3.1. Semi-Crystalline Polymers in (n-s)-CD-ICs and Coalesced from Their Stoichiometric ICs
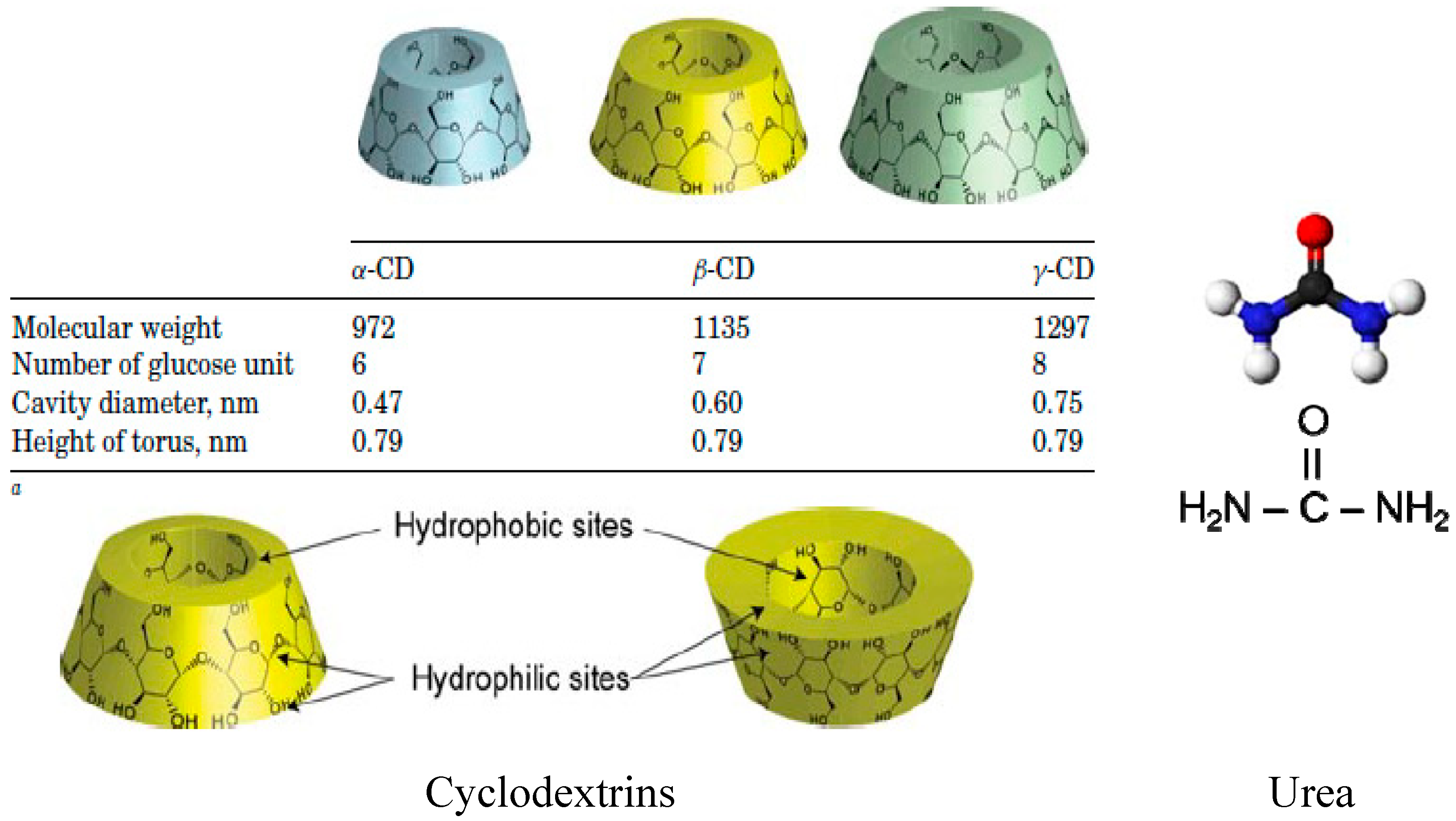
We begin by summarizing the formation and behaviors of (n-s)-ICs containing guest nylon-6 samples of various molecular weights and α- and γ-CD hosts [
8,
9]. Nylon-6 (N-6) samples with
MW = 30,000 and 60,000 Da were obtained from BASF (Ludwigshafen, Germany), while those with
MW = 300,000 and 600,000 Da were obtained by anionically initiated ring-opening polymerization of ε-caprolactam [
15]. It should be noted that single and two side-by-side nylon-6 chains [
16] are, respectively, threaded through α- and γ-CDs.
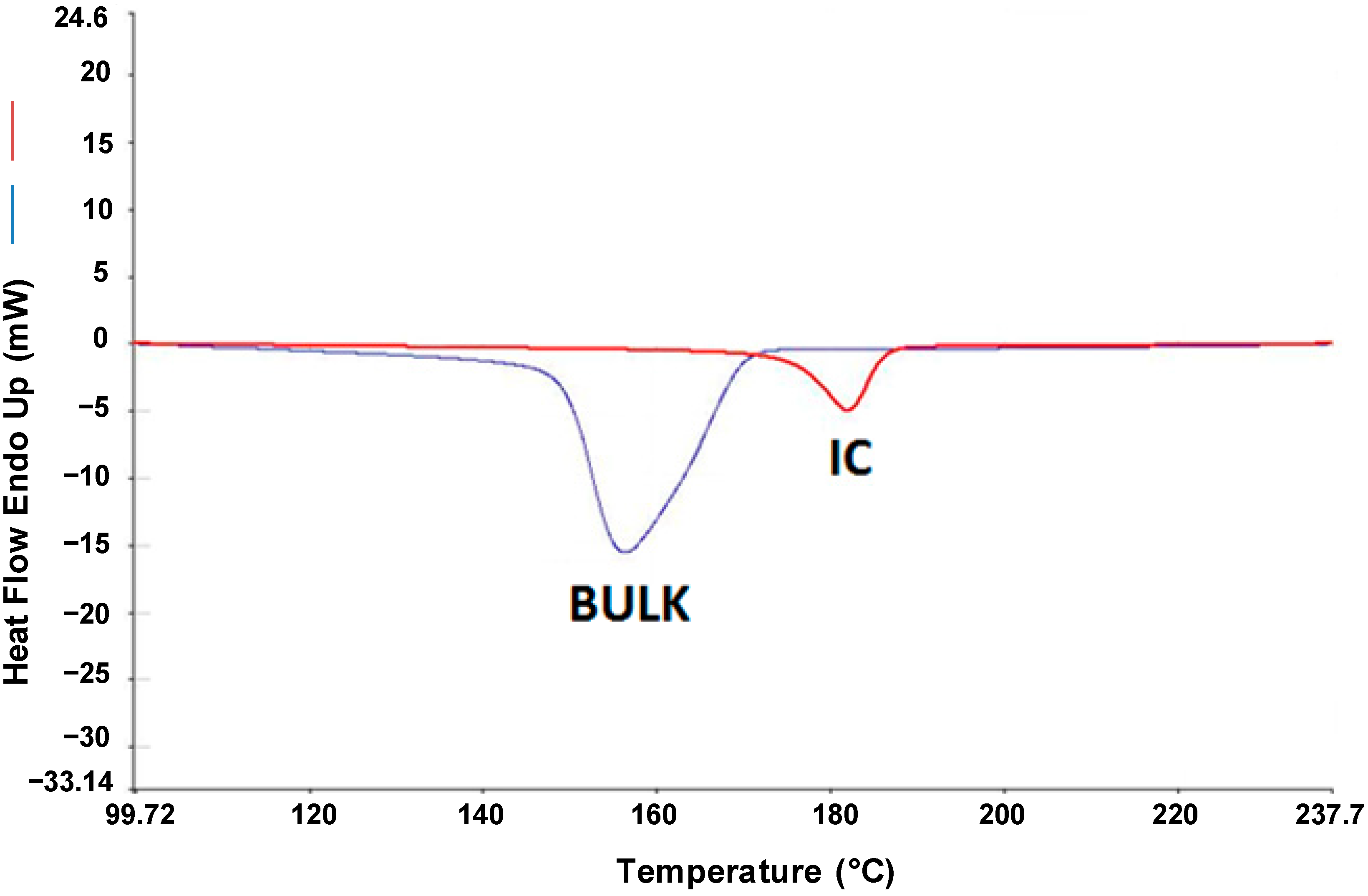
The melt crystallization behavior shown in
Figure 3 for the 3:1 (n-s)-N-6-γ-CD-IC cooled at 10 °C/min in the DSC is typical for all the (n-s)-N-6-CD-ICs examined. Note that, upon cooling from the melt, the un-included chain portions in the (n-s)-IC sample crystallize before,
Tc ~30 °C higher, than the neat bulk sample. It was also observed that a higher fraction or proportion of the un-included N-6 chain portions “dangling” from the 3:1 (n-s)-α-CD-IC crystals are able to crystallize compared to the neat N-6 melt. Qualitatively similar observations were made for 6:1 N-6-γ-CD-IC and bulk samples, with N-6 of
MW = 600,000. As the N-6
MW increased, the constraints on the “dangling” chains in 3:1 (n-s)-N-6-α-CD-ICs increasingly facilitate their crystallization in comparison to their neat melts, which, to the contrary, generally crystallize less readily as their MWs increase. Also note that the “dangling” chain portions in the (n-s)-nylon-6-γ-CD-ICs showed elevated
Tcs as well.
Figure 3.
Differential Scanning Calorimetry (DSC) cooling scans from the melts of 600,000 Da bulk N-6 and its 3:1 (n-s)-γ-CD-IC [
8].
Figure 3.
Differential Scanning Calorimetry (DSC) cooling scans from the melts of 600,000 Da bulk N-6 and its 3:1 (n-s)-γ-CD-IC [
8].
Observations that the un-included, uncovered N-6 chain portions in both 3:1 α-CD- and 6:1 γ-CD-ICs show
Xcs and
Tcs that are increased in comparison to the 2:1 N-6-α-CD- and 4:1 N-6-γ-CD-ICs are in agreement with the results reported for (n-s)-poly(butylene succinate) (PBS)-α-CD-ICs [
6,
17]. Actually N-6 chains dangling from (n-s)-N-6-α- and γ-CD-ICs with stoichiometries less than 3:1 and 6:1, respectively, were observed to crystallize less readily than their neat bulk samples.
A more germane measure of the crystallizability of bulk and un-included “dangling” N-6 chains was suggested to be provided by comparing “dangling” N-6 chains of the same length (
MW) as the bulk N-6. As an example, the N-6 chains “dangling” from the 4:1 N-6 (600,000)-γ-CD-IC crystals should on average have a length characterized by
MW = 300,000, because the 2:1 N-6-γ-CD-IC is stoichiometric, with no un-included “dangling” chain portions. The 2:1 N-6 (600,000)-α-CD-IC should also have “dangling”chains of average
MW = 300,000. Bulk N-6 (300,000); 2:1 N-6 (600,000)-α-CD-IC; and 4:1 N-6 (600,000)-γ-CD-IC showed, respectively,
Xc,
Tc = 0.23, 174; 0.15, 173; and 0.16, 162 °C,. Thus, N-6 chains “dangling” from both (n-s-)-CD-ICs did not show enhanced crystallizability compared with bulk N-6 with
MW = 300,000. Except for those between the chains “dangling” from 3:1 and 6:1 N-6-α- and γ-CD-ICs and bulk N-6 with the same
MWs, this observation held for other like comparisons. In opposition to observations reported [
17] for (n-s)-PBS- and poly-(ε-caprolactone) (PCL)-α-CD-ICs, however, as the N-6:CD ratio increased, the crystallinity
Xc (of the un-included chain portion) increased.
Even though Δ
Hc [(n-s)-CD-IC]/Δ
Hc (bulk) =
Xc [(n-s)-CD-IC]/
Xc (bulk) increased with N-6
MW for 3:1 α- and 6:1 γ-CD-ICs, their values of (
Tc ((n-s)-CD-IC) −
Tc (bulk))s did not. In other words, as the
MW of N-6 increased, the levels of crystallinity achieved for the un-included yet constrained “dangling” chains in their (n-s)-CD-ICs increased in comparison to their neat melts. At the same time, even though they generally crystallized at temperatures higher than bulk samples, their rates of crystallization did not increase [
9].
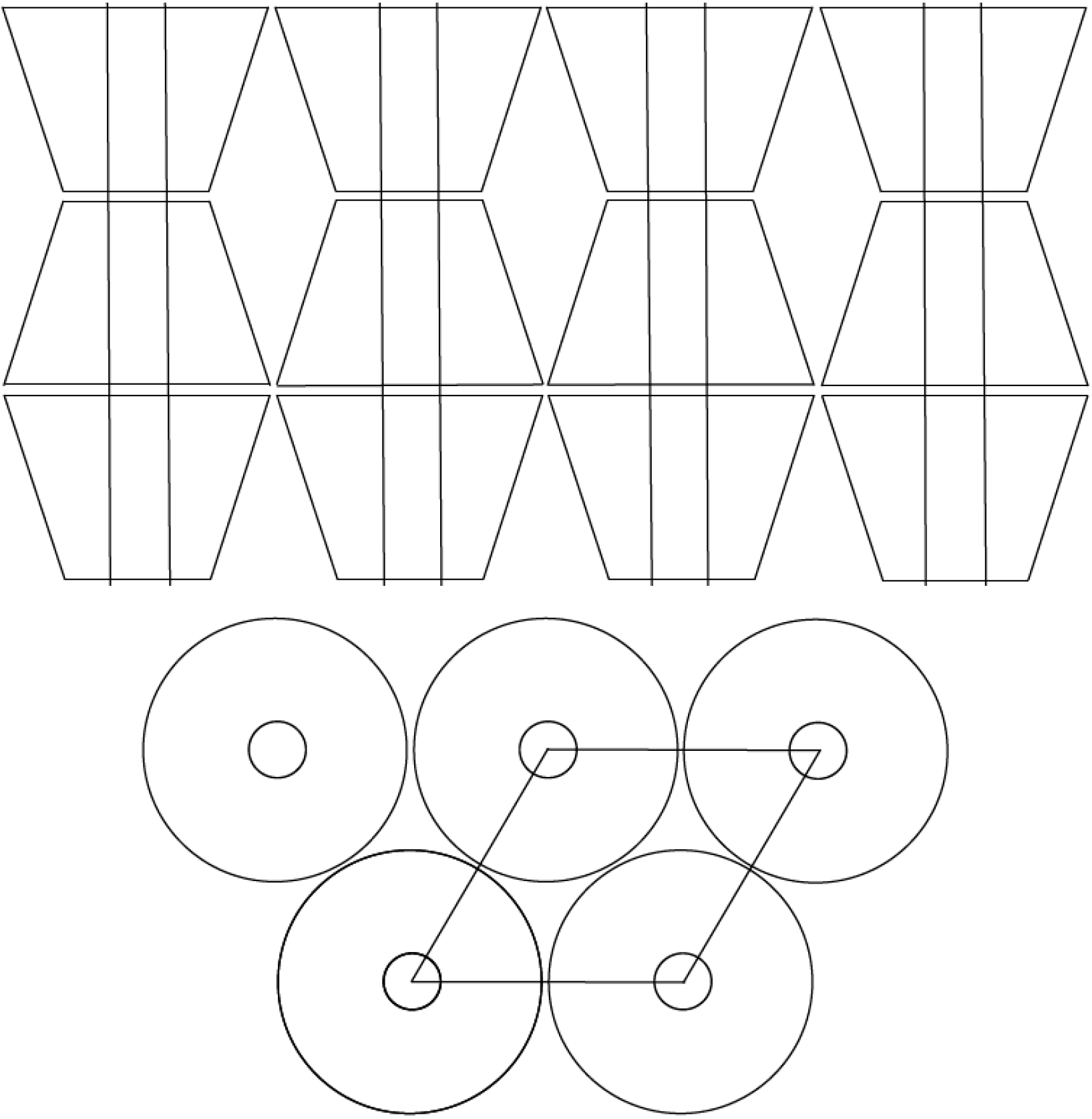
Consider the constraints placed on un-included N-6 chains “dangling” from their (n-s)-α-CD-ICs, as they emerge from ~0.5 nm channels separated by ~1.4 nm, as indicated below in
Figure 4, to those experienced by the pair of closely adjacent N-6 chains in each ~1 nm γ-CD-IC channel, separated by ~1.7 nm. The distinct behaviors observed for (n-s)-N-6-CD-ICs made with α- and γ-CDs—lower
Tcs and Δ
Hcs for (n-s)-N-6-γ-CD-ICs—can in part be attributed to the differences in the constraints imposed on their un-included chains “dangling” from their CD-IC channels. Because, in fact, they form dense brushes [
18] that do not significantly “dangle”, but are largely extended and parallel, and will be discussed further below.
Figure 4.
Channel structure of a columnar CD-IC, with 0.6 (α-CD), 0.8 (γ-CD) N-6 chains/nm
2 of CD-IC crystal surface. Threaded chains are ~1.5–1.8 nm apart, so the protruding N-6 chains form dense polymer brushes [
18] (For comparison, in the α-form bulk crystal there are 1.4 N-6 chains/nm
2) [
8].
Figure 4.
Channel structure of a columnar CD-IC, with 0.6 (α-CD), 0.8 (γ-CD) N-6 chains/nm
2 of CD-IC crystal surface. Threaded chains are ~1.5–1.8 nm apart, so the protruding N-6 chains form dense polymer brushes [
18] (For comparison, in the α-form bulk crystal there are 1.4 N-6 chains/nm
2) [
8].
Single and pairs of side-by-side N-6 chains included, respectively, in the nylon-α- and γ-CD-IC channels emerge from them immediately above their crystalline CD-IC surfaces, with initial channel densities of ~4 and ~3 nylon-6 chains/nm2 of channel, respectively. Relative to the entire crystalline surface, or farther above the surface, the density of N-6 chains protruding above them is only modestly greater for the (n-s)-γ-CD-ICs (0.8 (γ) vs. the 0.6 (α) nylon-6 chains/nm2 of IC crystal surface).
We observed the N-6 chains protruding from their 6:1 (n-s)-γ-CD-ICs to be only minimally more crystallizable (higher Tcs, but comparable ΔHcs) than bulk N-6, while significantly less crystallizable than the N-6 chains protruding from their 3:1 (n-s)-α-CD-ICs. Apparently the pairs of side-by-side N-6 chains emerging from each (n-s)-γ-CD-IC channel are less able to crystallize than the single chains emerging from each (n-s)-α-CD-IC channel.
The density of an emerging pair of N-6 chains just above the surface of the (n-s)-γ-CD-IC crystals is much greater (~3/nm2 of γ-CD channel vs. 1.4/nm2 in the bulk crystal) than the N-6 chain density farther above the (n-s)-Υ-CD-IC crystal surfaces (0.6 (α)-0.8 (γ) chains/nm2), or for that matter in dense polymer brushes. This could result in strong associations, such as hydrogen bonding, between pairs of emerging side-by-side N-6 chains. This may retard their interactions/crystallization with other such closely and strongly interacting pairs of N-6 chains emerging from nearby γ-CD-IC channels.
This suggestion can also be made based on the comparison of two chain “one-dimensional N-6 nano-crystals” with multi-chain “three-dimensional N-6 micro-crystals”, with the former resulting from pairs of side-by-side chains included and initially emerging from the γ-CD-IC channels. If viable, then as the lengths (MWs) of the chains protruding from N-6-(n-s)-γ-CD-ICs are reduced, the constraint opposing crystallization produced by the pairs of N-6 chains emerging from the γ-CD-IC channels should increase making their crystallization more difficult.
As the lengths of N-6 chains or their
MWs increase, generally their melt-crystallization temperatures
Tc decrease/increase and the levels of crystallinity
Xc decrease/increase, respectively, for bulk and (n-s)-CD-IC samples. For neat molten polymers, the increased entanglement between higher
MW chains and the structural defects they produce are generally cited [
19] as reasons that they may crystallize more slowly and not as completely as samples with lower
MWs. Since the opposite behavior is observed for the un-included “dangling” yet constrained chains in (n-s)-N-6-CD-ICs, their degree of entanglement, if any, is apparently not important to their ability to crystallize. Their relative center-of-mass diffusion is limited by the constraining CD-IC crystals from which they emerge. Thus, entanglement and un-entanglement of the dangling chains, would be expected to be significantly retarded in comparison to that in unconstrained neat molten N-6 samples. DSC observations of (n-s)-N-6-CD-ICs, such as those presented in
Figure 3, but not presented here, remain unchanged upon subsequent repeated heating/cooling scans, which suggests the absence of CD de-threading.
We believe the major factor contributing to the enhanced melt crystallizability observed for the protruding N-6 chains (higher
Tcs and larger Δ
Hcs and
Xcs), as their
MWs increase, is likely due to the concomitant increase in their extension and orientation with respect to the CD-IC crystal surfaces. Though seemingly discordant with the well-known behavior of most polymer brushes [
18], whose chains lose orientation perpendicular to and extension from their surfaces as their
MW increases, and which initiates a brush-to-mushroom transition, it must be remembered that polymer brushes are generally formed with non-crystallizable polymers and at lower surface densities than the 0.6–0.8 N-6 chains/nm
2 emanating from the (n-s)-N-6-CD-IC crystal surfaces. Furthermore, and probably more important, many of the un-included chain portions in (n-s)-CD-ICs both begin and terminate in CD-IC crystals, and so their constraints would correspond to dense polymer brushes covalently bonded to parallel separated solid surfaces.
At higher brush densities, similar to those expected for (n-s)-polymer-γ-CD-ICs (see
Figure 1,
Figure 2 and
Figure 4), it has been observed [
18,
20] that even the chains in non-crystallizing brushes are highly extended and, thus, highly aligned, perpendicular to the surfaces of their attachment, and increasingly so as
MW increases. Particularly for the 3:1 α- and 6:1 γ-CD-ICs, this could explain the observed enhancement of their crystallizability, both kinetically and thermodynamically, with increased
MW of the protruding N-6 chains. Whether the protruding N-6 chains begin crystallizing near the CD-IC crystalline surfaces or farther removed and nearer to their “free” chain ends [
9] each would favor the enhancement of their crystallization as their
MWs increase.
It is apparent that portions of N-6 chains that are un-included and protrude from their (n-s)-CD-ICs crystallize more readily (faster at higher temperatures, over a narrow temperature range, and to greater extents) than bulk N-6 chains. However, the melting temperatures,
Tm, of their resulting crystals are very similar.
Tm = Δ
Hm/Δ
Sm, is the ratio of the enthalpy to the entropy of melting. Apparently this ratio is very similar for N-6 chains in bulk and those constrained by and protrude from (n-s)-CD-IC crystals. This is surprising because we expect in the melt that N-6 chains protruding from the surfaces of their (n-s)-CD-ICs likely constitute a high density brush with largely extended chains, which are expected to possess less conformational entropy than an unconstrained neat N-6 melt. The Δ
Sm of polymers is generally dominated by the difference in the intramolecular conformational entropies, Δ
Sconf, of their randomly coiling molten and highly extended and conformationally restricted crystalline chains [
21,
22,
23,
24,
25,
26,
27]. Thus, we would expect the melting of the crystals formed by the nylon-6 chains protruding from their (n-s)-CD-ICs to have smaller Δ
Sconfs ~ Δ
Sms, and therefore, higher
Tms than neat nylon-6 crystals.
The melting of N-6 chains protruding from their (n-s)-CD-ICs is apparently accompanied by ΔHms that are similarly reduced, because they show Tms close to neat samples. The greater extension and orientation of chains expected in their dense brush-like arrangement in the melt, which likely increases the interactions between molten un-included N-6 chains, in comparison to their neat melts, would serve to lower the difference in enthalpies (ΔHm) between crystalline and molten N-6 chains protruding from the surfaces of their (n-s)-CD-IC crystals.
Though crystallization and melting data for high density brushes composed of semi-crystalline polymers is sparse [
28,
29,
30,
31], it appears that their
Tcs and
Tms are much closer and similar, respectively, to those of their bulk samples than have been observed for the un-included constrained chain portions of the polymer chains in their (n-s)-CD-ICs. This may at least be partly a consequence of the fact that at least some of the un-included polymer chains in (n-s)-CD-IC are constrained at both ends, because they thread into and out of the same or two different CD-IC crystals, causing, in the latter case, “Shape-Memory” behavior.
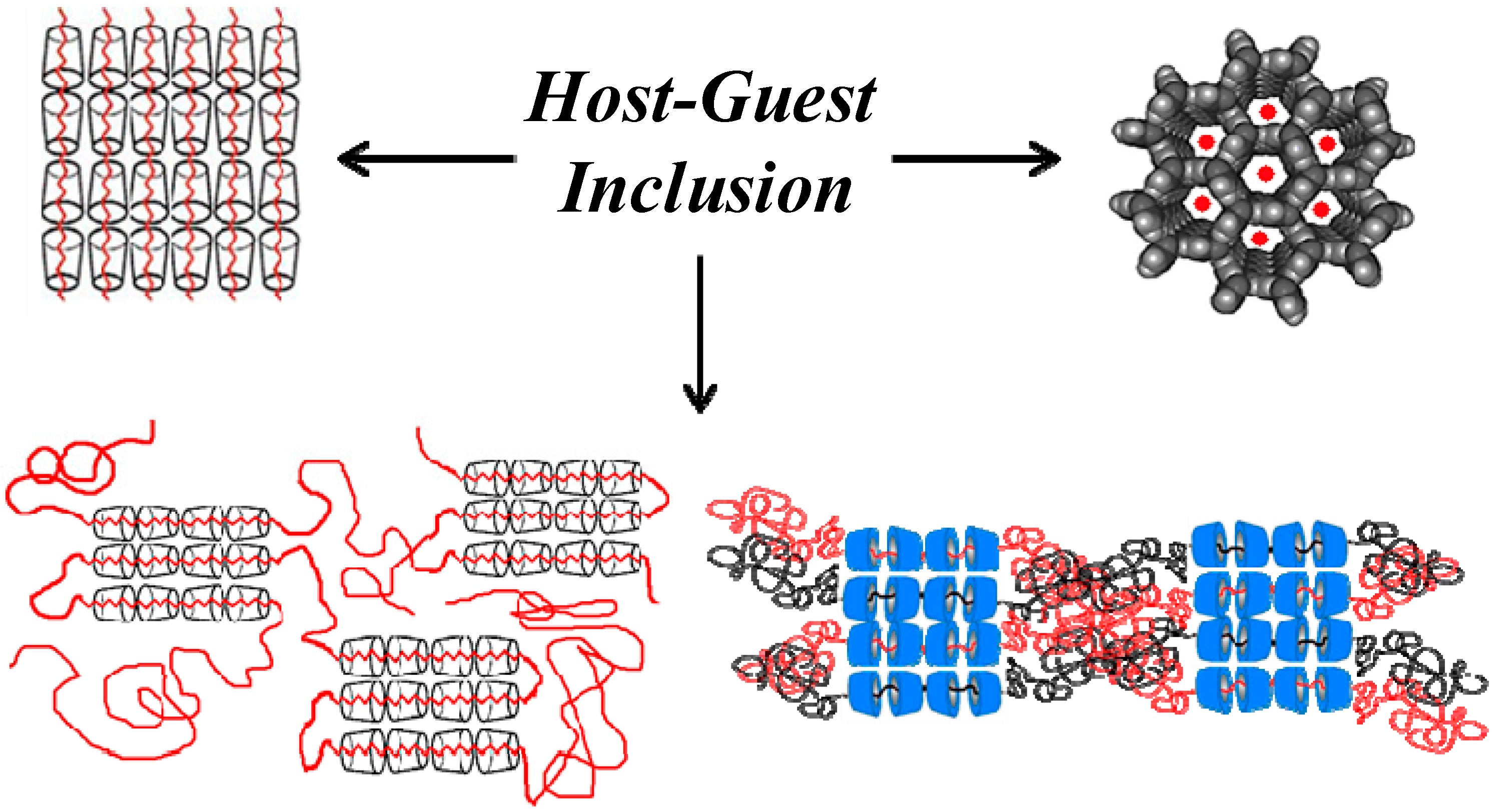
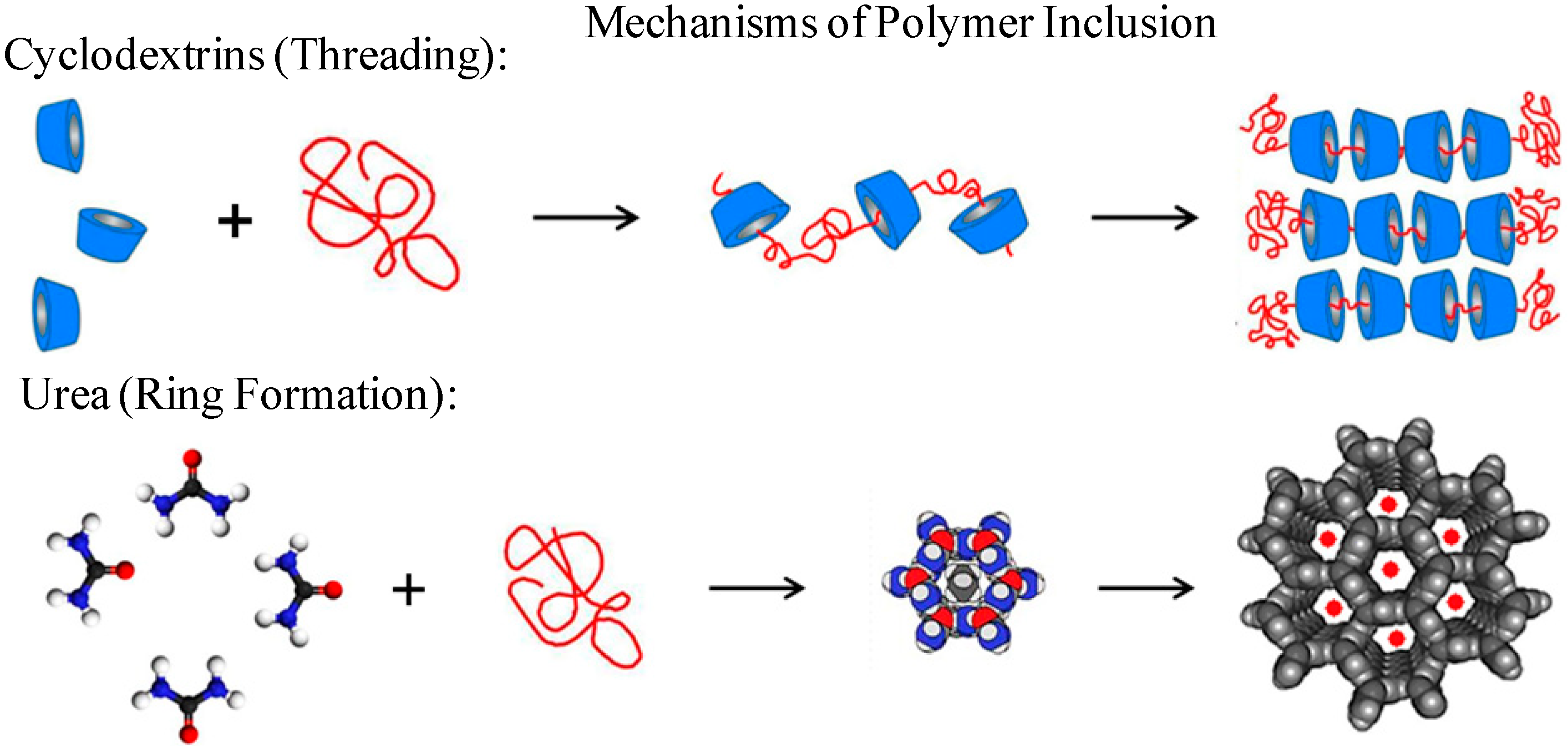
Similar to (n-s)-polymer-CD-ICs, it has been observed [
4,
9,
12] that guest polymers coalesced from their ICs (see
Figure 5 and
Figure 6) upon careful removal of the host crystalline lattice also crystallize more rapidly and at higher temperatures from their melts than neat as-received bulk samples of the same polymer. Consequently, they, along with (n-s)-polymer-CD-ICs, may be used as effective self-nucleants [
9,
10,
11,
12,
13]. Melt nucleation of as-received bulk polymers with small amounts of the same coalesced polymer has an important advantage over (n-s)-polymer-CD-ICs. Self-nucleation with coalesced polymers does not introduce any CDs, and therefore serves as a “stealth” means of nucleation and provides polymer samples that may be used for medical implantation and that are easily recycled, as well.
Figure 5.
Mechanisms of polymer-IC formation with CD and U hosts [
32].
Figure 5.
Mechanisms of polymer-IC formation with CD and U hosts [
32].
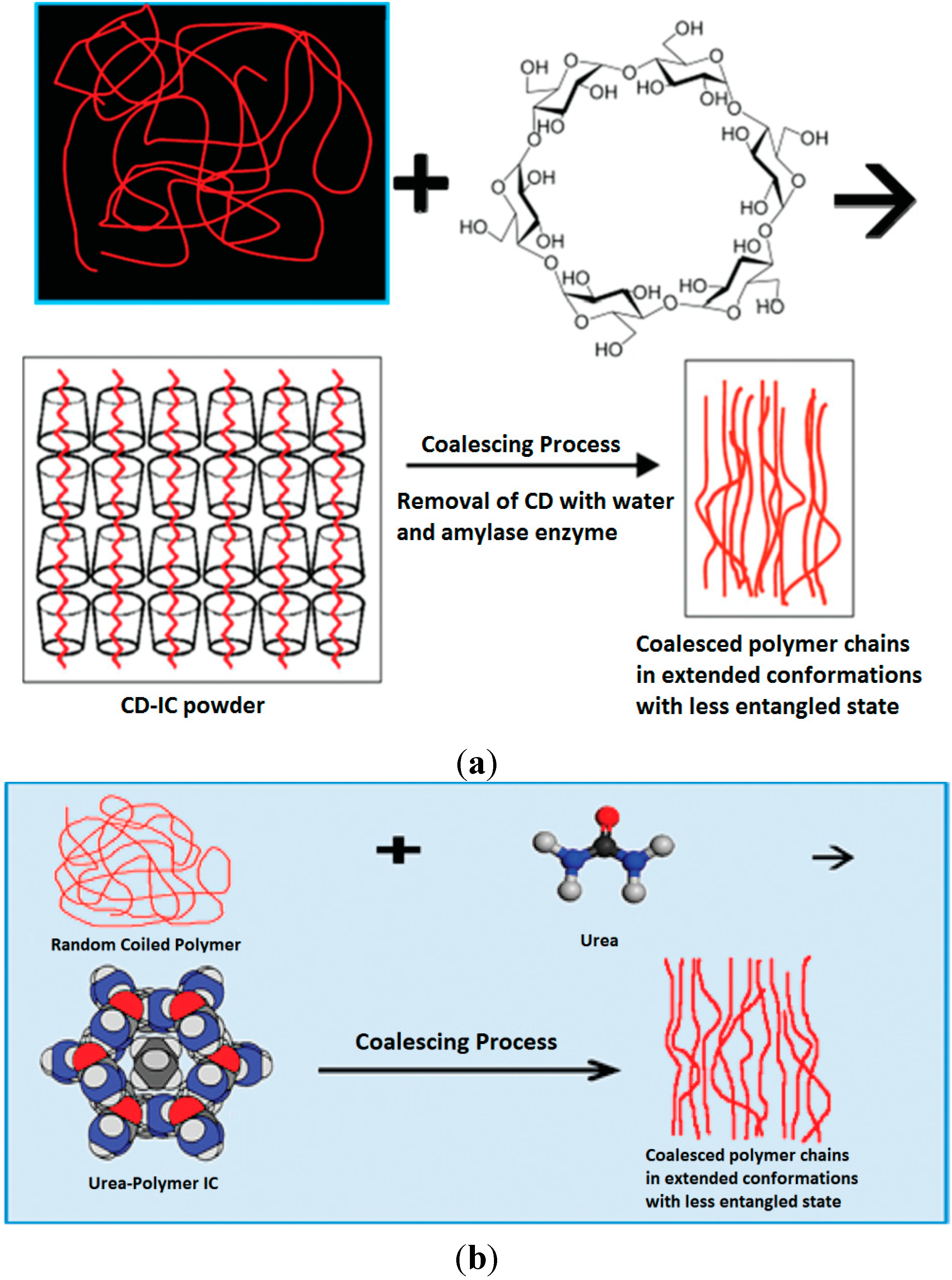
Figure 6.
Formation and coalescence from CD (
a) and U (
b) polymer ICs [
32].
Figure 6.
Formation and coalescence from CD (
a) and U (
b) polymer ICs [
32].
3.2. Amorphous Polymers Coalesced from Their Stoichiometric and (n-s)-CD-ICs
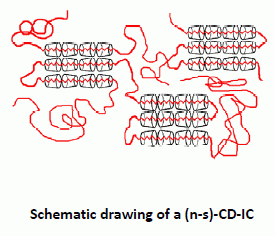
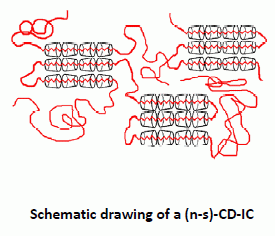
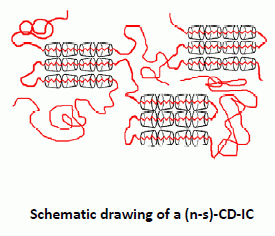
Behaviors of amorphous polymers threaded through and coalesced from their (n-s)-CD-ICs and stoichiometric CD- and U-ICs (see
Figure 7) have also been blended/mixed and examined [
32,
33,
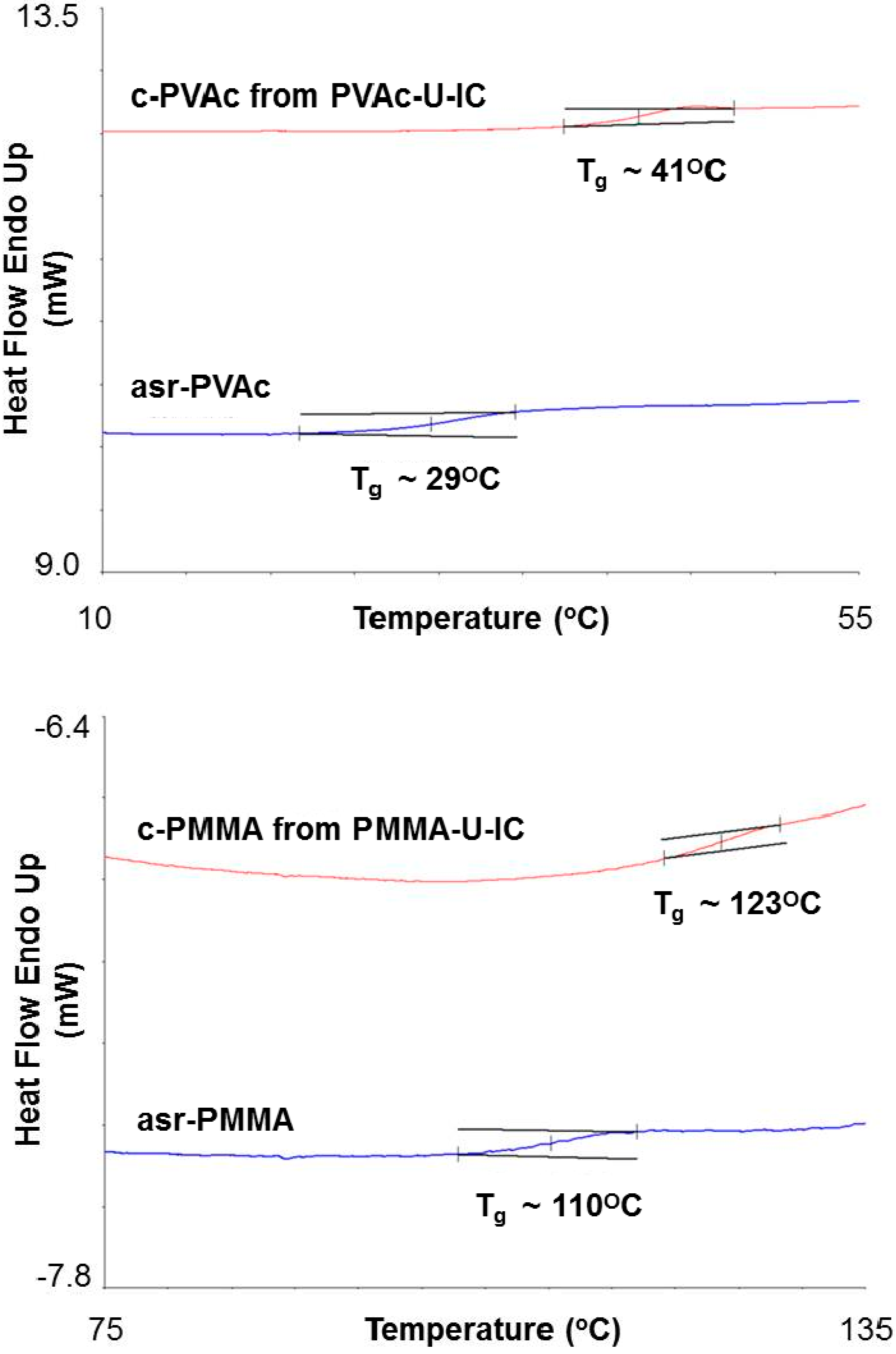
34]. In general, the glass transition temperatures,
Tgs, of amorphous guest polymers coalesced from their stoichiometric CD- and U-ICs and (n-s)-CD-ICs and those un-included chain portions in the (n-s)-CD-ICs exhibit significantly elevated
Tgs. This is demonstrated [
34] for atactic PVAc and PMMA in
Figure 8. In addition, as observed and noted above for semi-crystalline guest polymers coalesced from their CD- and U-ICs, the
Tgs of amorphous guest polymers coalesced from their CD- and U-ICs also remain elevated above those of their as-received samples even upon being annealed considerably above
Tg for long periods of time. The
Tgs of coalesced (c)-poly(vinyl acetates) (PVAcs) annealed at 70 °C for 0, 8, 14, and 28 days were 41.5, 41.7, 41.5, and 41.2° C, respectively [
34].
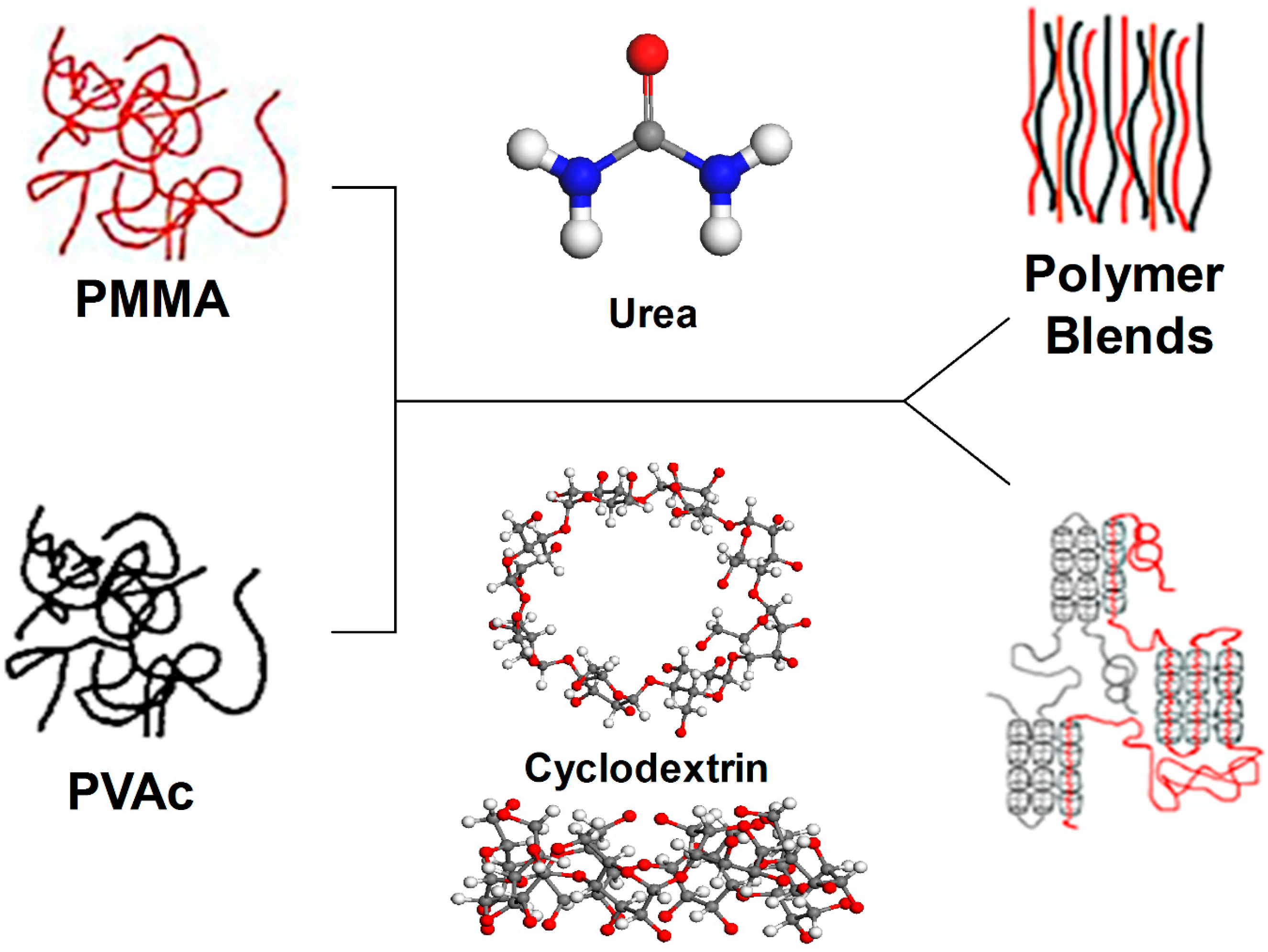
Figure 7.
Comparison of blending poly(methyl methacrylate) (PMMA) and poly(vinyl acetate) (PVAc) via coalescence from stoichiometric U-IC and formation of (n-s)-γ-CD-ICs [
34].
Figure 7.
Comparison of blending poly(methyl methacrylate) (PMMA) and poly(vinyl acetate) (PVAc) via coalescence from stoichiometric U-IC and formation of (n-s)-γ-CD-ICs [
34].
Mention should be made that the
Tgs observed for asr- and c-PMMA are 83 and 97 °C and 110 and 124 °C, respectively, for samples with molecular weights of 15 and 350 kDa that were coalesced from their γ-CD- and U-ICs. Since these molecular weights are considerably below and above the entanglement molecular weight of PMMA, respectively [
35], it is not surprising that the
Tgs observed for their asr-samples differ by 27 °C. It does seem somewhat surprising, however, that their coalesced samples also have
Tgs that differ by 27 °C. If the agreement of the
Tg differences between asr- and c-PMMA samples with very divergent molecular weights is not fortuitous, then it may possibly indicate that the molecular weight dependence of polymer glass-transition temperatures is not sensitive to whether or not they are entangled, but rather to the concentration of their chain-ends [
36]. The basis for this suggestion arises because c-polymer samples are believed to be comprised of largely extended and un-entangled chains, while the chains in asr-samples are randomly-coiling and entangled (see
Figure 6). This is supported by the higher densities measured for c-PVAcs presented in
Table 1. Judging from the virtually identical densities measured for PVAcs coalesced from their γ-CD- and U-ICs both below and above their
Tgs, it appears that the resulting organization and behavior of c-PVAcs do not depend on the host crystalline lattice from which they were coalesced.
Figure 8.
Differential scanning calorimetry (DSC) heating scans of c-PVAc and coalesced (c)-PMMA (from their U-ICs) and as-received (asr)-PMMA and asr-PVAc [
34].
Figure 8.
Differential scanning calorimetry (DSC) heating scans of c-PVAc and coalesced (c)-PMMA (from their U-ICs) and as-received (asr)-PMMA and asr-PVAc [
34].
Table 1.
Densities of asr- and c-PVAcs measured below and above their
Tgs [
34].
Table 1.
Densities of asr- and c-PVAcs measured below and above their Tgs [34].
| Sample | Density at 25 °C (g/cm3) (below Tg) | Density at 58 °C (g/cm3) (above Tg) |
|---|
| asr-PVAc | 1.093 | 1.040 |
| c-PVAc (γ-CD) | 1.156 | 1.077 |
| c-PVAc (U) | 1.154 | 1.076 |
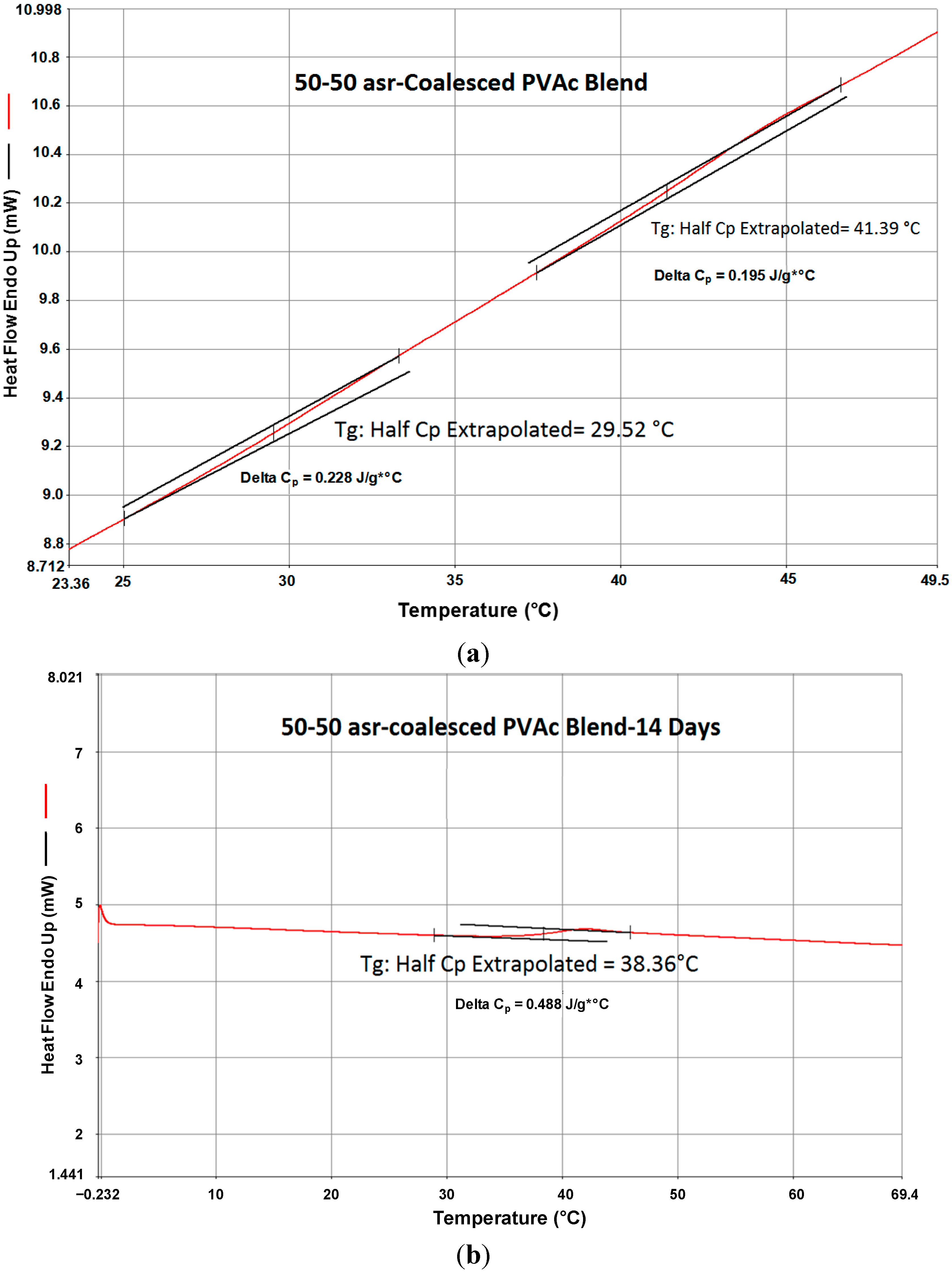
In a preliminary study, we made a 50:50 (wt:wt) mixture of asr- and c-PVAcs and recorded its DSC heating scan, which is shown in
Figure 9. There the
Tgs of both asr- and c-PVAcs can be seen. The sample pan was then removed from the DSC instrument and annealed in an oven at 70 °C for two weeks. The DSC heating scan for the annealed 50:50 asr/c-PVAc mixture was then measured and is also presented in
Figure 9. A single
Tg is observed and it is close to that of c-PVAc. It appears that long-term annealing above
Tg has resulted in the reorganization of the asr-PVAc portion of the mixture into that similar to the c-PVAc portion. The observation that the single
Tg observed for the apparently well-mixed blend is close to that of c-PVAc, rather than asr-PVAc, is surprising. In other words, the blend is likely not characterized by completely entangled randomly-coiling PVAc chains, but rather by an organization of PVAc chains more similar to that of the initially coalesced neat c-PVAc (γ-CD). These preliminary DSC observations are supported by the densities that were measured for the asr- and c-PVAcs and their 50:50 blend after annealing at 70 °C for two weeks, which were 1.044, 1.077, and 1. 076 g/cm
3, respectively.
Figure 9.
DSC scans of an initial 50/50 physical mixture of asr-PVAc/c-PVAc (
a) before and (
b) after annealing for 14 days at 70 °C [
34].
Figure 9.
DSC scans of an initial 50/50 physical mixture of asr-PVAc/c-PVAc (
a) before and (
b) after annealing for 14 days at 70 °C [
34].
Currently we are annealing further the 50:50 mixture and have also begun annealing an 80:20 asr-PVAc:c-PVAc mixture at 70 °C to see if it also eventually is transformed/reorganized to that of a completely coalesced PVAc sample.
3.3. (N-S)-ICs with Amorphous Guest Polymers
(N-S)-CD-ICs were formed with amorphous PVAc and PMMA and examined by DSC [
34].
Table 2 presents the
Tgs measured for (n-s)-PVAc-γ-CD-ICs with different stoichiometries, which lead to distinct lengths of uncovered un-included PVAc chain portions, as well as those of asr- and c-PVAcs. There it is seen that the constrained un-included PVAc chain portions in (n-s)-γ-CD-ICs have higher
Tgs than neat asr- and c-PVAc samples. Furthermore, as the lengths of the un-included chain portions are shortened it appears that their
Tgs increase as a consequence of the increasing constraint placed on them by their included chain portions. Similar observations were made on (n-s)-, asr-, and c-PMMAs [
34].
Table 2.
Tgs of (n-s)-PVAc-γ-CD-ICs [
34].
Table 2.
Tgs of (n-s)-PVAc-γ-CD-ICs [34].
| Sample | PVAc (g) | γ-CD (g) | Tg (°C) |
|---|
| asr-PVAc | – | – | 28 |
| c-PVAc | – | – | 41 |
| 2:1 (n-s)-IC | 0.369 | 0.928 | 49 |
| 3:1 (n-s)-IC | 0.369 | 0.615 | 49 |
| 6:1 (n-s)-IC | 1.107 | 0.928 | 44 |
Very recently Mano
et al. [
37,
38] reported (n-s)-α-CD-ICs made with amorphous atactic guest poly
l,
d-(lactic acid) (PLDLA). For a ~7:1 (n-s)-PLDLA-α-CD-IC they reported a
Tg for the un-included portions of the guest PLDLA chains that was only ~5 °C above that of their neat PLDLA sample. On the other hand, we observed [
34] and have mentioned above, that the
Tgs of the un-included chain portions of atactic PVAc and PMMA in their (n-s)-γ-CD-ICs to be at least as high as those of their coalesced bulk samples, which in turn are more than 10 °C above their asr-samples. Because Mano
et al. [
37,
38] used 2-, 3-, and 4-fold excesses of PLDLA over the stoichiometric amount for full coverage by α-CD, they likely ended up with both excess free PLDLA and stoichiometric PLDLA-α-CD-IC in their sample, which would explain the nearly identical
Tgs they reported for their IC and neat PLDLA samples.
There are two ways of producing partially compatible PVAc/PMMA blends. The first involves formation of stoichiometric common γ-CD- or U-ICs [
33,
39] and the second the formation of common (n-s)-γ-CD-ICs [
34]. Coalescence from common stoichiometric ICs yields neat blends, while in common (n-s)-γ-CD-ICs the blending occurs between the un-included portions of PVAc and PMMA chains. Previously PMMA/PVAc intimately mixed blends were obtained by formation and coalescence of their common γ-CD-ICs [
37,
38]. This was done using lower molecular weight PVAc (~13 kDa) and PMMA (~15 kDa). These coalesced blends showed some miscibility above the
Tg of PVAc, where PVAc chains are able to diffuse in to PMMA domains.
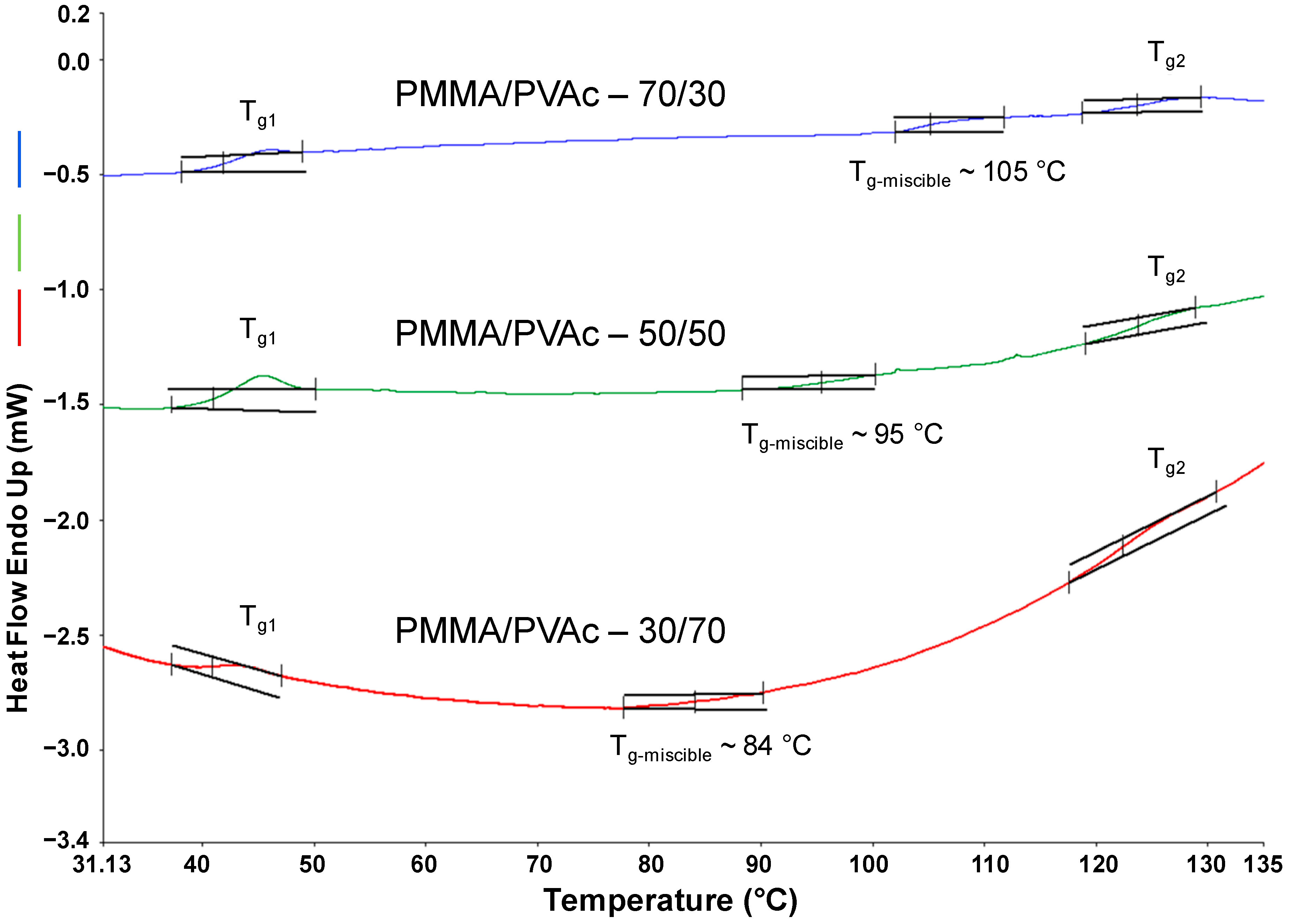
In
Figure 10 DSC data for PMMA/PVAc blends coalesced from their common U-ICs with varying initial PMMA:PVAc (70/30, 50/50, and 30/70) mol% ratios are presented, where all heating scans exhibit three
Tgs. The lower and higher
Tgs (
Tg1 and
Tg2) correspond to those of neat PVAc and PMMA, respectively, coalesced from their U-ICs. This indicates that the polymer chains in the coalesced blends are largely aligned similar to the coalesced homopolymers processed with U. In between these two T
gs, there is an intermediate
Tg-miscible, which corresponds to the miscible phase.
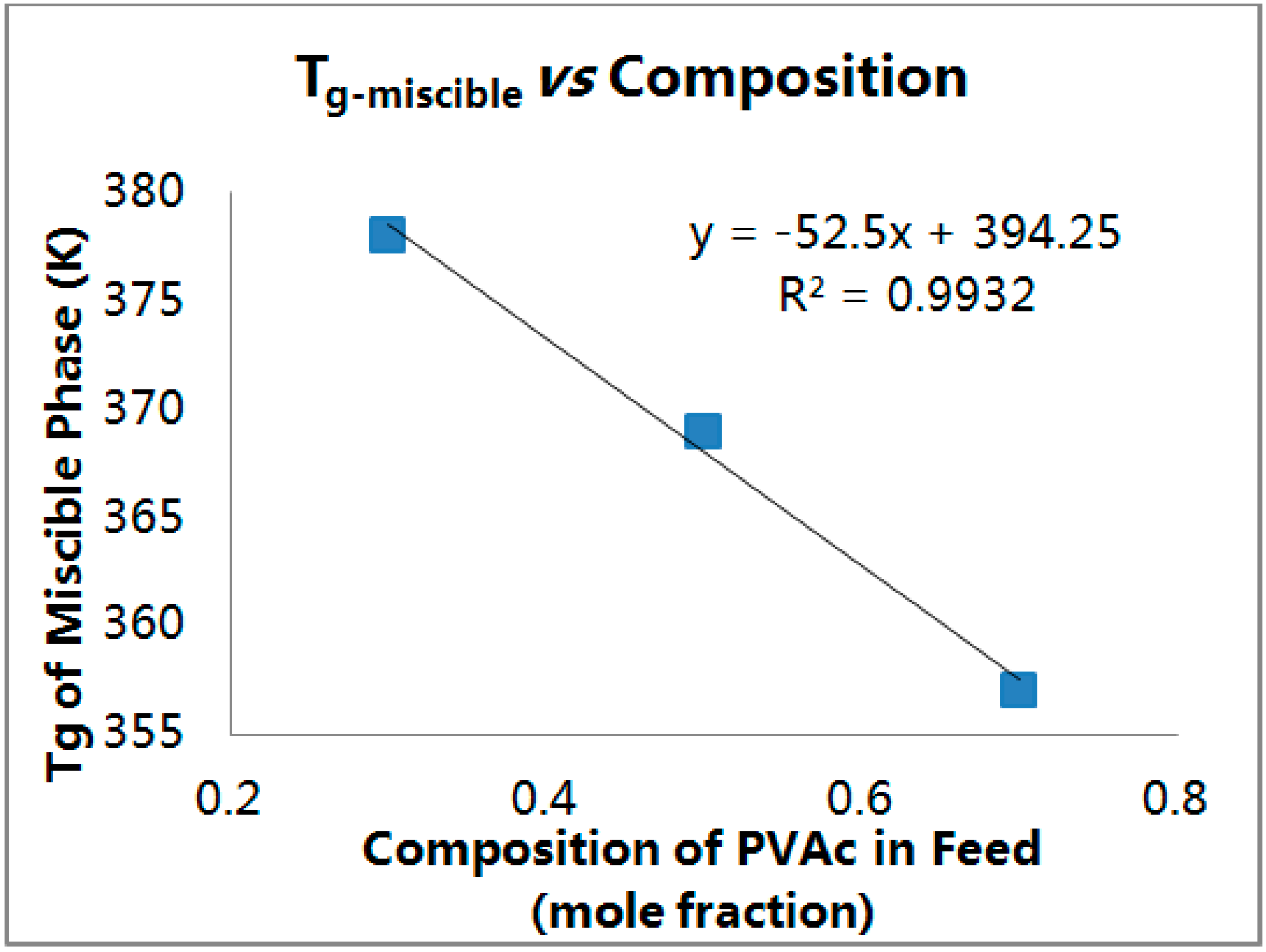
From the DSC scans it can be concluded that
Tg-miscible moves toward the
Tg of PVAc with increasing initial PVAc content. The compositions of the phases corresponding to
Tg-miscible was estimated using the Fox Equation [
40,
41,
42] (below) and are presented in
Table 3 and
Figure 11. This analysis confirms preferential inclusion of PMMA over PVAc [
34,
37,
38] and indicates that in order to get an approximate equimolar mixture in a coalesced PMMA/PVAc blend an intial PVAc/PMMA composition of 70/30, or close to 2:1, in the feed is required:
Figure 10.
DSC heating scans of PMMA/PVAc blends coalesced from their stoichiometric U-ICs [
34].
Figure 10.
DSC heating scans of PMMA/PVAc blends coalesced from their stoichiometric U-ICs [
34].
Table 3.
Tg-miscibles of PMMA/PVAc blends coalesced from their stoichiometric U-ICs [
34].
Table 3.
Tg-miscibles of PMMA/PVAc blends coalesced from their stoichiometric U-ICs [34].
| Feed | Blend |
|---|
| PVAc/PMMA (mol%) | Tg-miscible (K) | PVAc (mol%) | PMMA (mol%) |
|---|
| 30/70 | 378 | 21 | 79 |
| 50/50 | 369 | 31 | 69 |
| 70/30 | 357 | 46 | 54 |
Figure 11.
Tg-miscibles observed (■) for PMMA/PVAc blends coalesced from their stoichiometric U-ICs [
34], using the Fox Equation [
40,
41,
42] to obtain the straight line.
Figure 11.
Tg-miscibles observed (■) for PMMA/PVAc blends coalesced from their stoichiometric U-ICs [
34], using the Fox Equation [
40,
41,
42] to obtain the straight line.
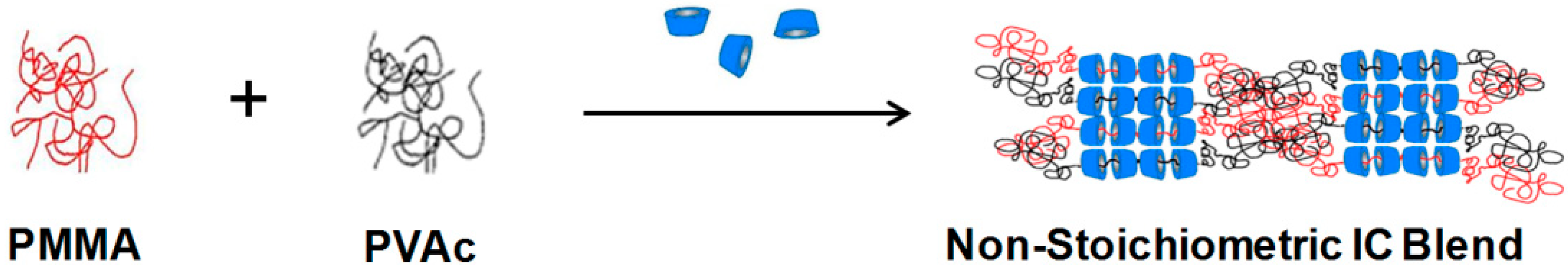
PMMA/PVAc can also be partially blended using γ-CD as a compatibilizer through formation of their common non-stoichiometric (n-s)-c-CD-ICs [
32], as shown in
Figure 12. We prepared (n-s)-γ-CD-ICs with 2:1, 3:1, and 6:1 ratios of the polymer pair to γ-CD and 1:1 and 1:2 PMMA:PVA stoichimetries, which were confirmed with
1H-NMR to be in agreement with the starting proportions of PVAc, PMMA, and γ-CD.
Figure 12.
Blending of un-included portions of PMMA and PVAc chains in their common (n-s)-γ-CD-ICs [
34].
Figure 12.
Blending of un-included portions of PMMA and PVAc chains in their common (n-s)-γ-CD-ICs [
34].
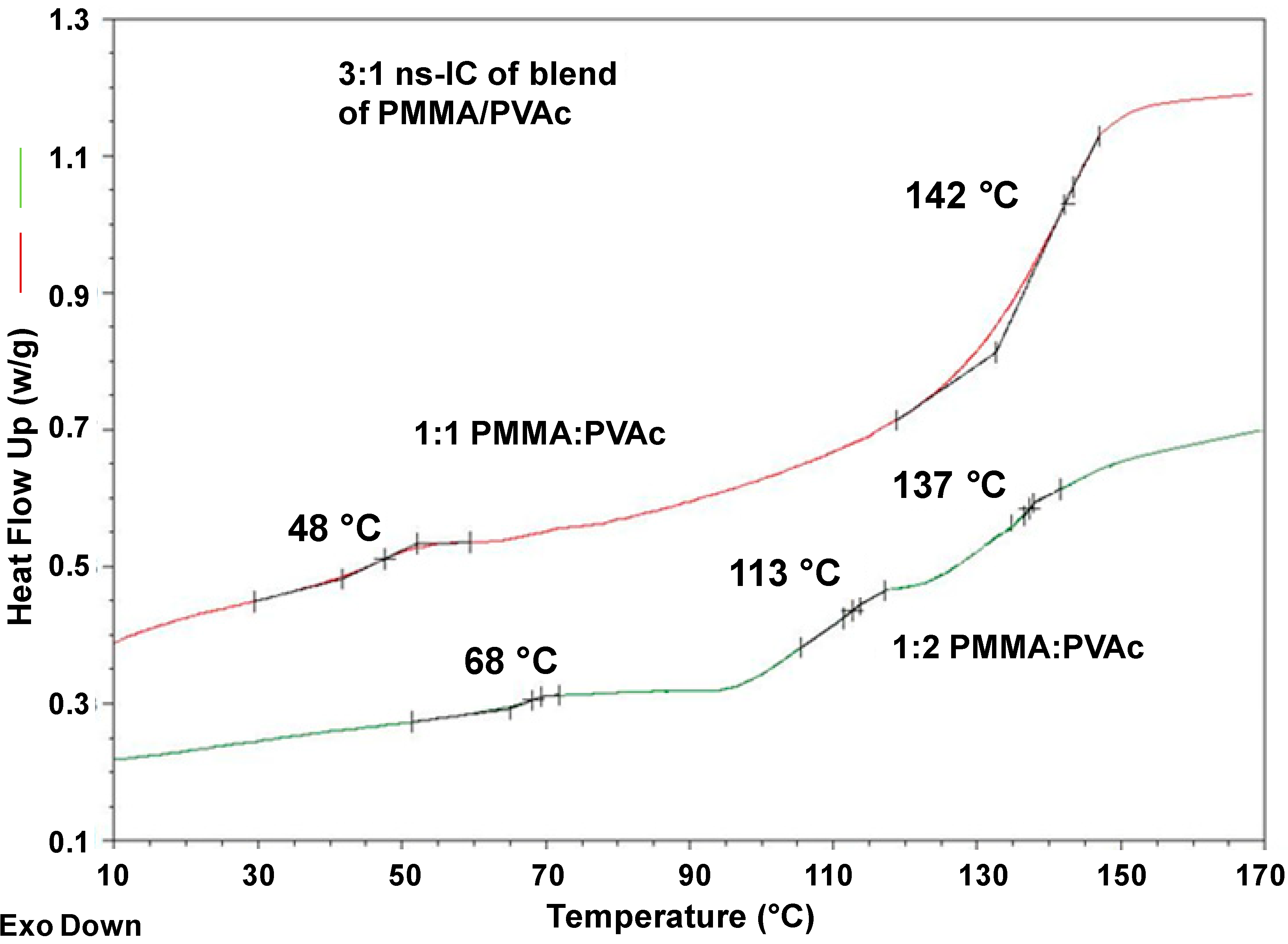
DSC results for the common 3:1 (n-s)-PVAc/PMMA-Υ-CD-IC blends are shown in
Figure 13 and summarized in the
Table 4. For the 1:1 PMMA/PVAc initial mixture, we observe the glass transition temperatures for PVAc and PMMA at 48 and 143 °C, respectively, similar to the
Tgs obtained for the neat 3:1 (n-s)-ICs of PVAc and PMMA. This indicates that the common 3:1 (n-s)-PMMA/PVAc(1:1)-γ-CD-IC contains distinct γ-CD-IC crystals including exclusively each of the neat guest polymers. We can also see in
Table 4, that unlike the
Tgs of the neat (n-s)-γ-CD-ICs, which simply shifted to higher temperatures, in the case of the common 3:1 (n-s)-PVAc/PMMA-γ-CD-IC, with 2:1 PVAc:PMMA stoichiometry, they move closer to each other, indicating a certain degree of mixing between the “dangling” un-included PVAc and PMMA chain portions. Although the mixing is far from complete, as indicated by a
Tg close to that of the neat 3:1 (n-s)-PMMA-γ-CD-IC, the difference in the other two
Tgs observed in relation to both the as-received and coalesced polymers is an indication of the presence of PMMA chains in PVAc-rich domains and
vice versa.
Figure 13.
Heating DSC scans of common 3:1 (n-s)-PVAc/PMMA-γ-CD-ICs [
34].
Figure 13.
Heating DSC scans of common 3:1 (n-s)-PVAc/PMMA-γ-CD-ICs [
34].
Table 4.
Tgs observed in common (n-s) PMMA/PVAc-γ-CD-ICs.
Table 4.
Tgs observed in common (n-s) PMMA/PVAc-γ-CD-ICs.
| (n-s)-IC Sample (Polymer: γ-CD) | PMMA/PVAc Molar Ratio | Tg1 (°C) | Tg2 (°C) | Tg3 (°C) |
|---|
| 3:1 | 1:1 | 48 | – | 142 |
| 3:1 | 1:2 | 68 | 113 | 137 |
Thus, we get hardening (higher Tg) of PVAc caused by the presence and partial mixing of PMMA, and softening (lower Tg) of the PMMA from the presence and partial mixing of PVAc, resulting in a decrease of its glass transition temperature, compared to both asr- and c-PMMAs. More interesting is the observation of an intermediate Tg at 113 °C, which is consistent with a well-mixed phase containing similar amounts of un-included portions of PVAc and PMMA chains.
At this point it may be useful to compare the softening temperatures (
Tgs) of the “dangling” un-included portions of amorphous guest polymer chains in (n-s)-CD-ICs with those in high density brushes tethered to solid surfaces. Before making the comparison we must emphasize that at least some of the un-included portions of guest polymer chains in (n-s)-CD-ICs are constrained at both ends as they emerge from one and enter another or a nearby CD-IC crystal channel (see
Figure 2 and
Figure 12). This suggests that host CDs thread over both ends of the guest chains during IC formation. In polymer brushes, on the other hand, polymer chains are covalently bonded to solid surfaces at only one chain end. However, as mentioned previously, the surface densities of chains in polymer brushes and the un-included chains emanating from the CD-IC crystal surfaces in (n-s)-CD-ICs can be similar [
7,
18,
20].
Above, we observed elevations as large as ~15–20 °C in the
Tgs of un-included PVAc and PMMA chain portions in their (n-s)-γ-CD-ICs above the
Tgs of their neat bulk samples. Dense PMMA [
20] and polystyrene (PS) [
43] brushes covalently attached to silicon wafers both show elevated glass-transition temperatures (
Tgs ~ 10–25 °C) higher than the values observed for their neat bulk samples), but only for thin brushes, with vertical heights in the ranges <50–100 nm. The elevations in glass-transition temperatures over those of their amorphous neat bulk samples observed for polymer chain portions un-included and constrained in (n-s)-CD-ICs and in thin dense brushes tethered to silicon wafers appear to be similar.
Based on their
MWs (167,000 and 350,000, respectively) and assuming full chain extension, the un-included chain portions in 2:1 (n-s)-PVAc- and PMMA-CD-ICs are on average ~250 and 450 nm. This is much longer than the <50–100 nm thickness of thin tethered PMMA and PS brushes that show elevated
Tgs [
20,
43]. Tethered PMMA and PS brushes with thicknesses of 250–400 nm comparable to the lengths of un-included PVAc and PMMA chains in their (n-s)-γ-CD-ICs do not show elevated
Tgs. This suggests that the constraints placed on both ends of un-included PVAc and PMMA chains emerging from and entering into different CD-IC crystals are more severe than the packing constraints placed on high density brushes more than 50–100 nm above the surface of their attachment.
The importance of the constraints placed on both ends of at least some un-included chains portions in (n-s)-CD-ICs receives additional support from their necessary contribution to the “shape-memory” of these materials [
4,
5,
6,
7,
9]. This strongly suggests that somewhere in the middle portions of the doubly tethered un-included chains there exit regions where they are not highly extended, but “randomly-coiled”, as in singly tethered high density polymer brushes far above their surface of attachment, and are able to be readily extended. Nevertheless, the likely presence of these less extended, less densely packed regions do not cause a reduction in their
Tgs to those of their bulk samples, as is the case in high density, singly tethered, thick polymer brushes. With respect to the glass-transition temperatures of amorphous polymers in their (n-s)-CD-ICs, apparently the constraint of double-attachment/tethering of their un-included portions outweighs the effect of extensional flexibility in their “randomly-coiling” regions.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




