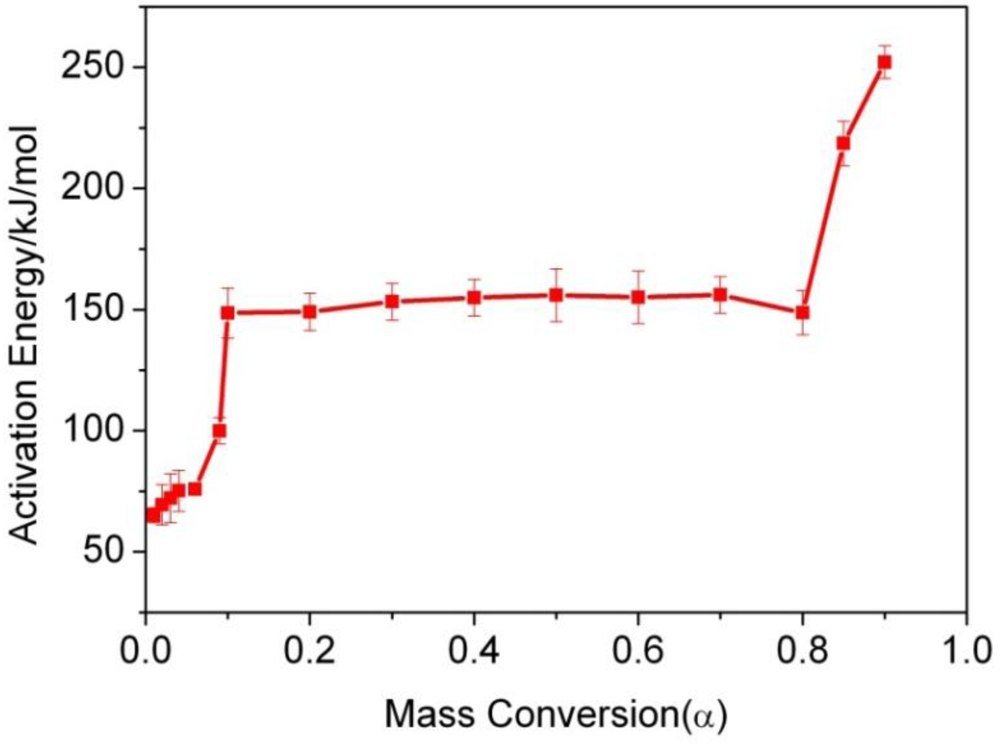
3.3. Thermogravimetric Analysis of CTs


| α/% | 1 | 2 | 3 | 4 | 6 | 9 |
| Ea(KJ/mol) | 65.0 ± 3.1 | 69.4 ± 8.3 | 72.1 ± 10.1 | 75.2 ± 8.5 | 75.9 ± 2.2 | 99.9 ± 5.4 |
| R | 0.9942 | 0.9603 | 0.9587 | 0.9487 | 0.9972 | 0.9843 |
| α/% | 10 | 20 | 30 | 40 | 50 | 60 |
| Ea(KJ/mol) | 148.6 ± 10.25 | 149.1 ± 7.79 | 153.3 ± 7.58 | 154.9 ± 7.58 | 156.0 ± 10.93 | 155.1 ± 10.93 |
| R | 0.9906 | 0.9957 | 0.9965 | 0.9965 | 0.9933 | 0.9923 |
| α/% | 70 | 80 | 85 | 90 | ||
| Ea(KJ/mol) | 156.1 ± 7.58 | 148.7 ± 9.16 | 218.6 ± 9.25 | 252.1 ± 6.72 | ||
| R | 0.9965 | 0.9947 | 0.9926 | 0.9966 |


| β(K/min) | 10 | 15 | 20 | 25 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mechanism | g(α) | E | R2 | E | R2 | E | R2 | E | R2 |
| A2 |  | 106 | 0.9903 | 116 | 0.9891 | 121 | 0.9916 | 127 | 0.981 |
| A3 |  | 67 | 0.9892 | 73 | 0.9881 | 77 | 0.9907 | 81 | 0.979 |
| A4 |  | 48 | 0.9879 | 52 | 0.9869 | 55 | 0.9898 | 58 | 0.9768 |
| R1 |  | 155 | 0.9875 | 168 | 0.9898 | 185 | 0.9936 | 207 | 0.9892 |
| R2 |  | 186 | 0.9744 | 193 | 0.9705 | 202 | 0.9848 | 217 | 0.9727 |
| R3 |  | 205 | 0.9979 | 220 | 0.9935 | 236 | 0.9971 | 243 | 0.9887 |
| D1 |  | 347 | 0.9922 | 359 | 0.9872 | 382 | 0.9875 | 407 | 0.9917 |
| D2 |  | 320 | 0.9966 | 396 | 0.9978 | 422 | 0.9919 | 441 | 0.9976 |
| D3 |  | 390 | 0.9974 | 407 | 0.9992 | 440 | 0.9983 | 467 | 0.9897 |
| D4 |  | 385 | 0.9878 | 421 | 0.9889 | 439 | 0.9856 | 473 | 0.9912 |
| F1 |  | 223 | 0.9921 | 242 | 0.9987 | 253 | 0.9993 | 267 | 0.9987 |
| F2 |  | 153 | 0.9974 | 156 | 0.9987 | 159 | 0.9992 | 160 | 0.9987 |
| F3 |  | 290 | 0.9914 | 300 | 0.9763 | 322 | 0.9668 | 328 | 0.9940 |


4. Conclusions
Acknowledgments
References
- Edgar, K.J.; Buchanan, C.M.; Debenham, J.S.; Rundquist, P.A.; Seiler, B.D.; Shelton, M.C.; Tindall, D. Advances in cellulose ester performance and application. Prog. Polym. Sci. 2001, 26, 1605–1688. [Google Scholar]
- Kwatra, H.S.; Caruthers, J.M.; Tao, B.Y. Synthesis of long chain fatty acids esterified onto cellulose via the vacuum-acid chloride process. Ind. Eng. Chem. Res. 1992, 31, 2647–2651. [Google Scholar]
- Heinze, T.; Liebert, T. Unconventional methods in cellulose functionalization. Prog. Polym. Sci. 2001, 26, 1689–1762. [Google Scholar]
- Crépy, L.; Chaveriat, L.; Banoub, J.; Martin, P.; Joly, N. Synthesis of cellulose fatty esters as plastics-influence of the degree of substitution and the fatty chain length on mechanical properties. ChemSusChem 2009, 2, 165–170. [Google Scholar]
- Huang, F.Y.; Yu, Y.; Wu, X.J. Characterization and Properties of Cellulose Oleate. Adv. Mater. Res. 2011, 197–198, 1306–1309. [Google Scholar]
- Heinze, T.; Liebert, T.F.; Pfeiffer, K.S.; Hussain, M.A. Unconventional cellulose ester: Synthesis, characterization and structure-property relations. Cellulose 2003, 10, 283–296. [Google Scholar]
- Granstrom, M.; Kavakka, J.; King, A.; Majoinen, J.; Makela, V.; Helaja, J.; Hietala, S.; Virtanen, T.; Maunu, S.L.; Argyropoulos, D.S.; Kilpelainen, I. Tosylation and acylation of cellulose in 1-allyl-3-methylimidazolium chloride. Cellulose 2008, 15, 481–488. [Google Scholar]
- Barthel, S.; Heinze, T. Acylation and carbanilation of cellulose in ionic liquids. Green Chem. 2006, 8, 301–306. [Google Scholar]
- Memmi, A.; Granet, R.; Gahbiche, M.A.; Fekih, A.; Bakhrouf, A.; Krausz, P. Fatty esters of cellulose from olive pomace and barley bran: improved mechanical properties by metathesis crosslinking. J. Appl. Polym. Sci. 2006, 101, 751–755. [Google Scholar]
- Satgé, C.; Verneuil, B.; Branland, P.; Granet, R.; Krausz, P.; Rozier, J.; Petit, C. Rapid homogeneous esterification of cellulose induced by microwave irradiation. Carbohyd. Polym. 2002, 49, 373–376. [Google Scholar]
- Joly, N.; Granet, R.; Branland, P.; Verneuil, B.; Krausz, P. New methods for acylation of pure and sawdust-extracted cellulose by fatty acid derivatives—Thermal and mechanical analyses of cellulose-based plastic films. J. Appl. Polym. Sci. 2005, 97, 1266–1278. [Google Scholar]
- Arni, P.C.; Gray, J.D.; Scougall, R.K. Chemical modification of wood. I. Use of trifluoroacetic anhydride in the esterification of wood by carboxylic acids. J. Appl. Chem. 1961, 11, 157–163. [Google Scholar]
- Roy, P.K.; Surekha, P.; Rajagopal, C.; Choudhary, V. Thermal degradation studies of LDPE containing cobalt stearate as pro-oxidant. Express Polym. Lett. 2007, 4, 208–216. [Google Scholar]
- Wang, H.M.; Tao, X.M.; Newton, E. Thermal degradation kinetics and lifetime prediction of a luminescent conducting polymer. Polym. Int. 2004, 53, 20–26. [Google Scholar]
- Morooka, T.; Norimoto, M.; Yamada, T. Dielectric properties of cellulose acylates. J. Appl. Polym. Sci. 1984, 29, 3981–3990. [Google Scholar]
- Sealey, J.E.; Samaranayake, G.; Todd, J.G.; Glasser, W.G. Novel cellulose derivatives. IV. Preparation and thermal analysis of waxy esters of cellulose. J. Polym. Sci. B Polym. Phys. 1996, 34, 1613–1620. [Google Scholar] [CrossRef]
- Jandura, P.; Riedl, B.; Kokta, B.V. Thermal degradation behavior of cellulose fibers partially esterified with some long chain organic acids. Polym. Degrad. Stabil. 2000, 70, 387–394. [Google Scholar]
- Huang, M.R.; Li, X.G. Thermal degradation of cellulose and cellulose esters. J. Appl. Polym. Sci. 1998, 68, 293–304. [Google Scholar]
- Sairam, M.; Sreedhar, B.; Mohan Rao, D.V.; Srinivasan, P. Synthesis and thermal degradation kinetics of cellulose esters. Polym. Adv. Technol. 2003, 14, 477–485. [Google Scholar]
- Hsu, W.P.; Levon, K.; Ho, K.S.; Myerson, A.S.; Kwei, T.K. Side chain order in poly(3-Alkyl Thiophanes). Macromolecules 1993, 26, 1318–1323. [Google Scholar]
- Ozawa, T.A. New method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar]
- Coats, A.W.; Redfern, J.P. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar]
- Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D.N. Thermal degradation kinetics of the biodegradable aliphatic polyester, poly(propylene succinate). Polym. Degrad. Stabil. 2006, 91, 60–68. [Google Scholar]
- Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D.N. Effect of molecular weight on thermal degradation mechanism of the biodegradable polyester poly(ethylene succinate). Thermochim. Acta 2006, 440, 166–175. [Google Scholar]
- Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D. Thermal degradation kinetics and decomposition mechanism of two new aliphatic biodegradable polyesters poly(propylene glutarate) and poly(propylene suberate). Thermochim. Acta 2010, 505, 59–68. [Google Scholar]
- Chrissafis, K.; Paraskevopoulos, K.M.; Papageorgiou, G.Z.; Bikiaris, D.N. Thermal decomposition of poly(propylene sebacate) and poly(propylene azelate) biodegradable polyesters: Evaluation of mechanisms using TGA, FTIR and GC/MS. J. Anal. Appl. Pyrol. 2011, 92, 123–130. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, F.-Y. Thermal Properties and Thermal Degradation of Cellulose Tri-Stearate (CTs). Polymers 2012, 4, 1012-1024. https://doi.org/10.3390/polym4021012
Huang F-Y. Thermal Properties and Thermal Degradation of Cellulose Tri-Stearate (CTs). Polymers. 2012; 4(2):1012-1024. https://doi.org/10.3390/polym4021012
Chicago/Turabian StyleHuang, Feng-Yuan. 2012. "Thermal Properties and Thermal Degradation of Cellulose Tri-Stearate (CTs)" Polymers 4, no. 2: 1012-1024. https://doi.org/10.3390/polym4021012
APA StyleHuang, F. -Y. (2012). Thermal Properties and Thermal Degradation of Cellulose Tri-Stearate (CTs). Polymers, 4(2), 1012-1024. https://doi.org/10.3390/polym4021012