4.1. Compatibilization Mechanism
Most physical blends of PLA with toughened secondary polymers (including PBSA) are thermodynamically immiscble [
29]. It is common practice to add compatibilizers in order to improve the compatibility of these immiscible blends. An addition of compatibilizer results in a reduction of interfacial tension due to the formation of either a block or graft copolymer at interfaces within the blend, depending on the kind of compatibilizer used [
13,
33]. For example, an addition of compatibilizers possessing reactive end groups will result in the formation of block copolymers (with a substantial increase in
Mn) [
34], while an addition of compatibilizers with reactive pendant groups (such as TPP) will generally result in the formation of graft/branched copolymers [
35].
Different researchers have undertaken studies on the effect of compatibilizers with reactive pendant groups (such as TPP) on polyester-based systems and have proposed two reaction mechanisms—one by Jacues et al. [
35] and the other by Aharoni et al. [
36]. These reaction mechanisms have a strong impact on the compatibilization of polymer blends and their properties. Hence, any understanding of how the addition of TPP influences the compatibilization of PLA and PBSA in this study needs to be taken into account. In both of the above-mentioned reaction mechanisms, the first step is the preferential reaction of hydroxyl end-groups of PLA/PBSA with TPP via the displacement of one of TPP’s phenoxy groups, as shown in
Figure 4a. This leads to the formation of an intermediate phosphorus-containing compound (intermediate alkyl diphenyl phosphite). The second step can be either of the two reaction mechanisms depicted in
Figure 4b,c.
In the first reaction mechanism, the second step involves a multi-substitution reaction of intermediate alkyl diphenyl phosphite whereby phenoxy groups are replaced with alkyl groups along with the elimination of phenol, as shown in
Figure 4b. It is highly likely that this reaction continues until phosphorus serves as a binding point for the occurrence of grafting/branching [
33]. In contrast, the second mechanism involves ester linkages from polymers, with phenoxy groups of intermediate product reacting with carboxyl groups of PLA/PBSA (instead of hydroxyl end-groups), leading to a chain extension without P atoms becoming part of the polymeric chain (
Figure 1c).
In all of the above-mentioned reaction schemes, chain extension and/or branching may occur. In our case, compatibilized blends show a marginal increase in molecular weight (
Table 3) compared to PLA, indicating that branching is a major reaction pathway. This has been observed in a previous study conducted by Jacues et al. [
35], where 2 wt % TPP was used to melt-blend PET/PBT in a ratio of 70:30 [
35]. The authors observed a small increase in
Mn, accompanied by branching of both polyesters, as proven by an increase in torque oscillations. Harada et al. [
37] observed a similar trend for compatibilized PLA–PBSA blends involving the use of lyscine triisocyanate as a coupling agent, with cross-linking behavior being reported and accompanied by a small increase in the
Mn of the PLA blends. Further studies involving multi-detector gel permeation chromatography GPC using viscometry and light scattering might be required to ascertain the exact nature of branching.
4.2. Crystallization & Melting Behavior
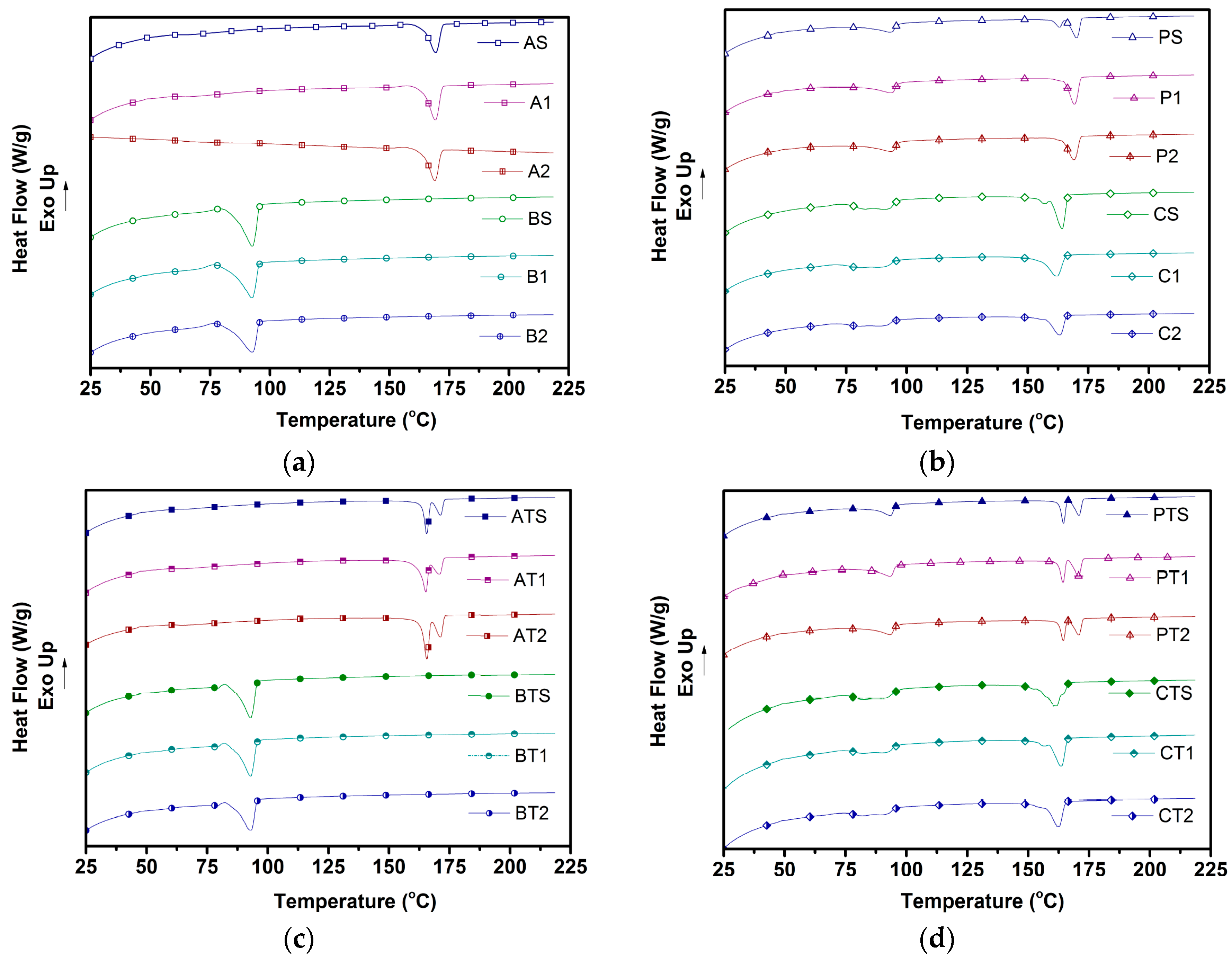
Typically, semi-crystalline polymers such as PLA and PBSA can exhibit three kinds of crystallization behaviors—melt crystallization, cold crystallization, and recrystallization—depending on the heating/cooling rate adopted. Melt crystallization refers to the formation of crystals during cooling. Cold crystallization is the ability of amorphous domains to crystallize during heating, while re-crystallization refers to the reorientation of crystals formed during melt/cold crystallization [
6,
38]. In our study, the observation during the first heating pertains to the behavior of injection-molded samples, which were typically subjected to high cooling rates (~200 °C/min), leading to insufficient time available for crystallization. The second heating cycle erases the prior thermal history of the samples while subjecting them to a low cooling rate in the first cooling cycle (5 °C/min), and is indicative of the behavior of the nascent material [
14]. Hence, the differences observed in the behavior of all samples between both heating cycles in this study, such as (1) the occurrence of cold crystallization only in the first heating cycle; (2) the presence of a single melting peak in the first cycle vs. double melting peaks in the second heating cycle (both corresponding to PLA) in few samples; and (3) an enhanced crystallinity of samples after the second heating cycle; all of which can be attributed to the stark difference in cooling rate.
The presence of
Tcc (corresponding to PLA) reported in
Table 4 and
Table 5 during the first heating cycle and its absence in the second heating cycle was because all amorphous molecular domains had crystallized in the first cooling cycle upon use of a slow cooling rate (5 °C/min). This is in good agreement with the observed increase in the crystallinity of blends from the first heating cycle to the second one, as it indicates that amorphous domains crystallized during the first cooling cycle. Pilla et al. [
14] observed similar behavior in the case of PLA/MWCNT (multi-wall carbon nanotubes) composites.
The absence of cold crystallization (corresponding to PBSA) in PBSA samples and blends could be due to several factors. First, PBSA molecules tend to undergo a faster rate of crystallization during cooling, leading to an absence of amorphous domains that could crystallize during reheating [
39]. Second, in the case of blend samples, the presence of stiff PLA chains hinders the cold crystallization of PBSA [
29], further making its occurrence impossible in blends. With regard to blends, the physical PLA/PBSA blends showed a reduction in
Tcc compared to the pure PLA samples, which could be attributed to the possible intermingling of chains of both polymers at the interfaces, resulting in the early onset of crystallization [
29]. A further decrease in
Tcc was observed for chemically compatibilized blends to ~71 °C, which could be attributed to the enhanced compatibility between PLA and PBSA chains [
27].
The reduction in melting temperatures in the compatibilized blend of around ~7 °C in both heating cycles was due to a stronger interaction between PLA and PBSA chain segments upon the addition of triphenyl phosphite (TPP), as TPP enhances the mobility of PLA chain segments [
27]. This finding is in good agreement with Ojijo et al. [
27], who observed a similar lowering in the
Tm (to ~152 °C) of compatibilized PLA/PBSA blends prepared via use of similar coupling agents.
Furthermore, solid blends (P-S and C-S) exhibited double melting peaks that were due to the melting of PLA crystals with different morphologies [
15]. Ojijo et al. [
29] had observed that PBSA in molten form has a nucleating effect on the crystallization of PLA, forming crystals of different sizes and morphologies. Hence, the observed double melting peaks was mainly due to the nucleating effect of PBSA. This is due to the inability of simultaneous crystallization of both polymers occurring due to the large difference in their melting temperatures. However, their foamed counterparts (P-1, P-2, C-1, and C-2) showed only one melting peak corresponding to a melting of PLA. This indicates that foaming had a strong impact on the reorientation of crystal structures, leading to the formation of highly ordered crystals, even as TPP induced strong compatibilization between PLA and PBSA.
The addition of talc also resulted in the obtainment of double melting peaks in PLA (AT-S, AT-1, and AT-2) and physical blends (PT-S, PT-1, and PT-2), which could be due to the heterogeneous nucleation effect of talc particles resulting in the obtainment of varying crystal sizes, which is in agreement with other literature [
40,
41,
42]. Interestingly, compatibilized blends showed only one melting peak upon the addition of talc—in stark contrast with the above-mentioned observation. This can be explained by the reinforcing effect of talc, which enhances bulk crystallinity without impacting crystal size, as observed by Tanniru and Misra et al. [
43] for CaCO
3-reinforced PE composites.
The crystallinity of foamed compatibilized blends was higher compared to the physically blended counterparts, a phenomenon also observed by Yang et al. [
44] on PLA–PBSA compatibilized blends, who attributed this to branching sites acting as nucleation points, leading to a higher probability of nucleation compared to the physical blends. This is in good agreement with our molecular weight results, measured by GPC, indicating a possible occurrence of grafting/branching. For both physical and chemically compatibilized blends, foaming resulted in a higher degree of crystallinity. This could also be attributed to the biaxial extensional flow of ScF affecting the orientation of polymer molecules around cell walls due to foaming, leading to strain-induced crystallization, which results in an increase in the final crystallinity, as observed by Ameli et al. [
45]. A similar trend was also observed by Zhai et al. [
46] in using chemical foaming agents to foam polycaprolactone. The addition of talc led to an increase in crystallinity for most samples, which could be attributed to the nucleating effect of talc.
4.3. Viscoelastic Behavior
The storage modulus is a measure of energy storage and recovery exhibited during cyclic deformation, reflecting the elastic moduli of a material. In general, the storage modulus of any given material can be altered via addition of fillers. Generally, an addition of inorganic fillers is known to enhance the storage modulus of PLA [
41,
42,
47]. However, the opposite trend was observed in the pure PLA in this study (A-S and AT-S), which could be due to the inability of talc to exhibit a reinforcing effect. In general, the reinforcing effect of talc is more pronounced in a material exhibiting less stiffness, as explained by Tanniru and Misra [
43], who have observed a similar effect of fillers on polymeric materials with reduced stiffness. The pure PLA used in this study exhibited a storage modulus of 3050 MPa at 40 °C, which is far higher than the storage modulus of both pure PLA (2450 MPa) and PLA containing 10 wt % of silane-treated wood fiber (2556 MPa) reported by Pilla et al. [
39]. This excessively high storage modulus of pure PLA used in our study might be a contributing factor towards the lack of any reinforcing effect of talc in the talc-filled PLA samples. However, the opposite trend was observed for both PBSA-based and blended samples due to the elastomeric nature and resultant lower stiffness of PBSA, resulting in an improvement in the storage modulus upon the addition of talc. Among solid blends, compatibilized blends showed a lower storage modulus vis-à-vis physical blends, primarily due to the hindrance in chain movement on account of the possible branching that prevented chain realignment/packing, as observed by Khonakdar et al. in crosslinked HDPE (High-density polyethylene) [
48]. Similar phenomena was observed by Ibrahim et al. [
49] for cross-linked PLA/PCL (poly(ε-caprolactone)) blends compared to physical PLA/PCL blends, and was attributed to the creation of voids in the system upon the formation of the crosslinking network. The compatibilized foamed blends showed a higher storage modulus in this study compared to their physically foamed counterparts, which could be attributed to the higher crystallinity (observed in
Table 5 and
Table 7) due to the synergistic effect of TPP and ScF on crystallinity.
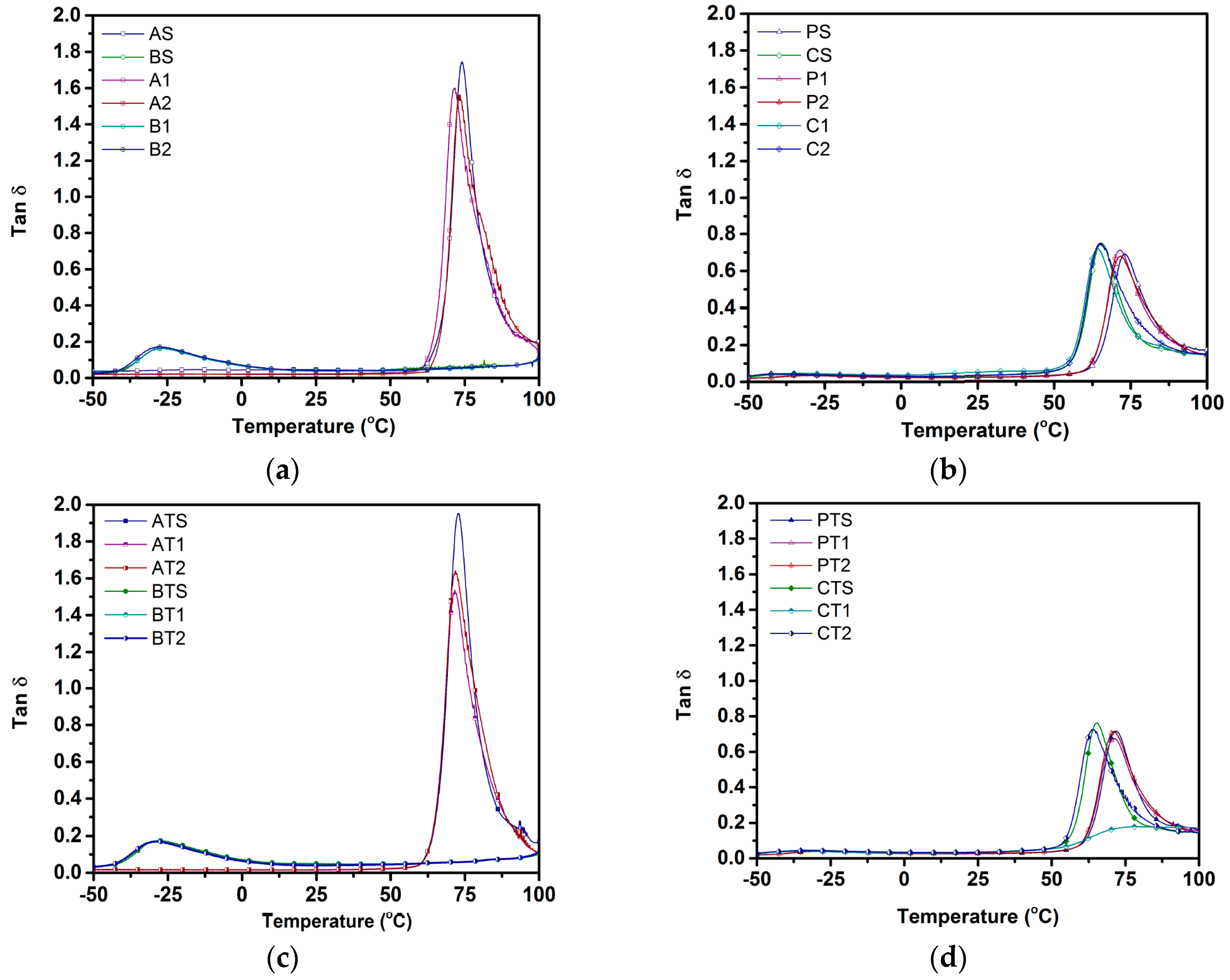
With regard to glass transition temperature, the absence of the plateau region in the storage modulus curve was observed for blend compositions, and can be attributed to the extremely low
Tg value of PBSA (~−40 °C). Similar observations have been made in another study by Ibrahim et al. [
49], where no plateau region was observed in the storage modulus curve of PLA/PCL blends on account of a low
Tg value of PCL (~−60 °C). Ojijo et al. [
29], in their study on PLA/PBSA blends, also observed similar trends, and attributed the absence of a plateau region to an increased mobility of PBSA chains above its
Tg (~−40 °C), leading to a lowering of blend stiffness.
Storage modulus was observed to undergo a sudden increase after a glass transition, corresponding to PLA, for all PLA-containing compositions. This increase was analogous to the cold crystallization from the first heating cycle of DSC, which is in accordance with Zhang et al. [
32] where cold crystallization was observed for both individual and blend compositions (PLA–PHBV (polyhydroxybutyrate-valerate)–PBS (poly(butylene succinate))) after glass transition. The appearance of T
cc can be explained by the fact that, for both individual and blend samples, the DMA (Dynamic Mechanical Analyzer) tests were undertaken on injection-molded samples that possessed low crystallinity levels due to the use of high cooling rates (as explained in
Section 4.2). Such low crystallinity levels indicated a significant presence of amorphous domains available for crystallization during heating in DMA, allowing them to crystallize post-glass transition, along with an associated sudden increase in storage moduli. With regard to blend compositions, the presence of molten PBSA as nucleating agents acted as an additional factor in enhancing the crystallinity and the subsequent jump in storage moduli [
29].
The trends observed for the glass transition temperature (
Tg) in storage moduli curves and tanδ curves were in good agreement with each other for all samples (
Figure 3 and
Figure 4). In the tanδ curve, a peak was observed in the region where, with increases in temperature, the rate of the decrease in storage modulus was higher than that of the loss modulus. Temperatures corresponding to the tanδ peak is often considered as
Tg. Interestingly,
Tg was not observed for the PBSA component in all blend samples due to the locking of PBSA chains by hard PLA segments, thus preventing their motion. Additionally, the use of a lower weight fraction of PBSA meant that a higher share of PBSA chains were restricted by PLA chain segments, ensuring that no
Tg corresponding to PBSA was observed for blend compositions [
29].
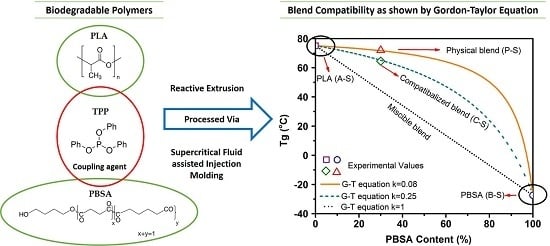
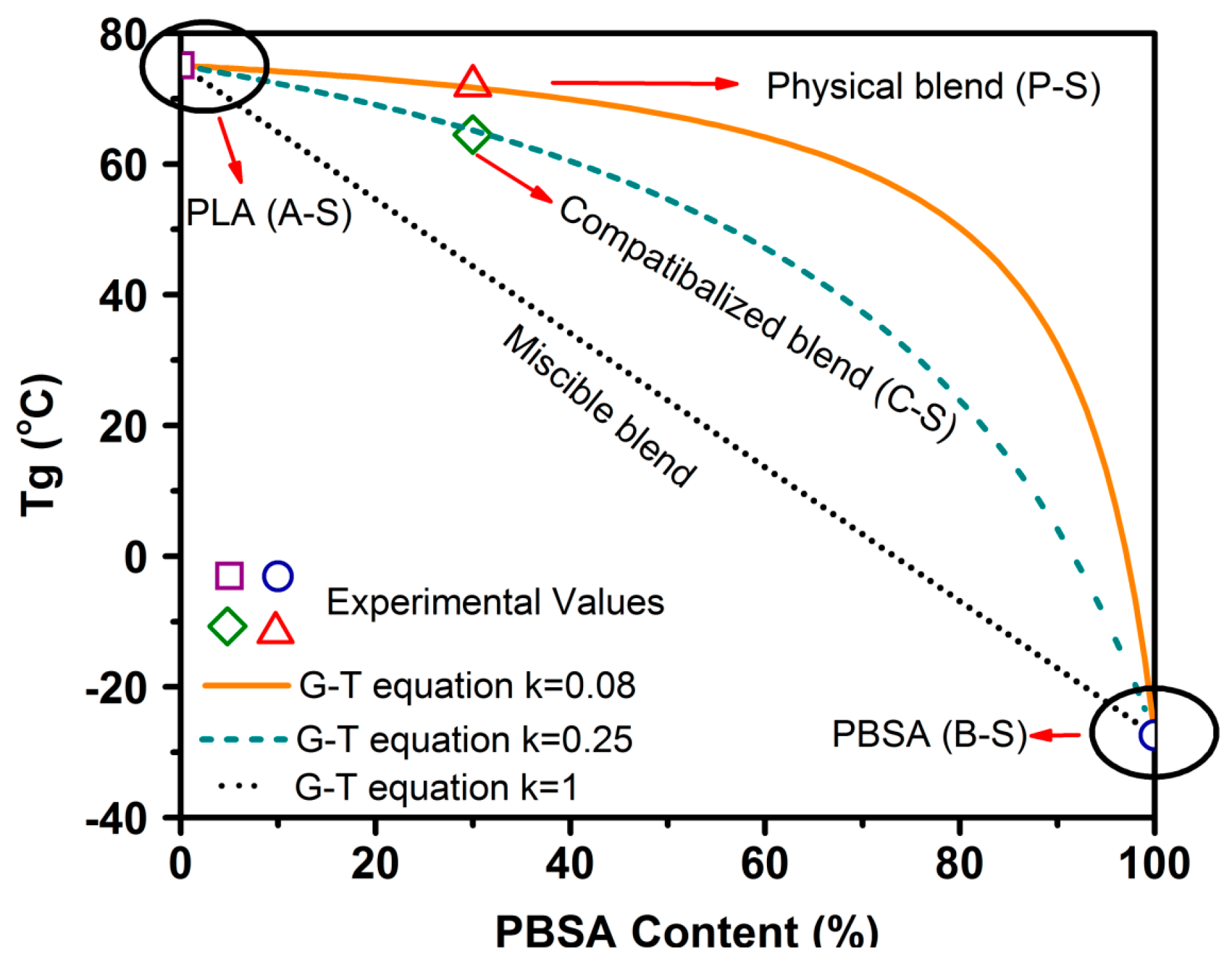
Glass transition temperature of blend samples gives us insight into the miscibility of pure polymers constituting the blends.
Tg is typically dependent on the polymer composition of blends, and lies between the
Tg values of pure constituents for a completely miscible blend [
50]. To obtain clarity on the miscibility and effect of TPP on PLA–PBSA blends, a simplified version of the Gordon–Taylor (G-T) equation (Equation (3)) [
51] was applied to
Tg obtained from tanδ.
Here,
and
are the glass transition temperatures of pure components PLA and PBSA, respectively, while
and
are the wt % of PLA and PBSA, respectively, and
is a curve-fitting factor representing the miscibility of the system, with
= 1 indicating the complete miscibility of the polymers and the lower/higher values of
k indicating poor miscibility.
Figure 5 depicts
of different blend compositions. Observed
values for A-S, P-S, and B-S (~75.1, ~72, and ~−27.4 °C) and A-S, C-S, and B-S (~75.1, ~64, and ~−27.4 °C) were plotted as the
Tg of the talc-filled and ScF-foamed blends, all of which were found to overlap (
Table 8). These observed values were closer to the G-T curve for
= 0.08 and
= 0.25, where the curve-fitting parameter
showed a value of 0.08 for the physical blends, indicating the poor miscibility of PLA and PBSA, as they are thermodynamically immiscible [
52]. However, an addition of 2 wt % TPP shifted the
of PLA–PBSA blends to around 64 °C, with the
value of 0.25 used to curve fit the G-T equation; this higher value of
indicates the possibility of enhanced compatibilization.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}