
Deformation-Induced Phase Transitions in iPP Polymorphs


Abstract
:
1. Introduction
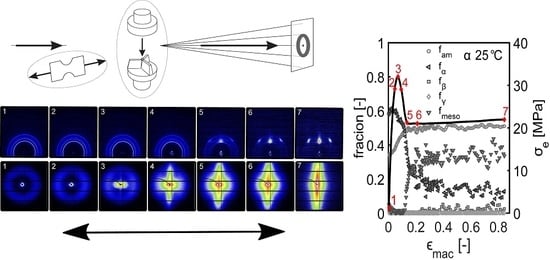
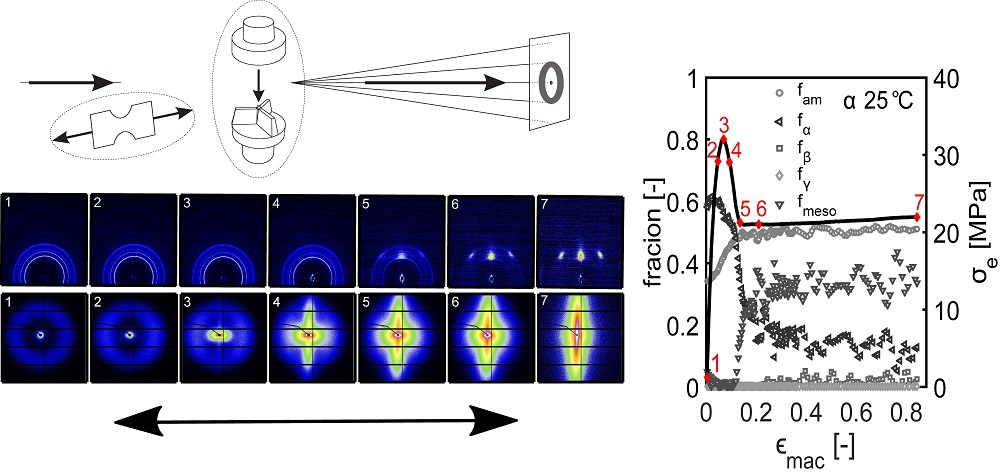
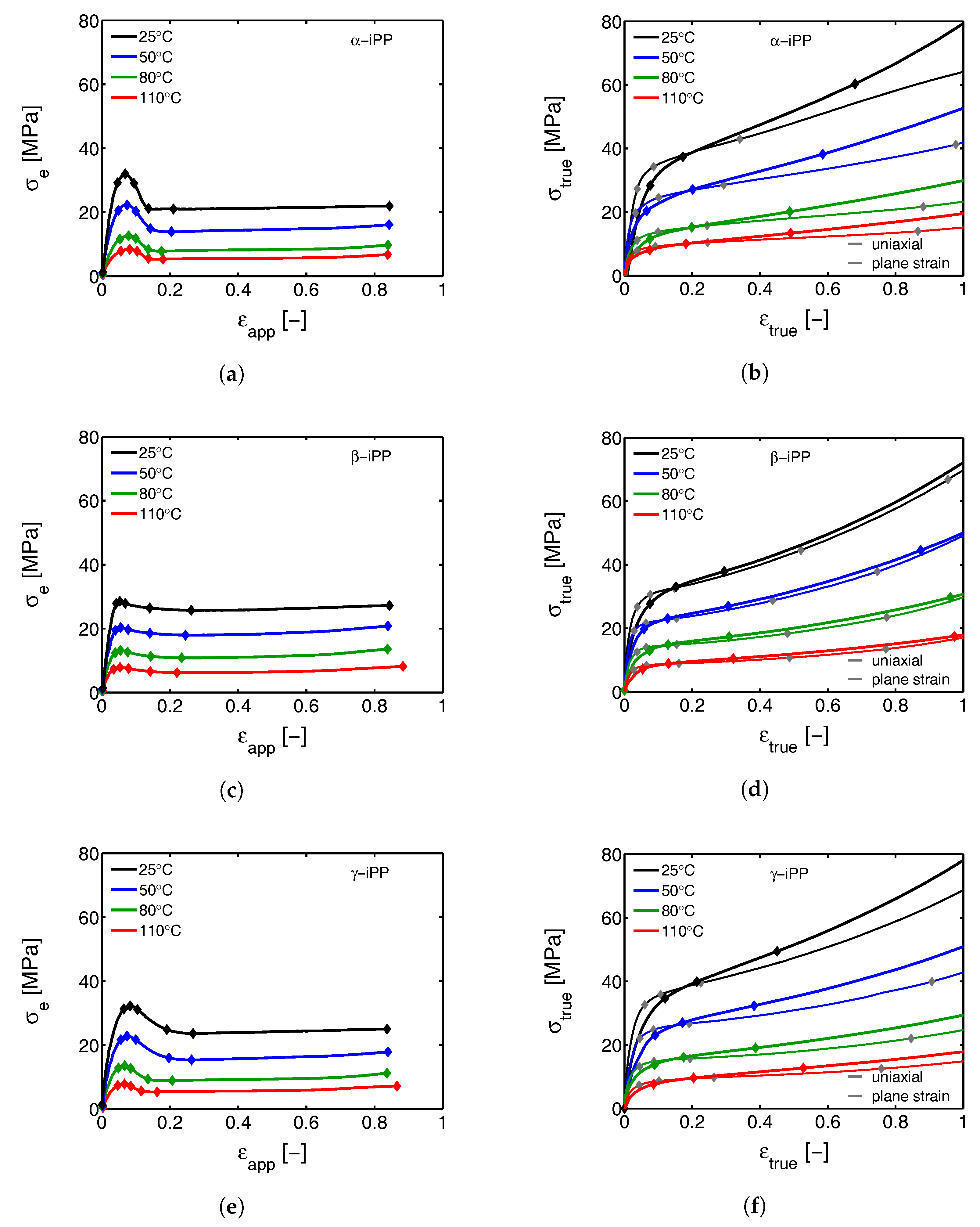
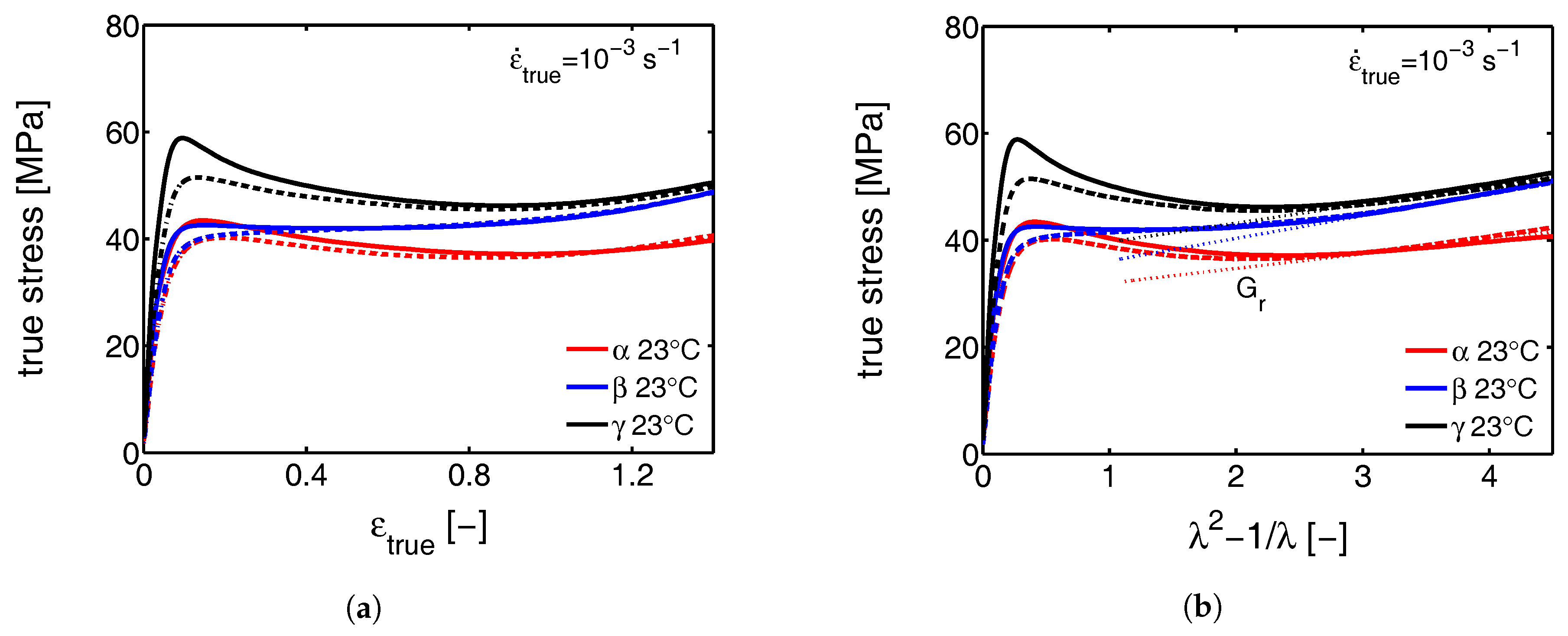
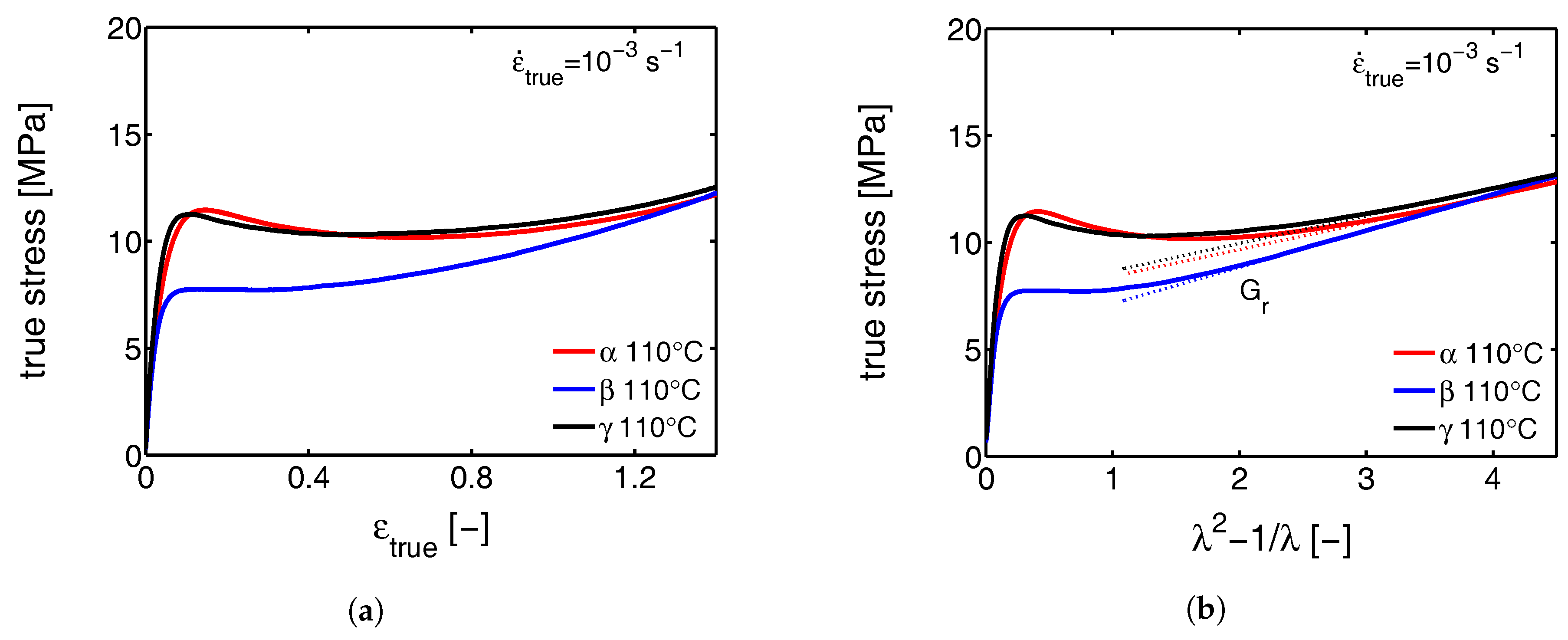
- First, the mechanical response obtained from the tensile experiments is shown, together with the calculated true stress as a function of the true strain. The behavior is discussed and compared with the intrinsic material response measured in uni-axial compression.
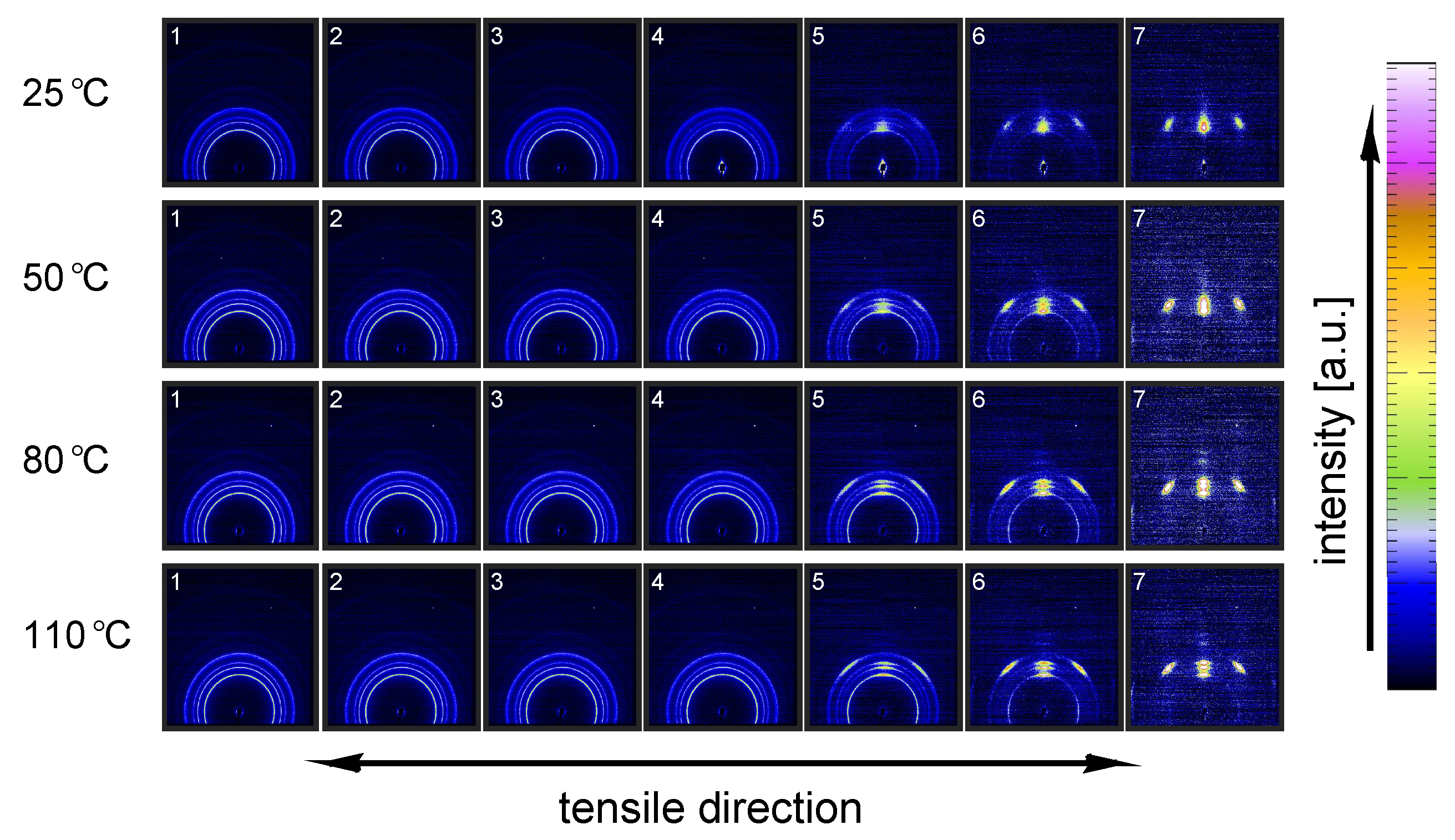
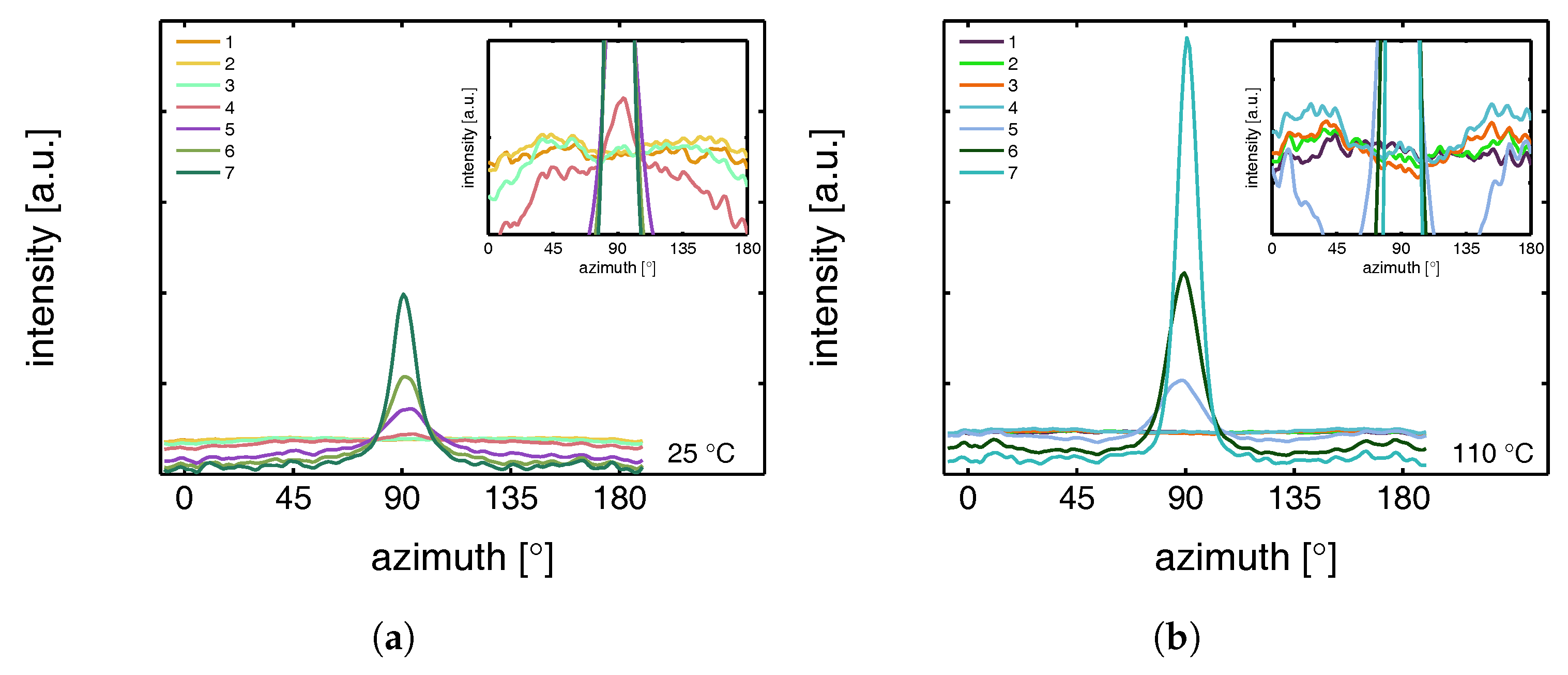
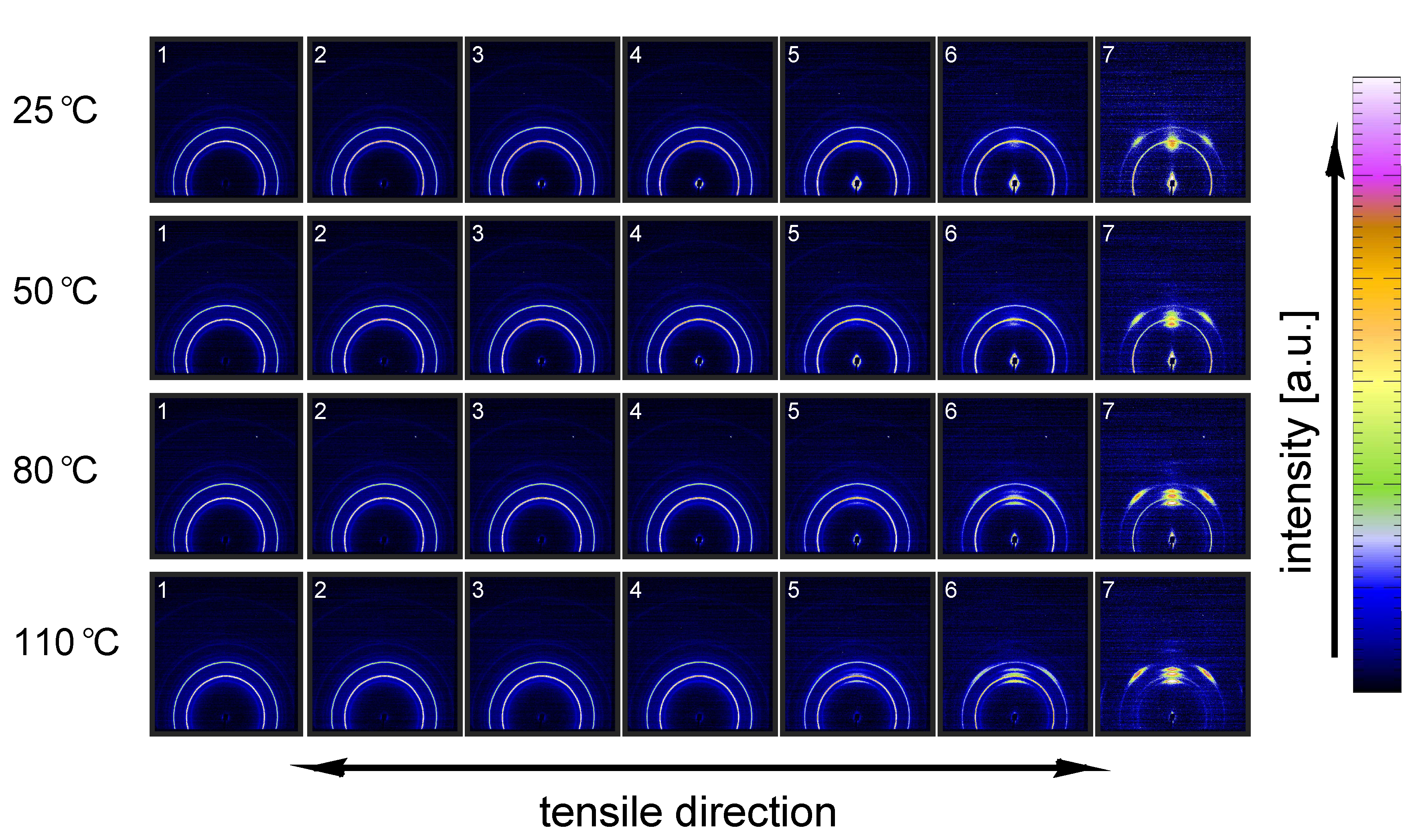
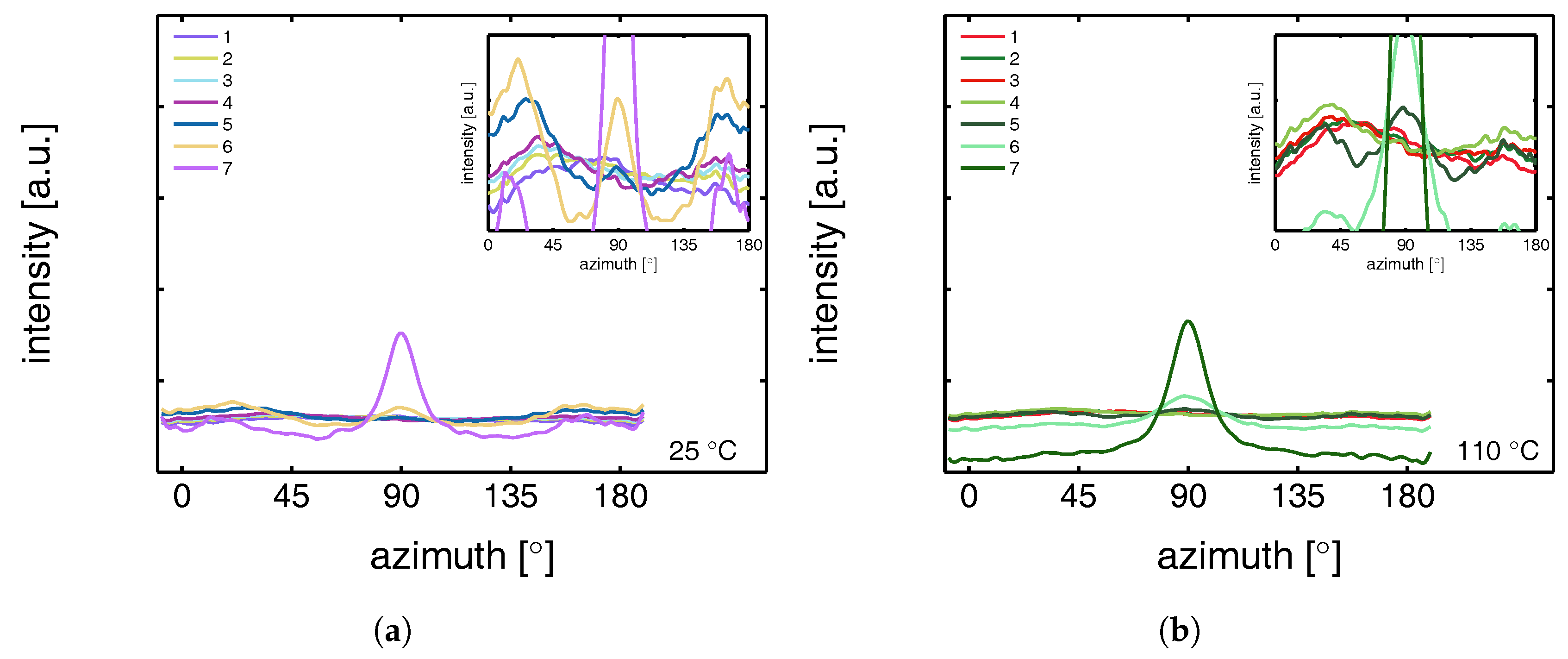
- The structural evolution in terms of phase transitions, crystal plane orientation and slip is revealed by WAXD experiments combined with uni-axial tensile tests. The crystal phase transitions are measured in compression experiments, as well, allowing one to compare these results with the ones obtained from tensile experiments (phase transitions) and couple them to the true strain.
- The combination of SAXS experiments with uni-axial tensile tests provided insights on the onset of voiding, lamellar orientation and evolution of amorphous and lamellar thickness. Furthermore, a transition of initially disk-like average void structure to a fibrillar structure was found.
2. Materials and Methods
2.1. Material and Sample Preparation
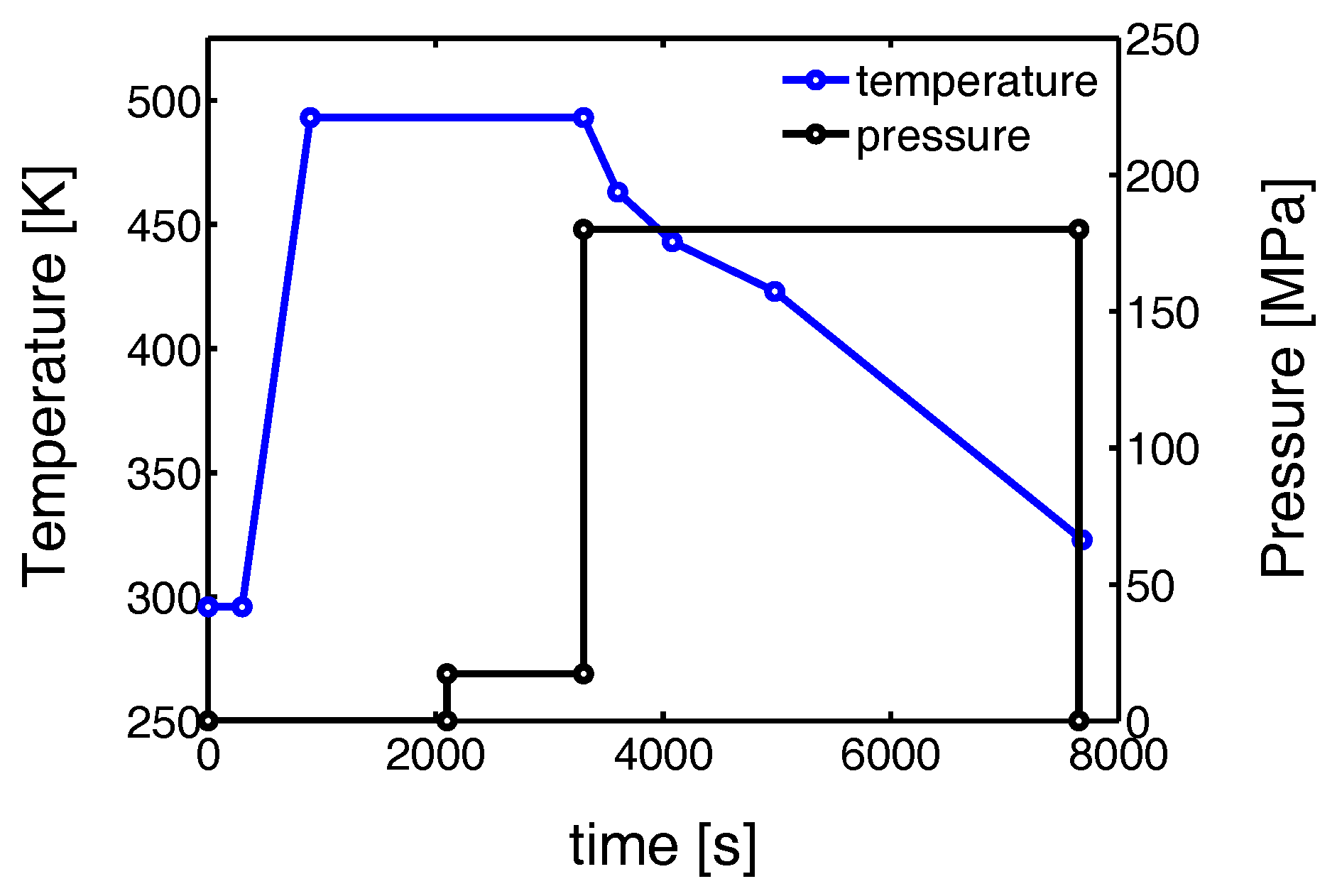
2.2. Mechanical Testing
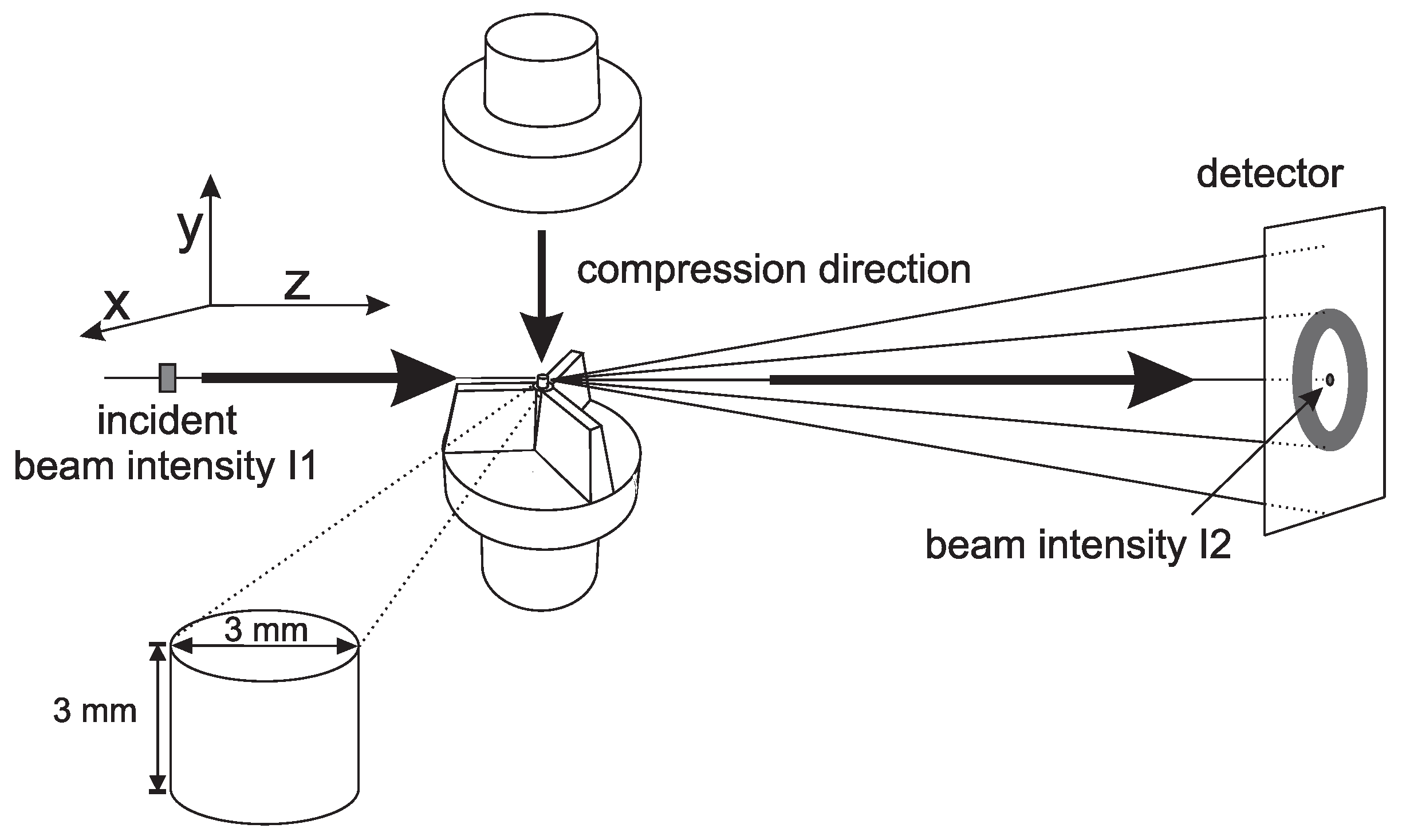
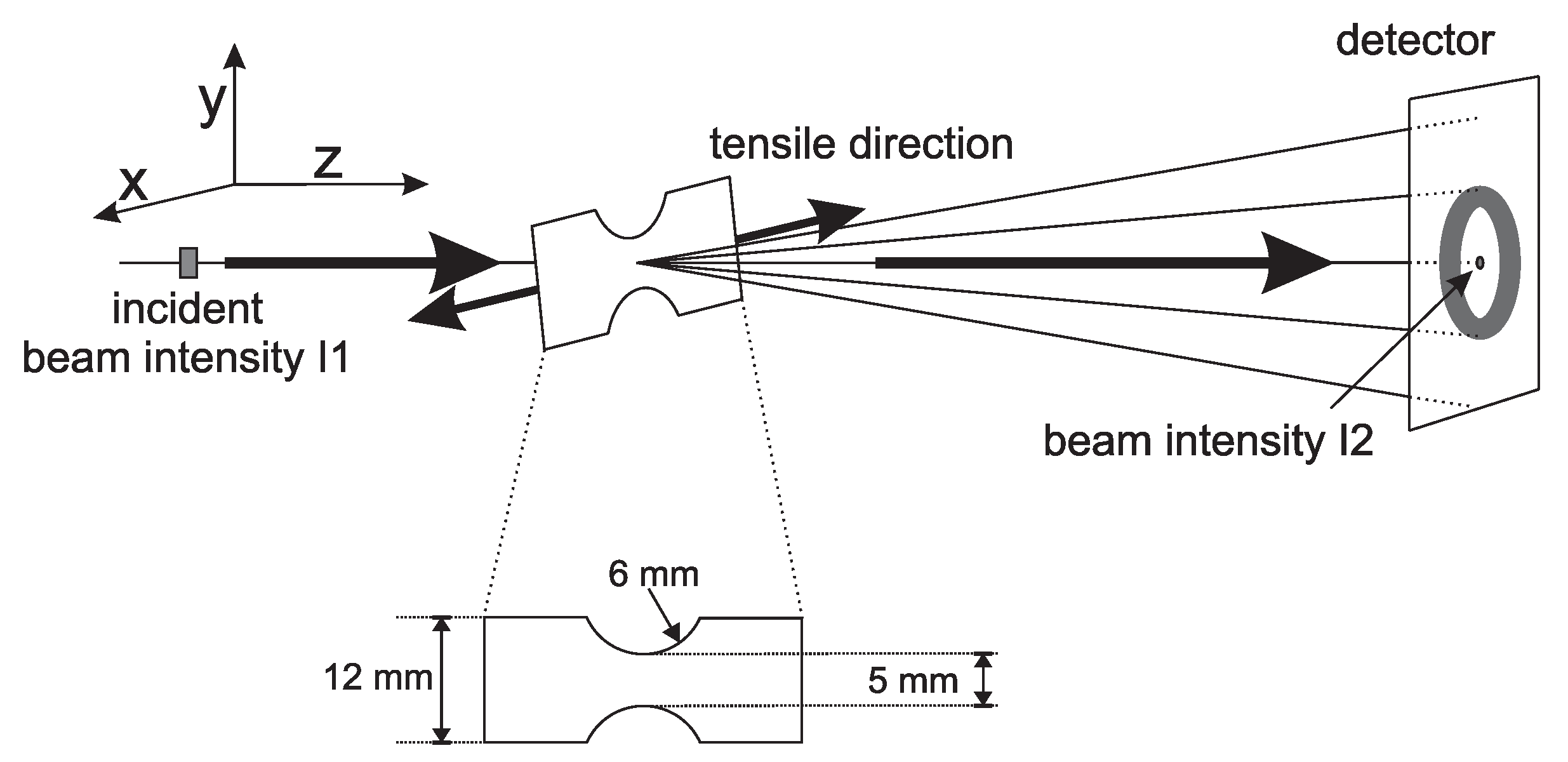
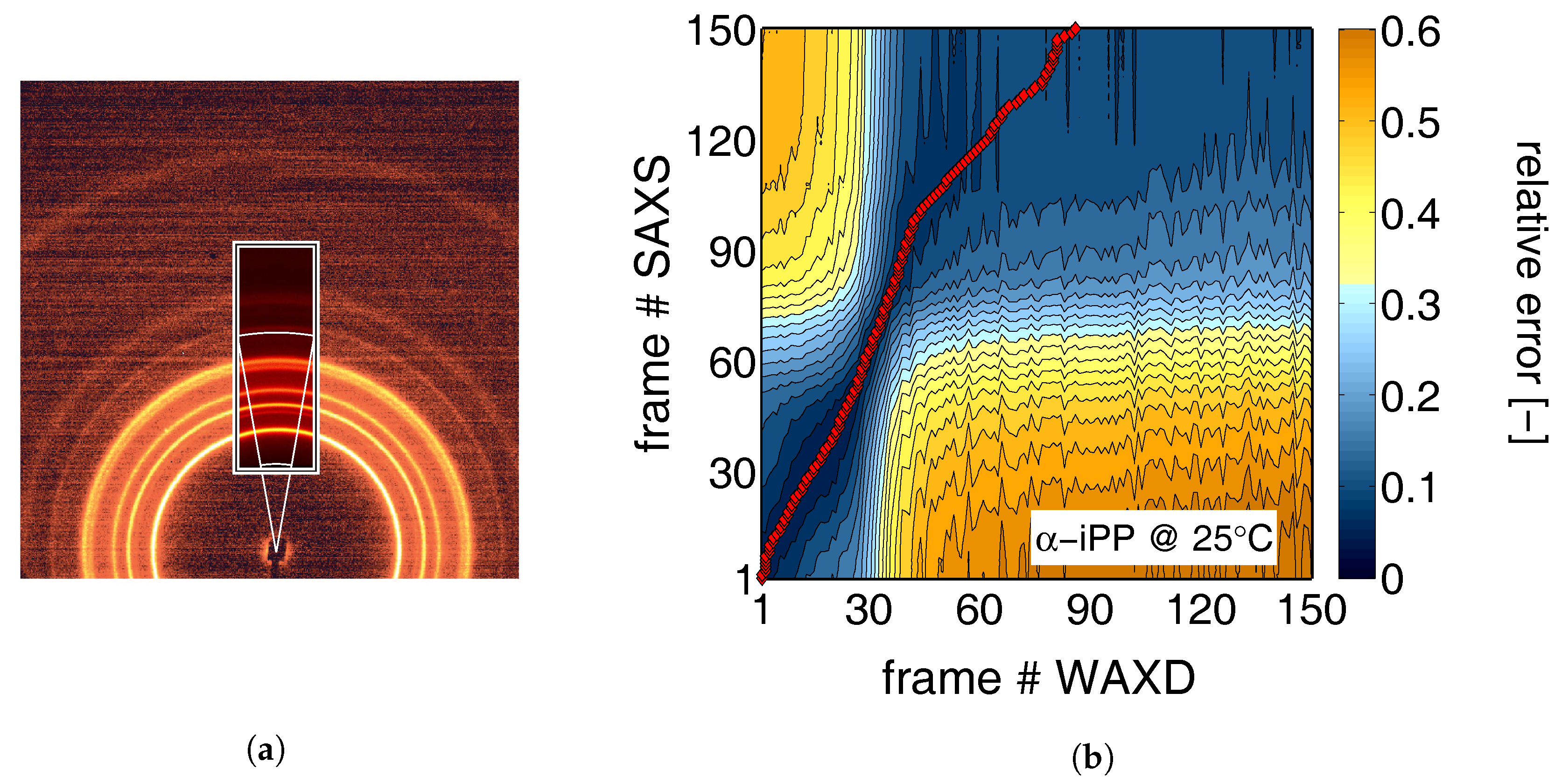
2.3. X-ray Techniques and Analysis
- The 2D WAXD patterns where recorded with a Frelon detector. This detector has a pixel size of 97.65 × 97.65 µm and was placed at approximately 140 mm from the sample. The acquisition time was 2.5 s.
- The 2D SAXS patterns where recorded with a Pilatus 1 M detector. Simultaneously with these patterns, a Pilatus 3 K was used to record WAXD patterns. From now on, we will refer to these patterns as 1D WAXD-patterns. The pixel size of the Pilatus 1 M and 1 K was 172 × 172 µm, with a sample to detector distance of approximately 2707 mm (for SAXS) and 283 mm (for WAXD). At this distance the Porod region is included in the SAXS data. Again the acquisition time was 2.5 s.
- Another set of 2D SAXS experiments were performed with a sample to detector distance of approximately 7258 mm, allowing one to obtain scattering data at low q-values. Again these frames where acquired simultaneously with 1D WAXD patterns, recorded with the Pilatus 3 K placed at a distance of approximately 183 mm, and an acquisition time of 2.5 s.
- During the compression experiments, in situ WAXD patterns where recorded using a Frelon detector. This detector is the same as the one used for the 2D WAXD experiments in tensile deformation, but was now placed at approximately 200 mm from the sample. In this case the acquisition time was 1 s.
2.3.1. Data Reduction
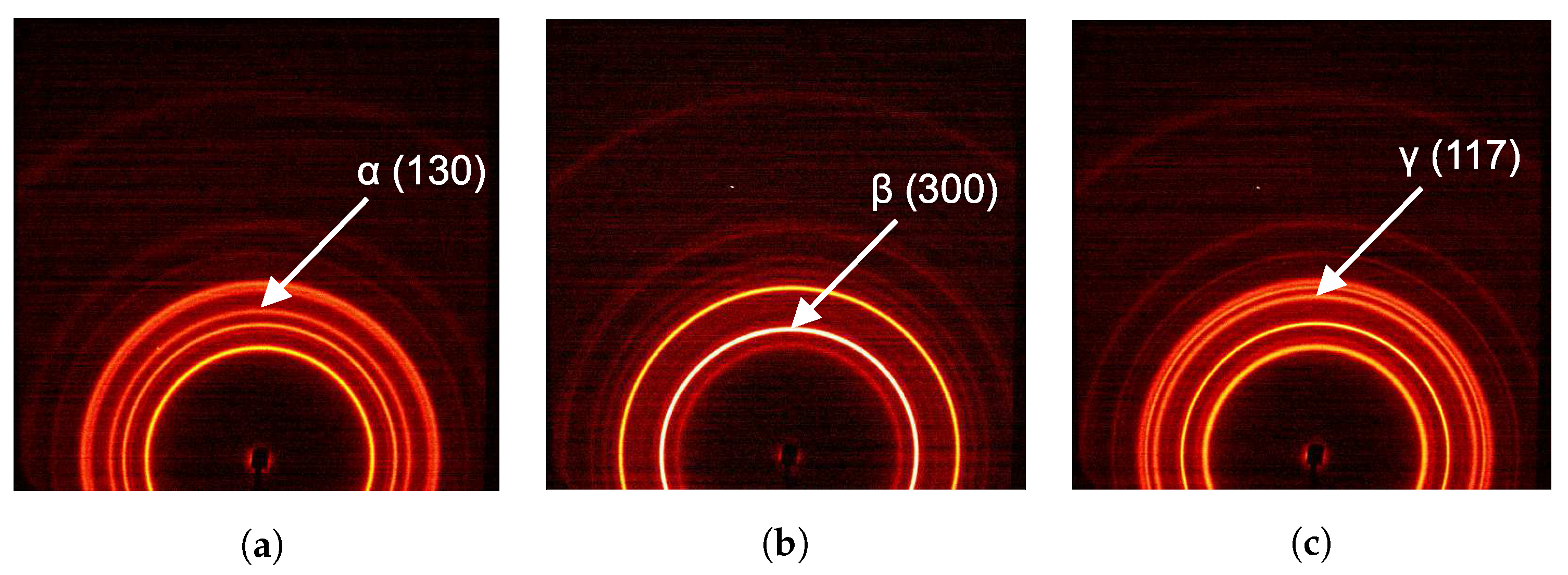
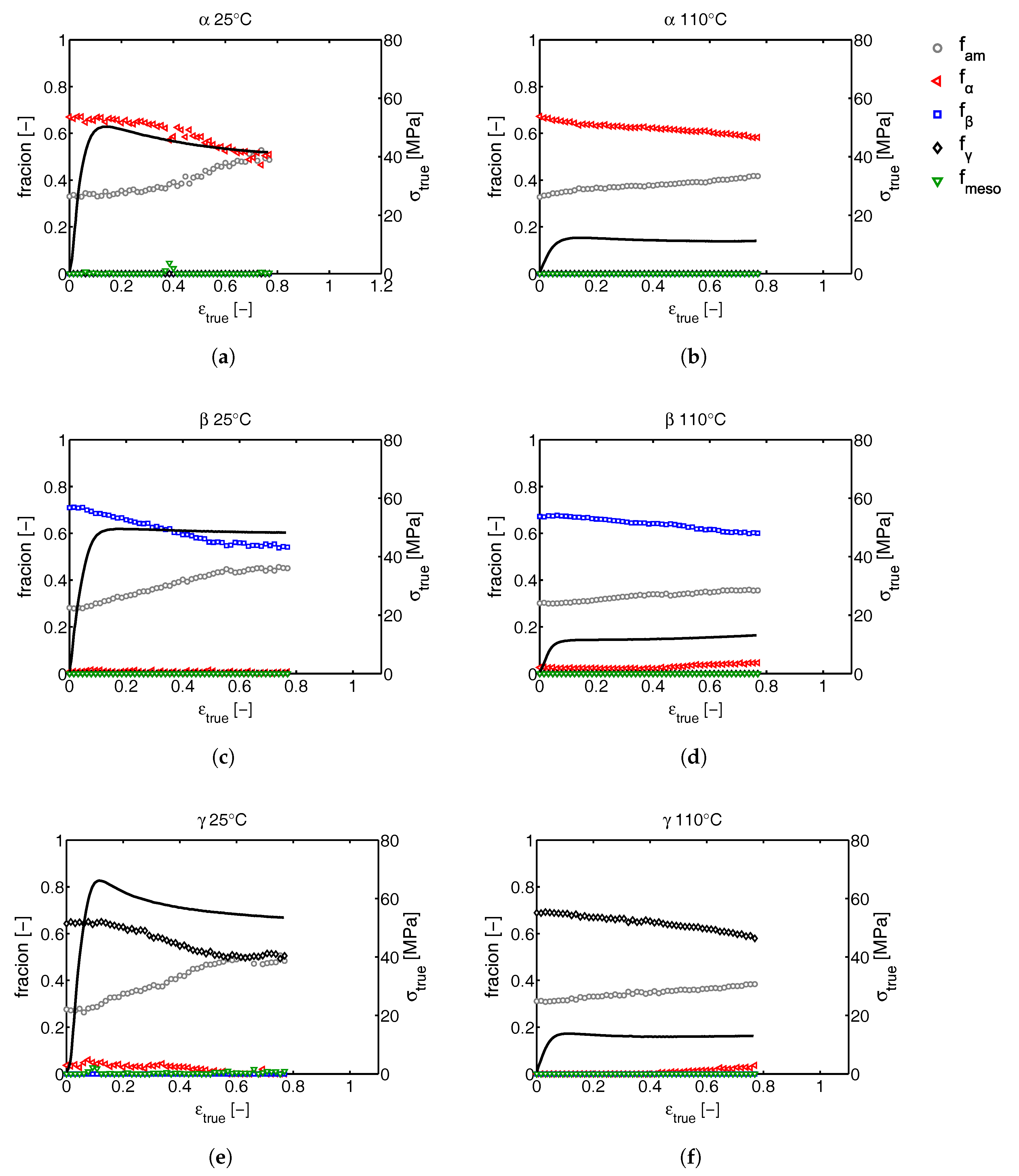
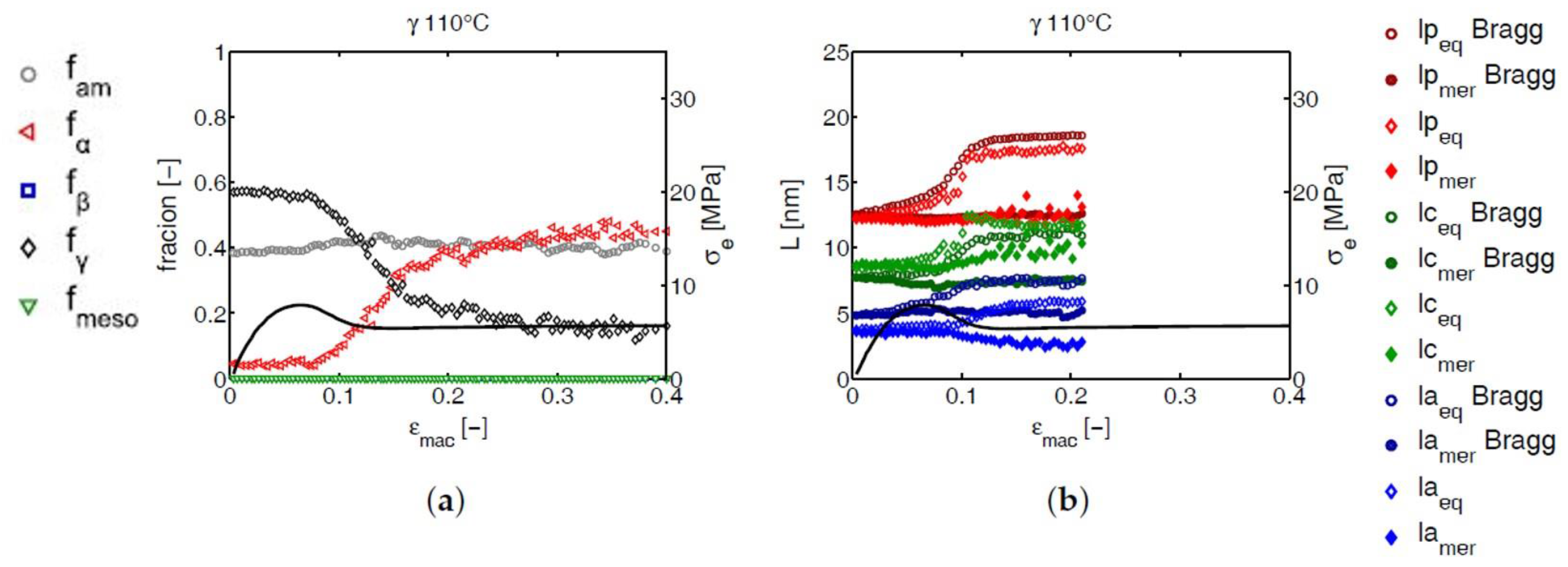
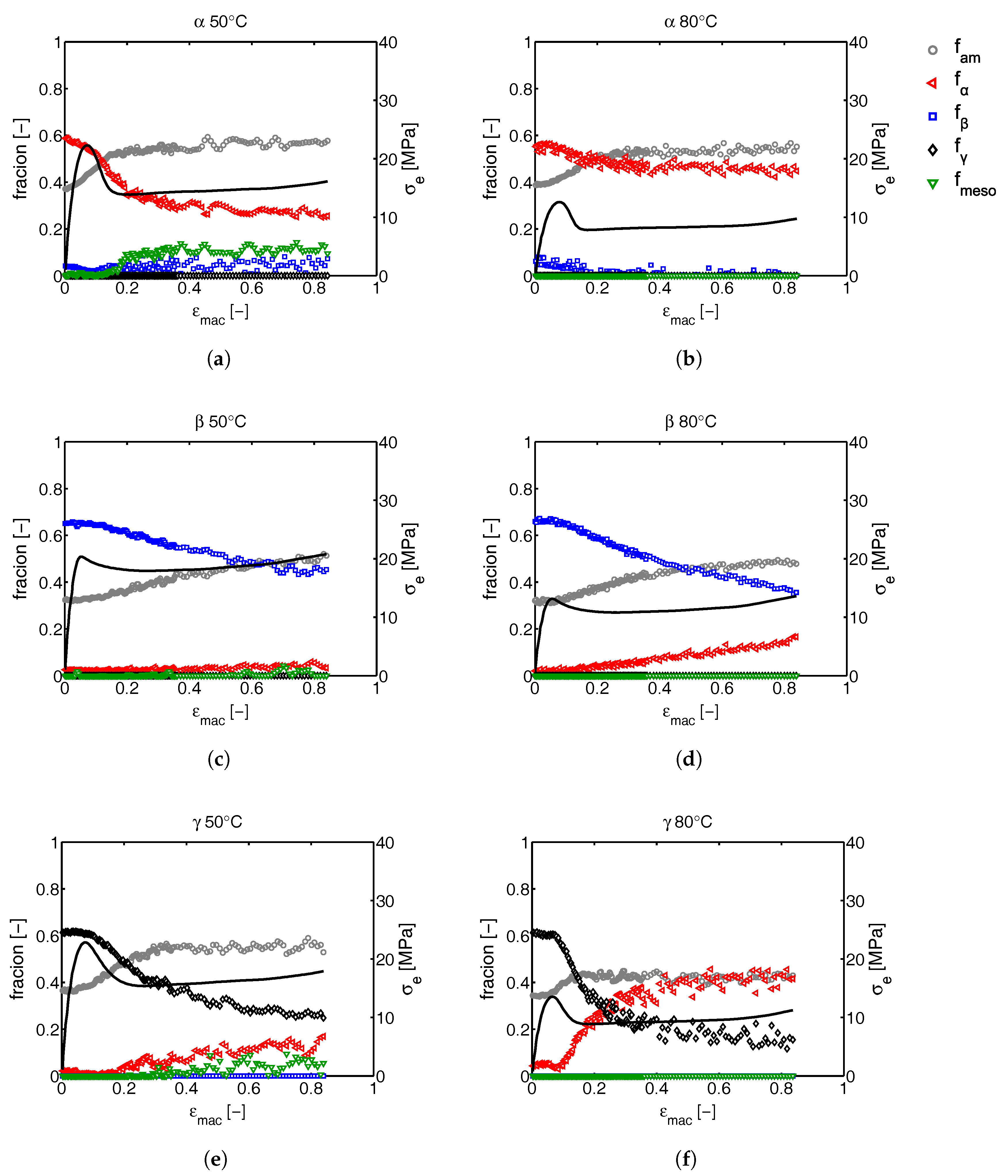
2.3.2. Phase Composition
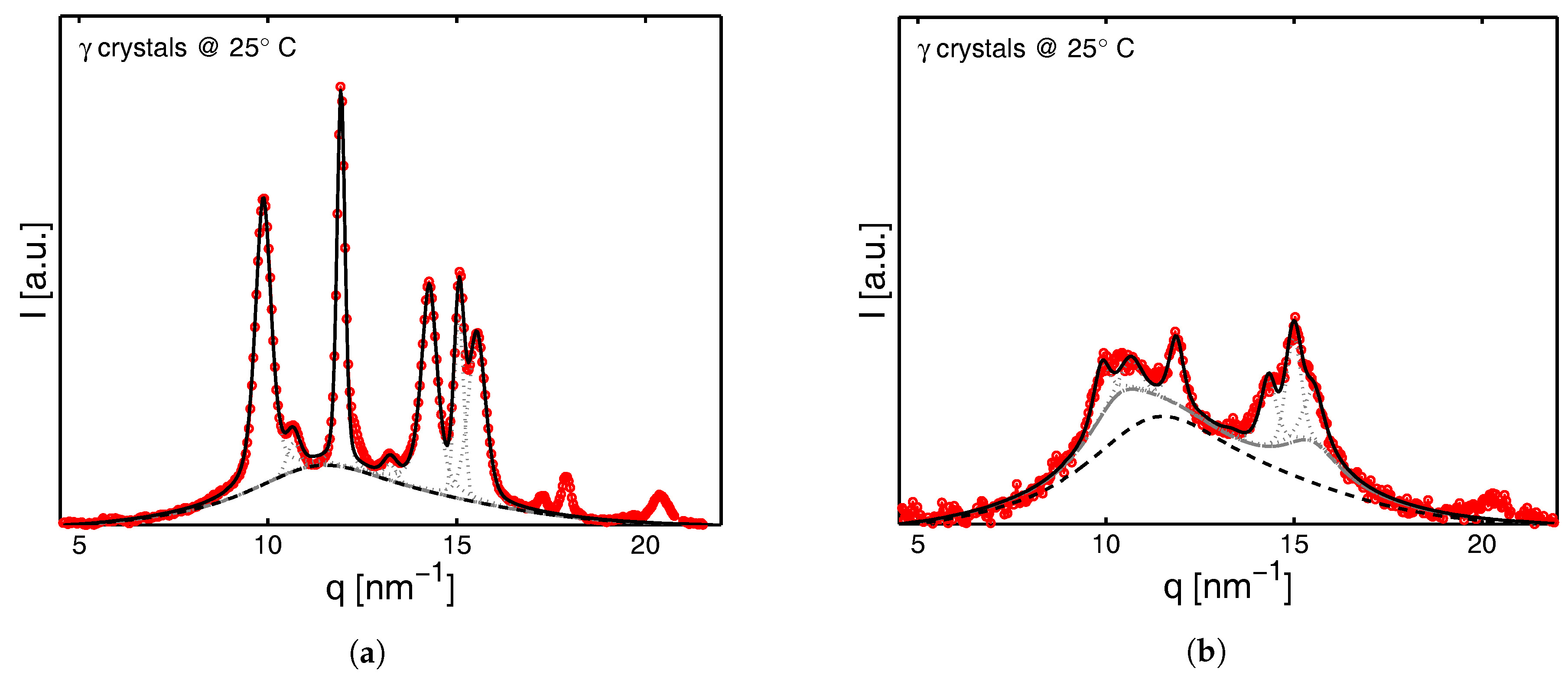
2.3.3. Determination of lp, lc and la
2.3.4. Determination of the Strain
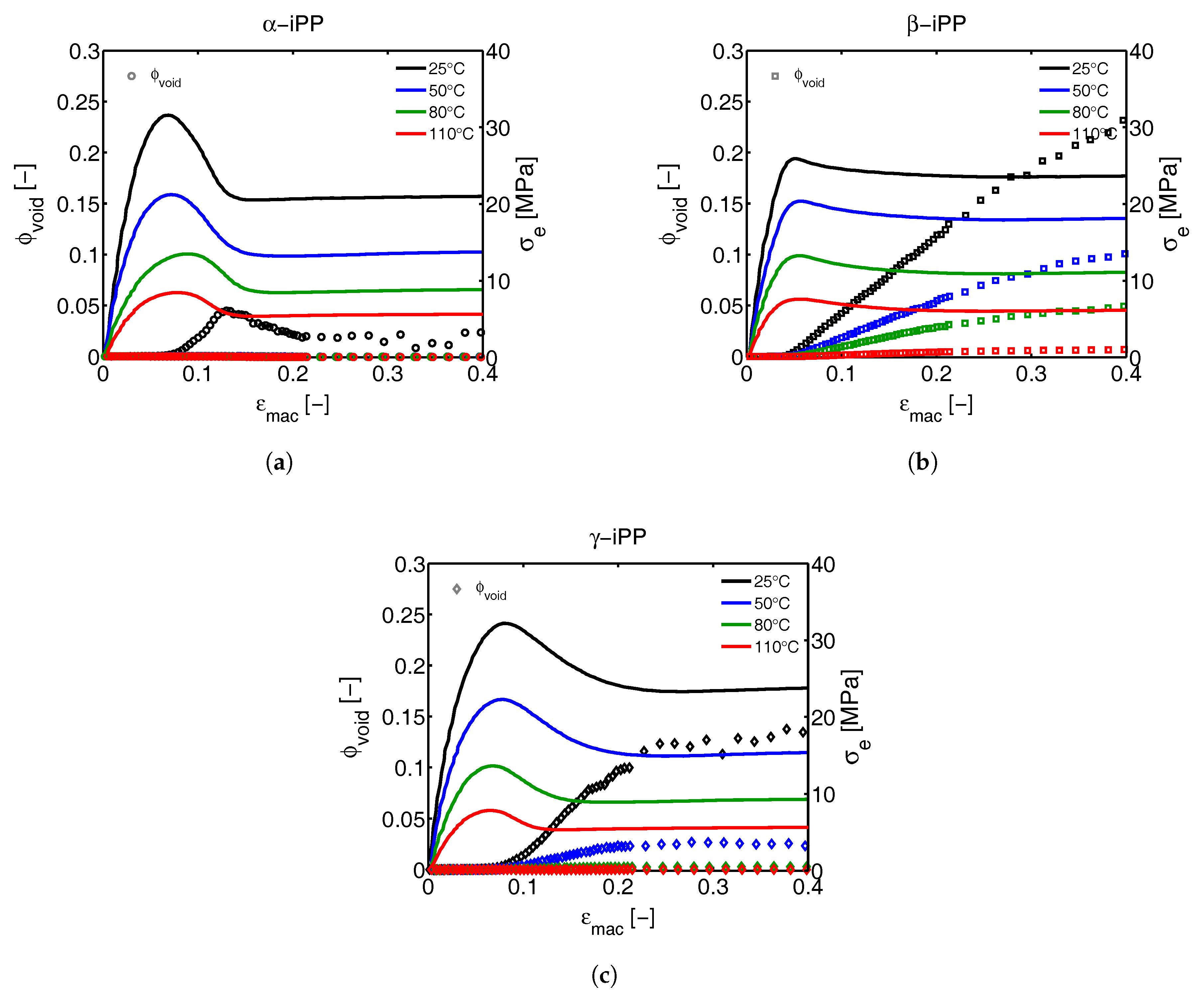
2.3.5. Void Fraction
3. Results and Discussion
3.1. The Mechanical Response
3.1.1. Tensile Tests (Mechanical)
3.1.2. Compression Tests (Mechanical)
3.2. WAXD Analysis
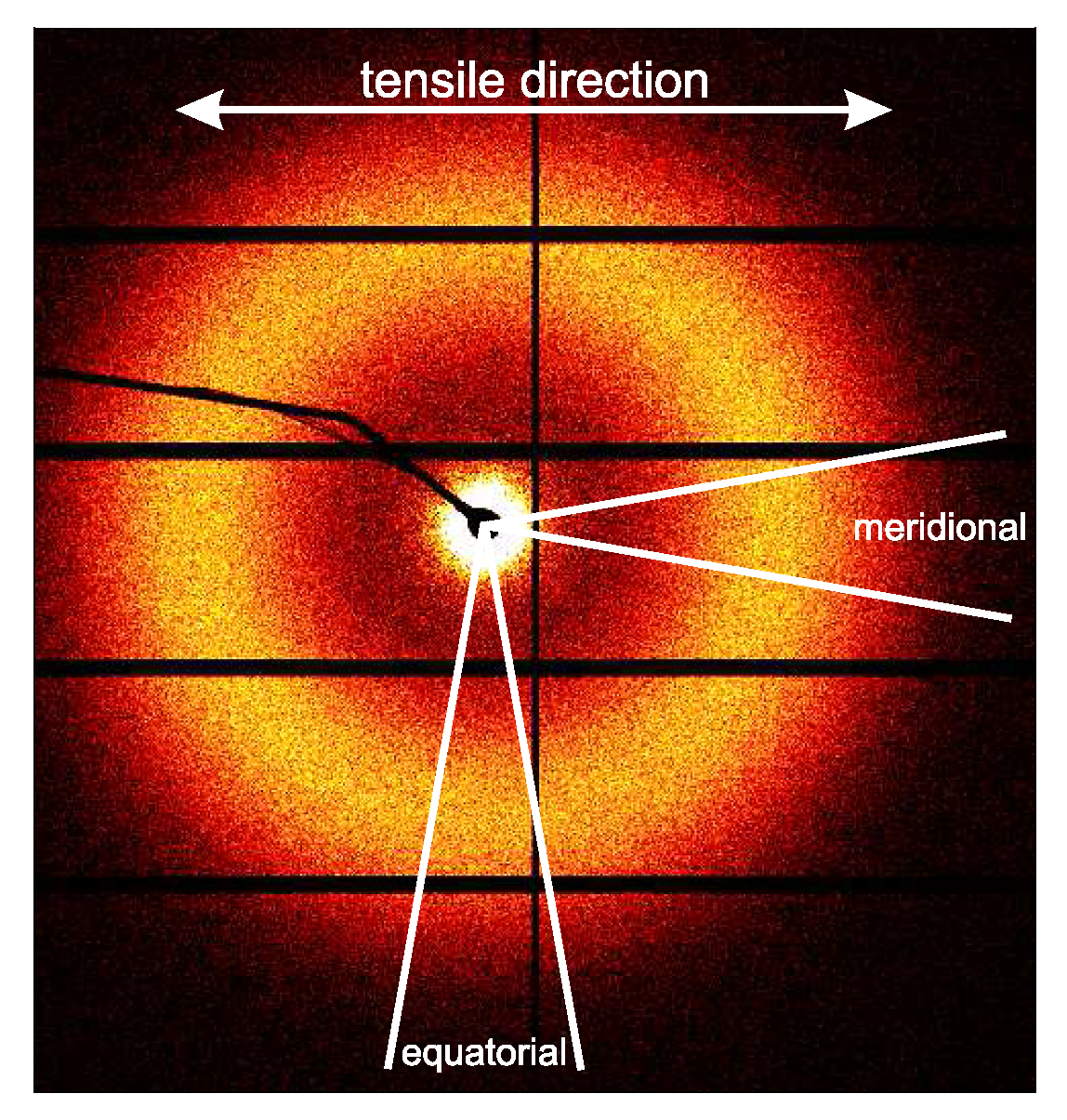
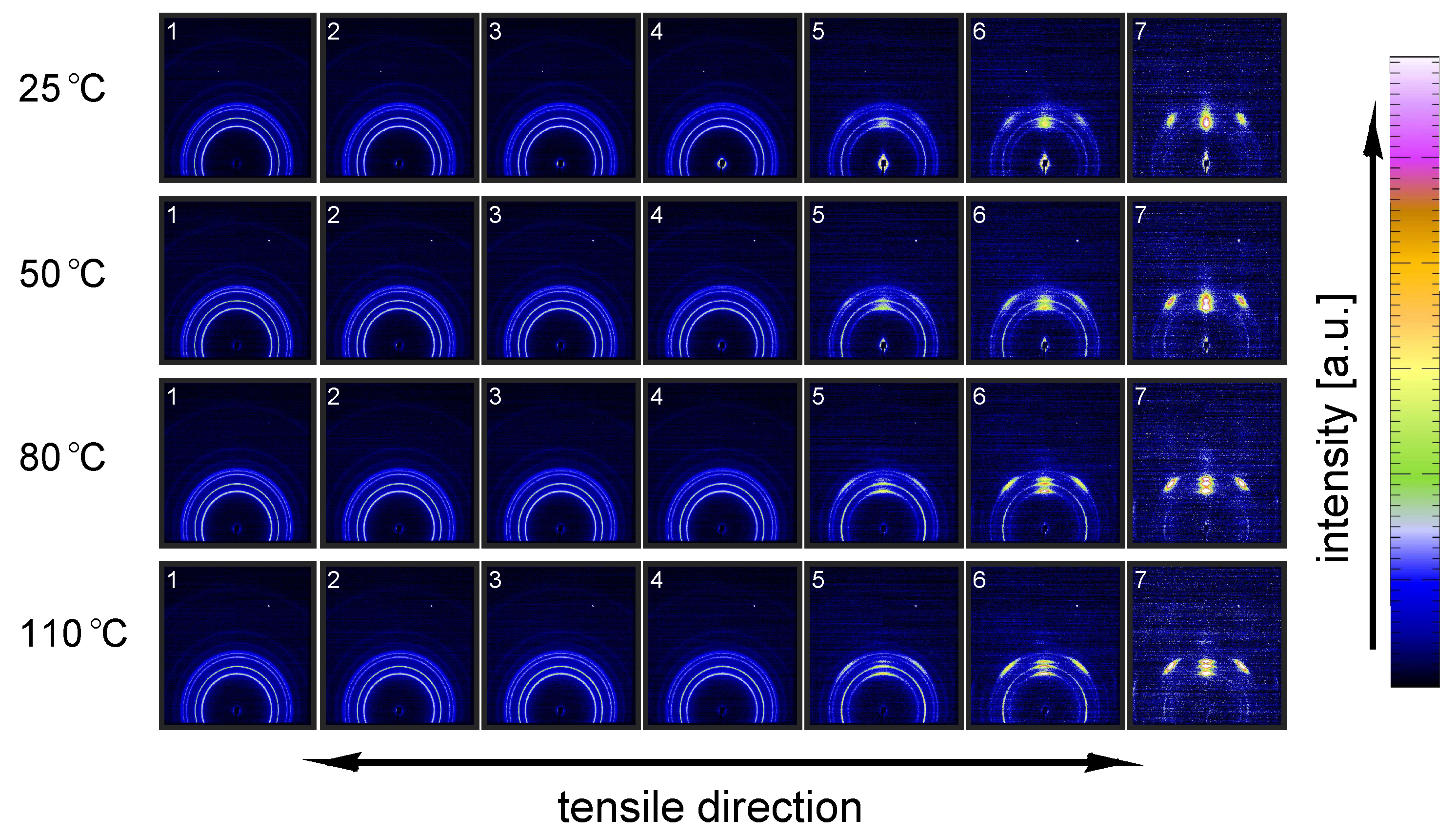
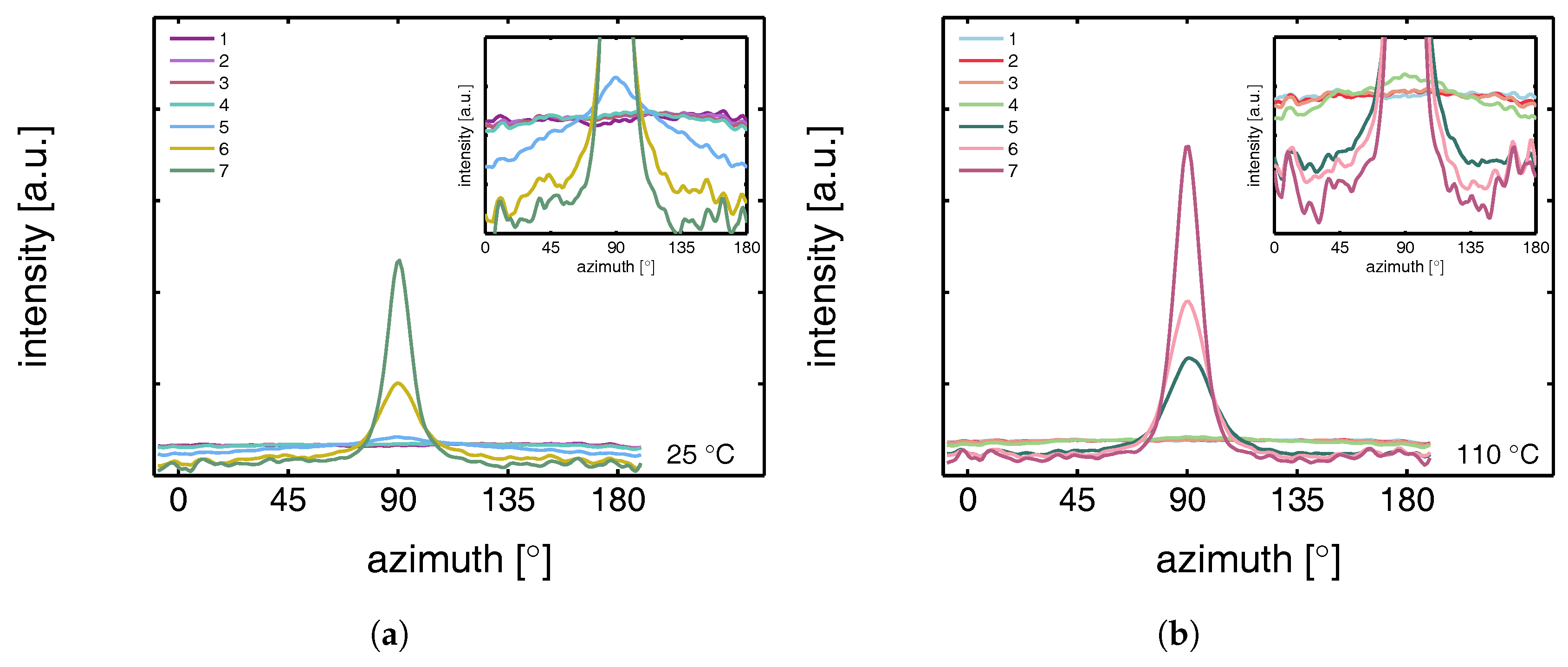
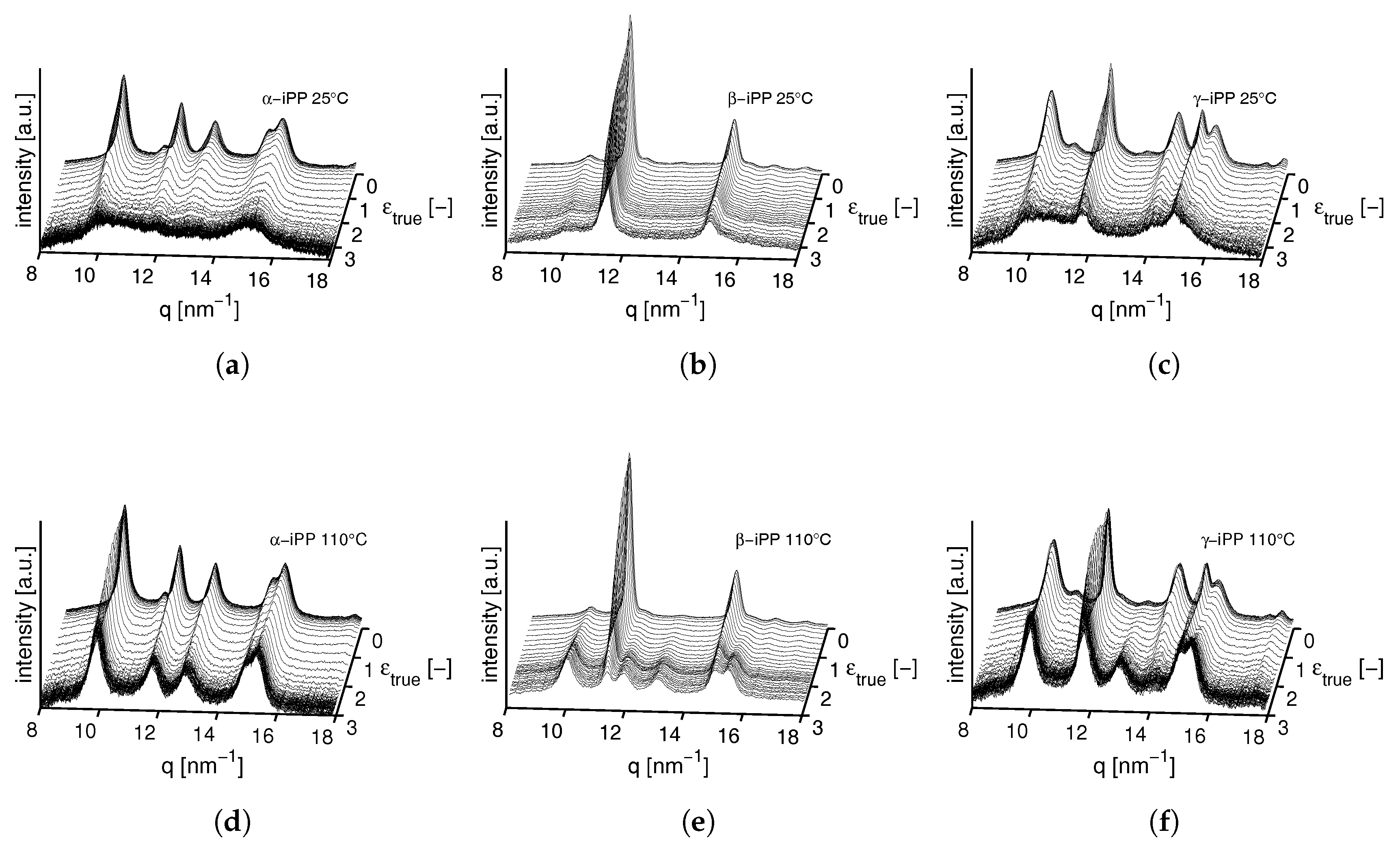
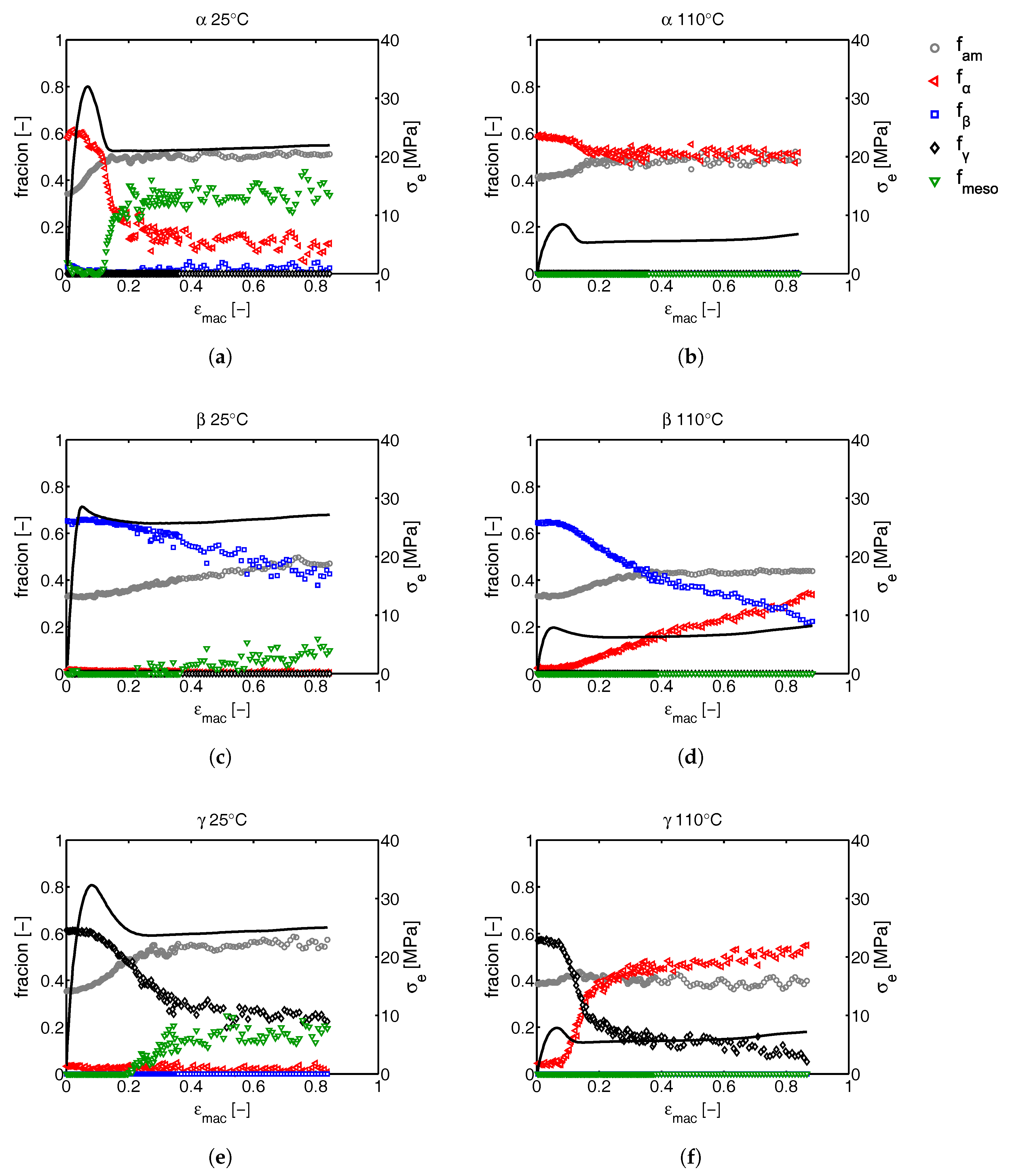
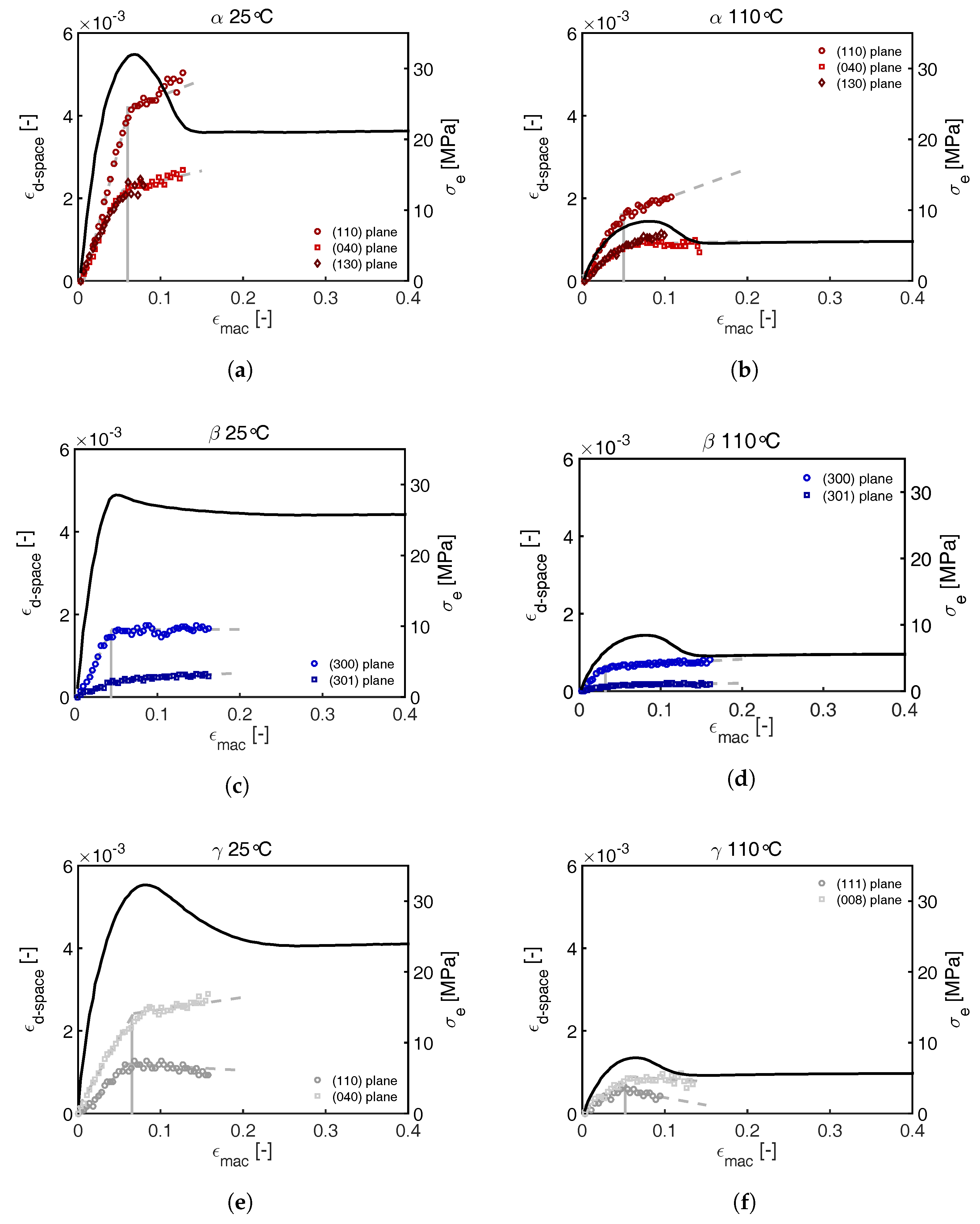
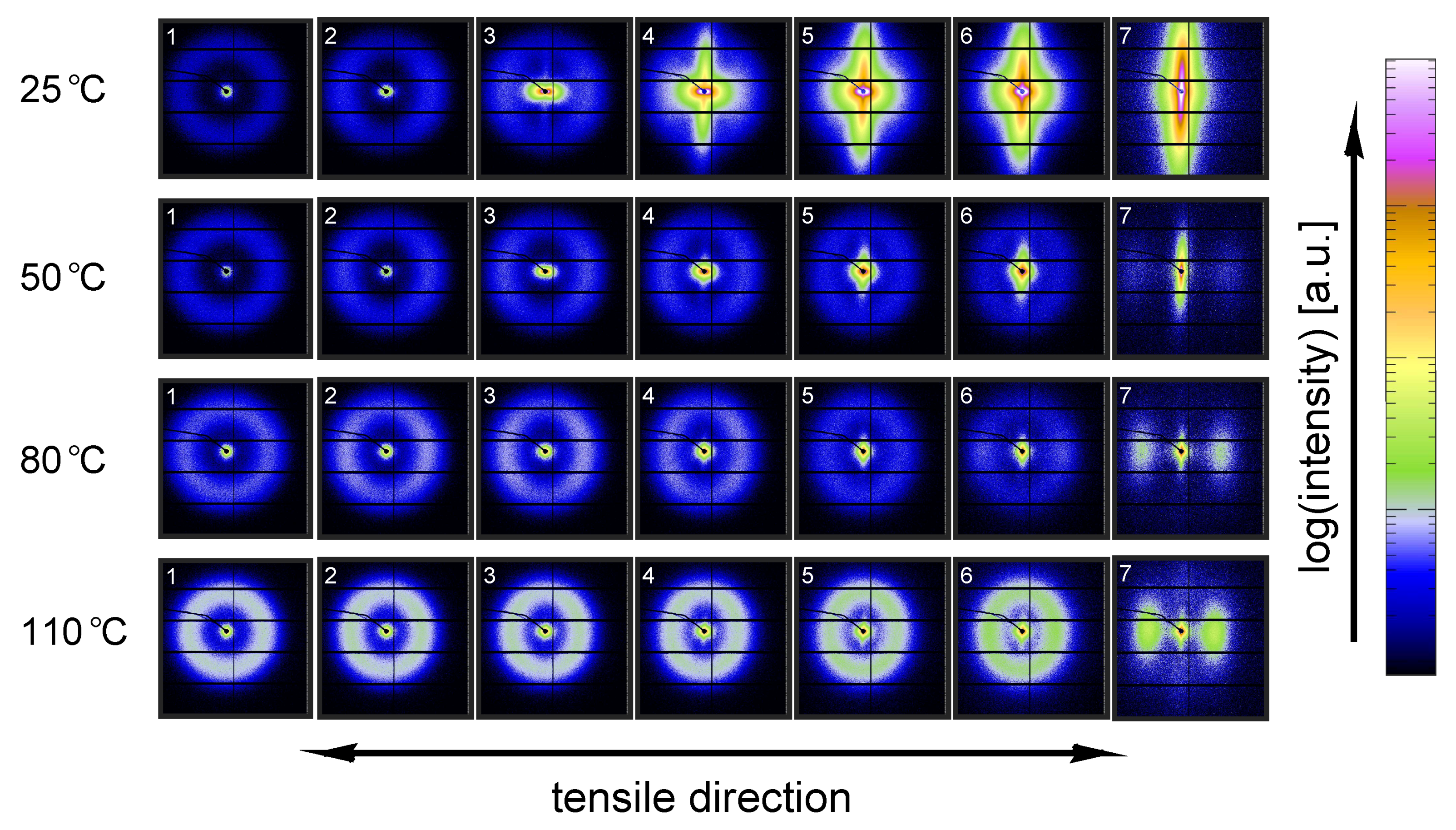
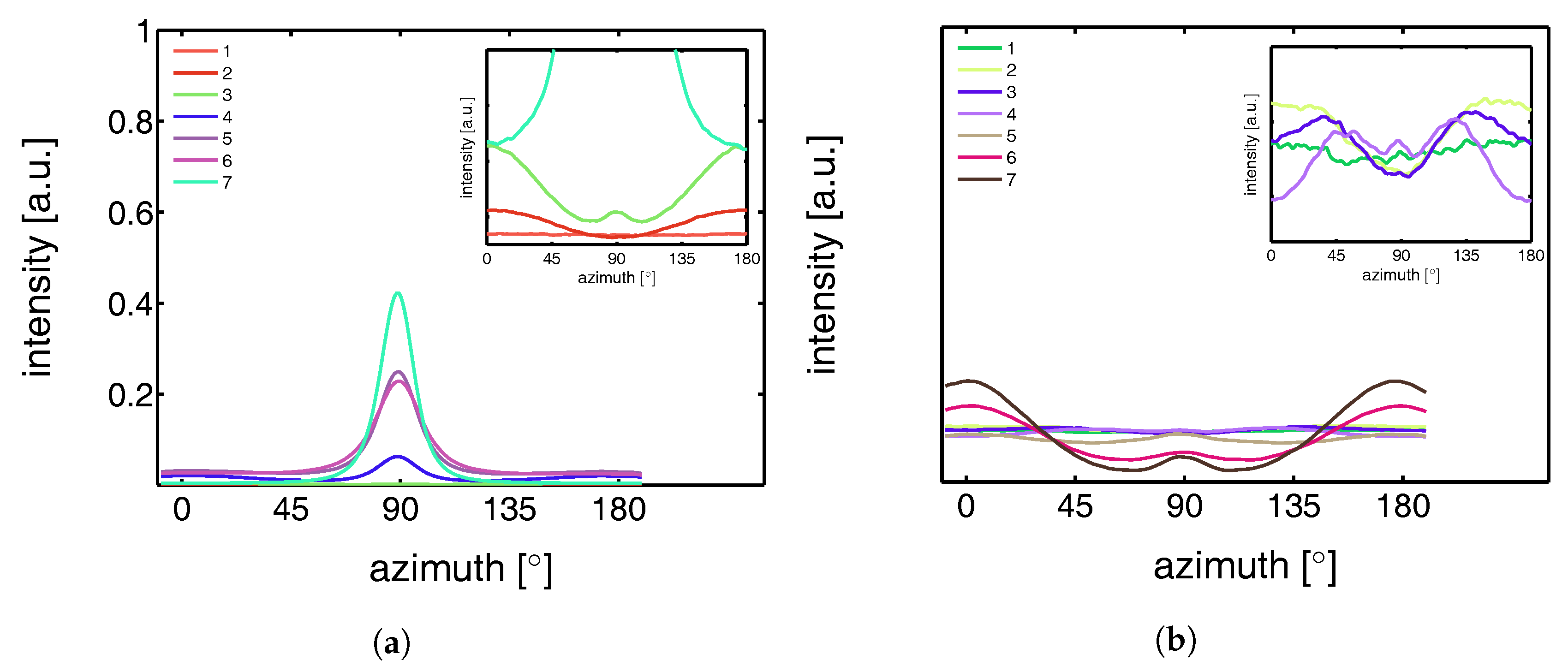
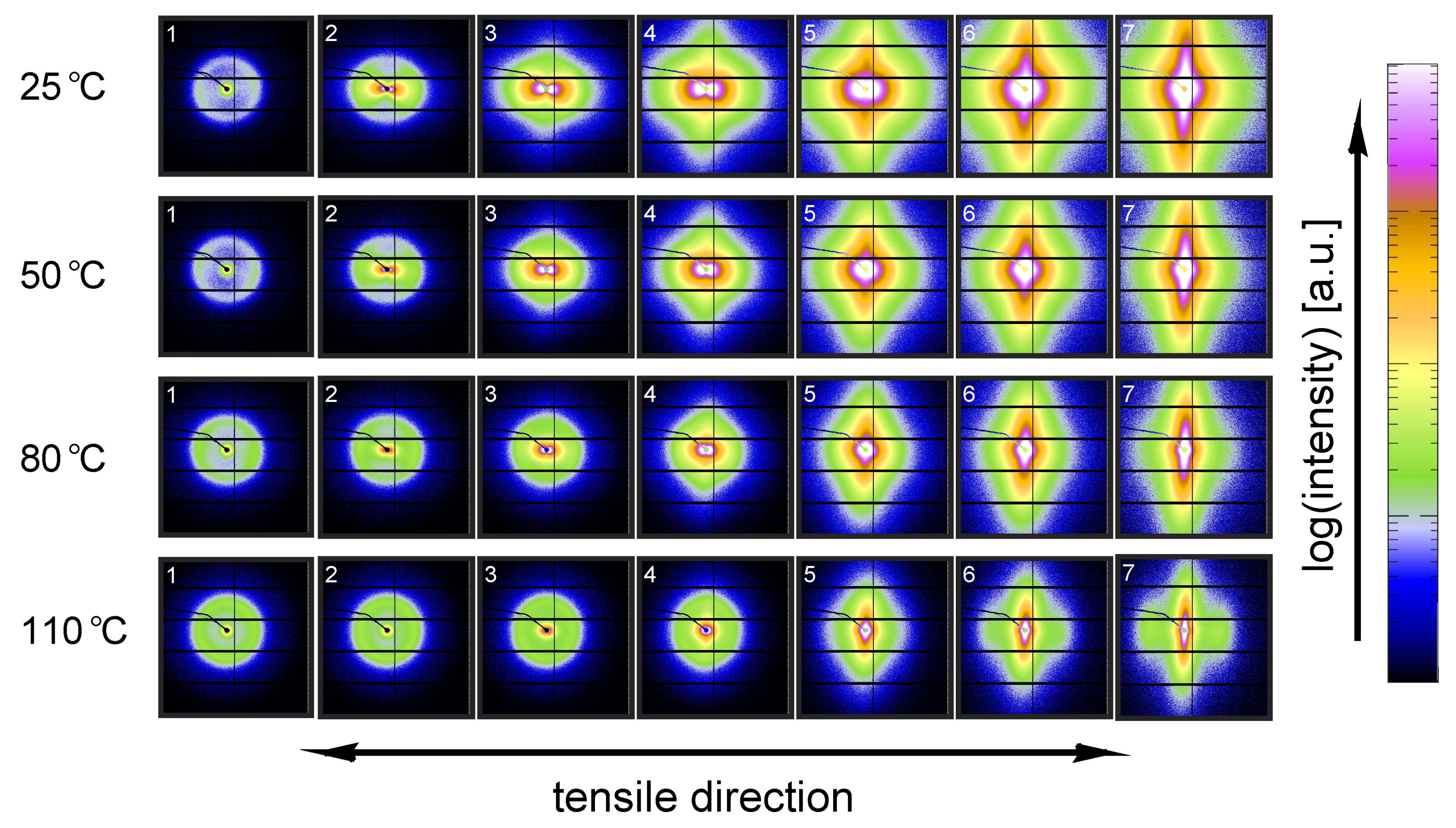
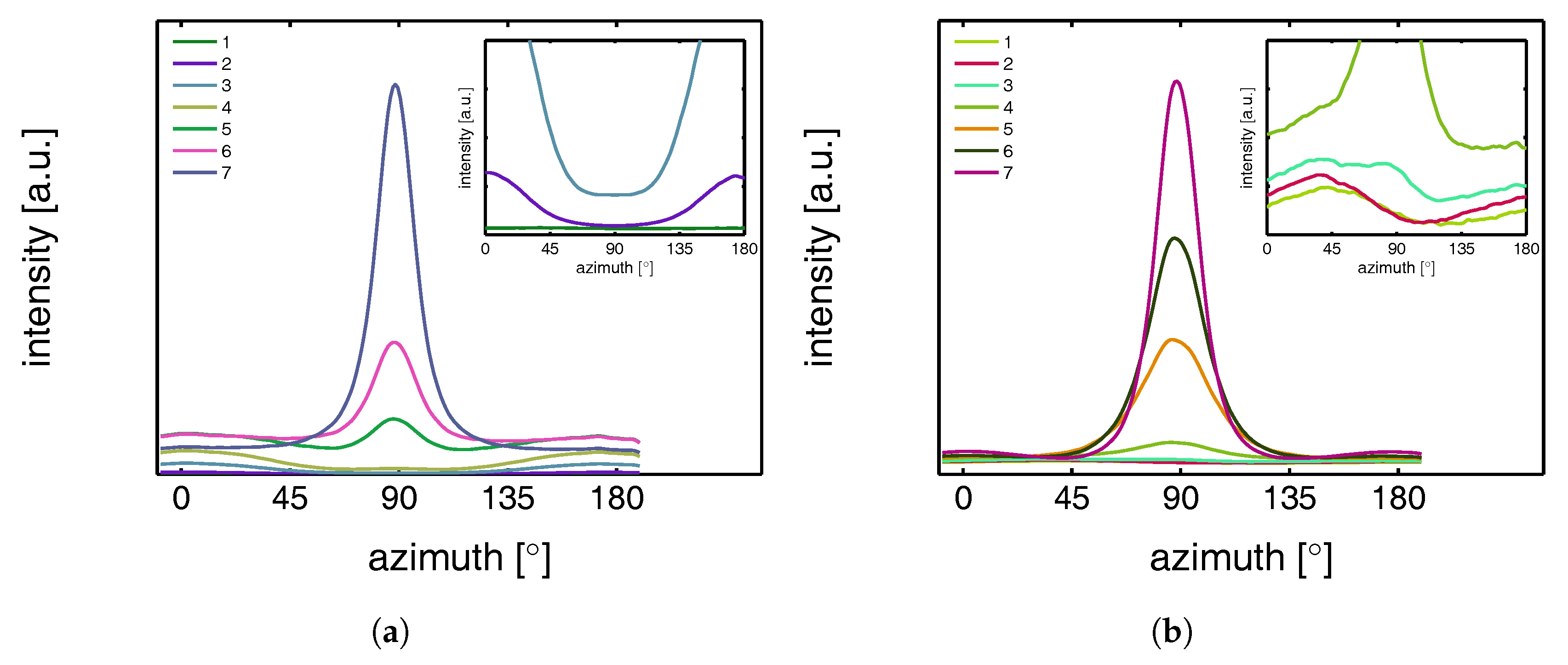
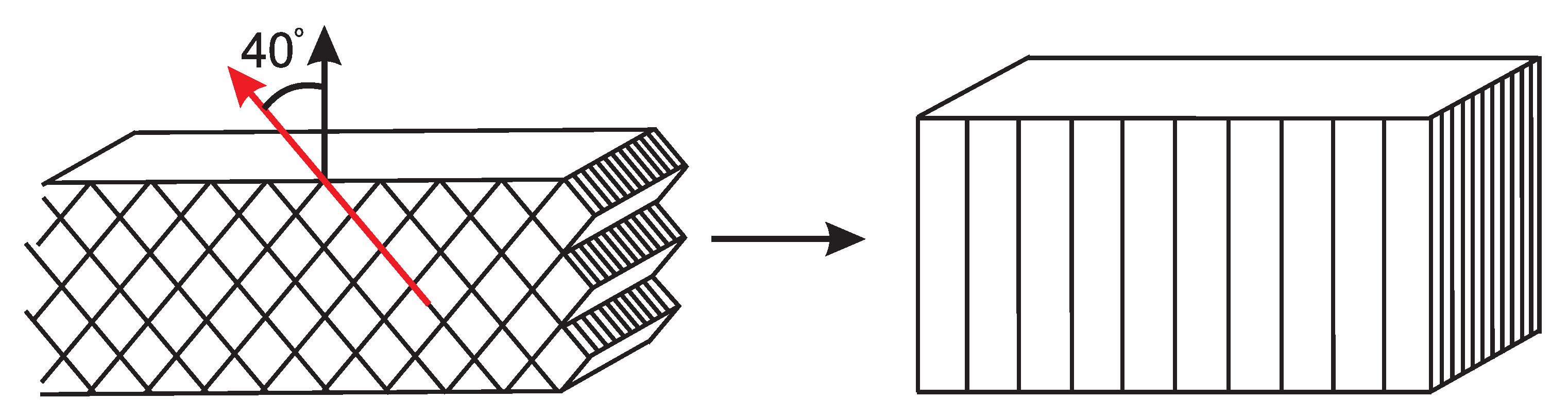
3.2.1. Tensile Tests (WAXD)
3.2.2. Compression Tests (WAXD)
3.3. SAXS Analysis
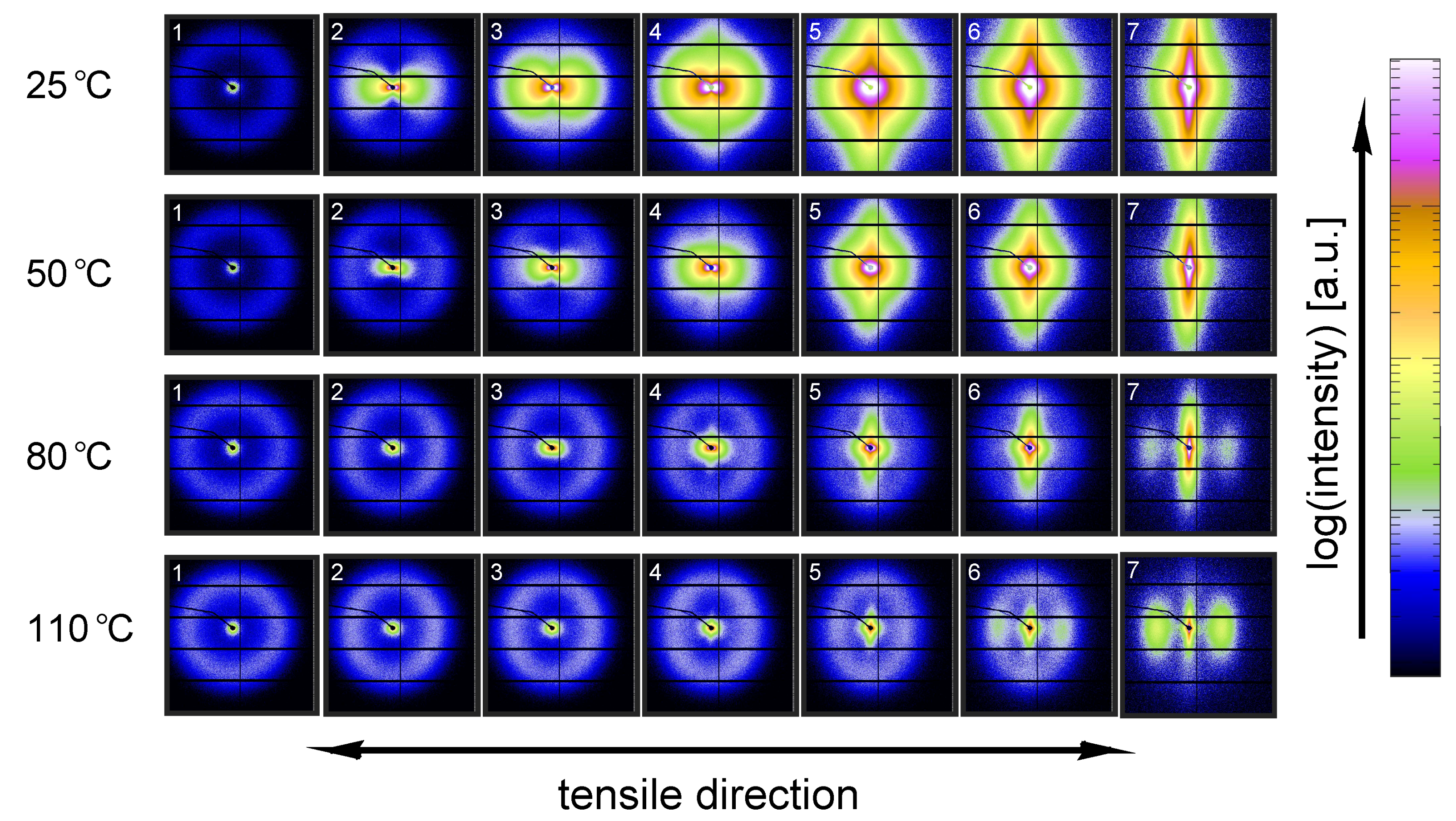
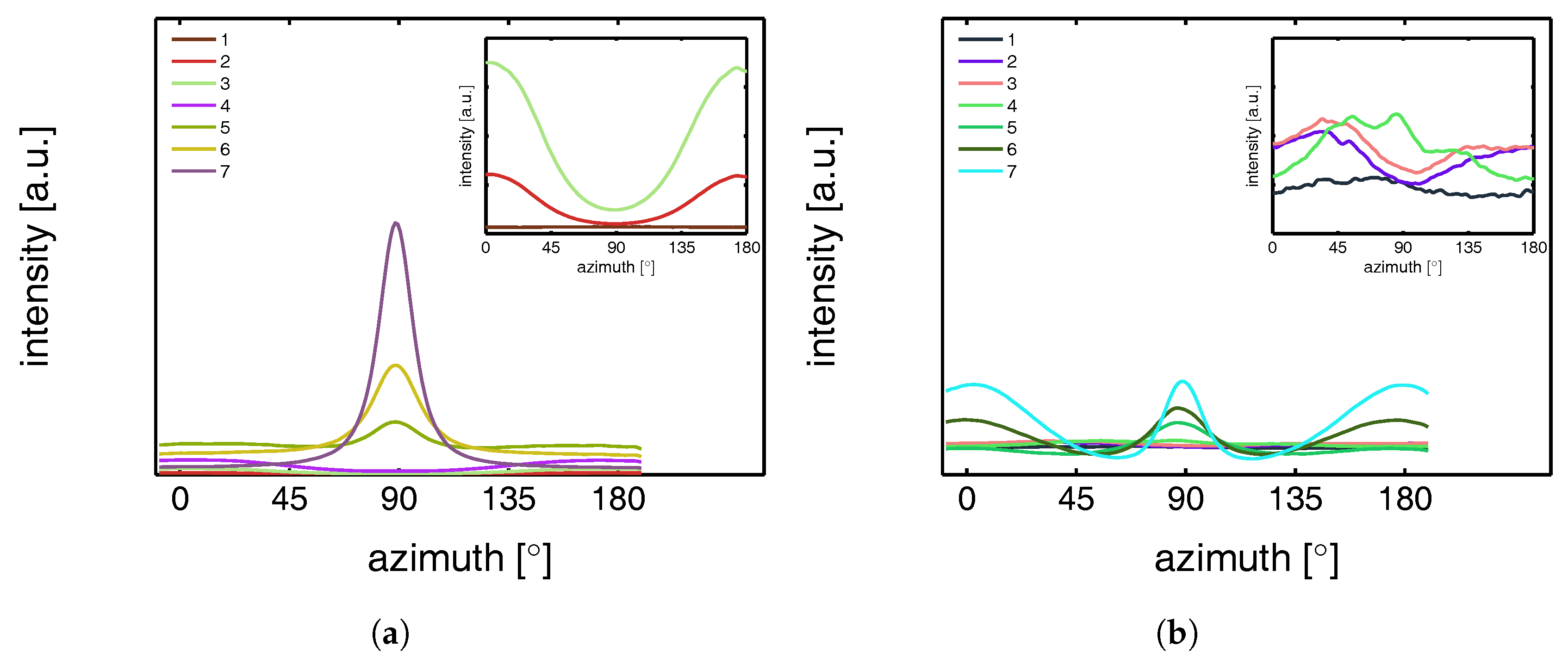
3.3.1. Tensile Tests (SAXS)
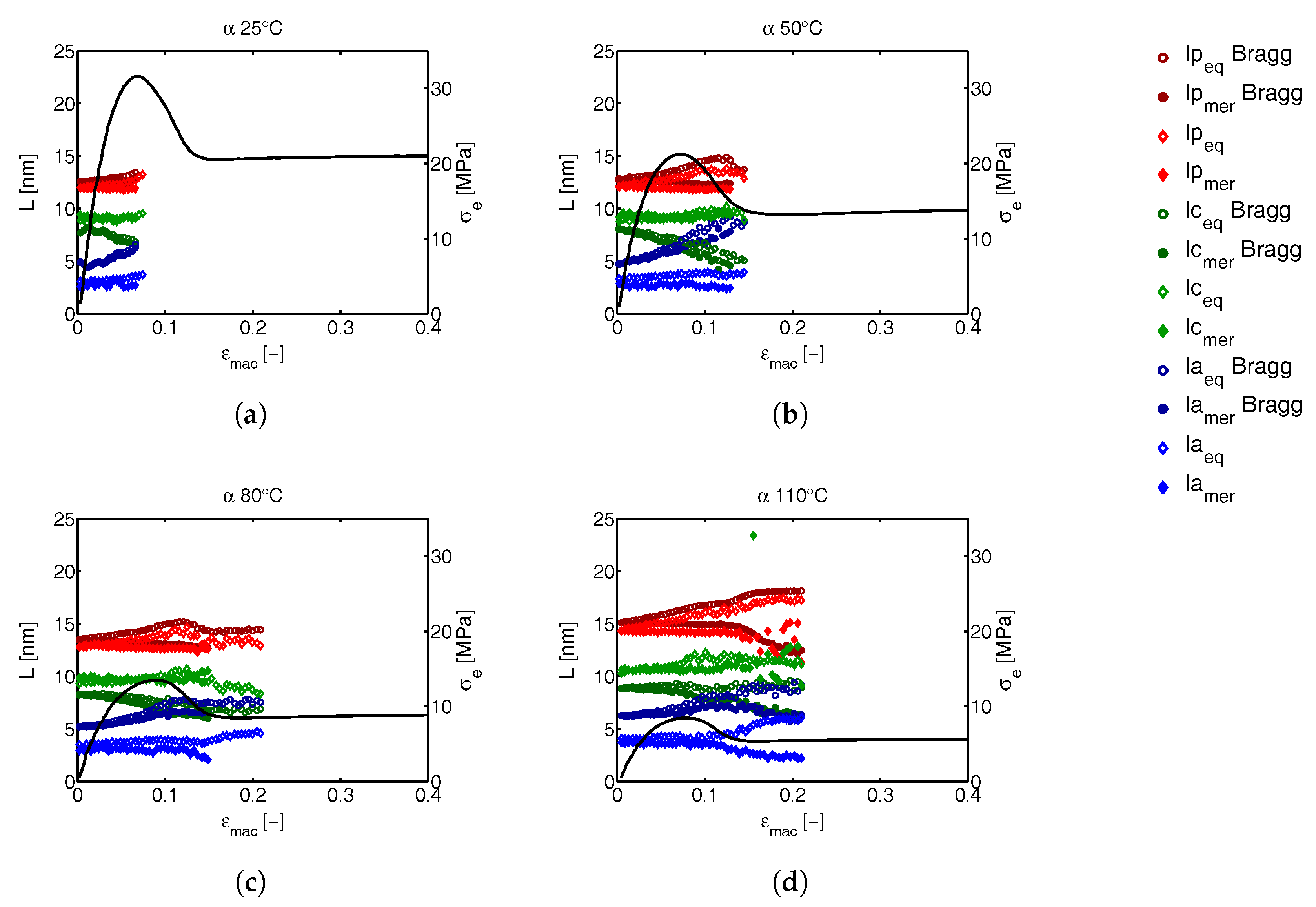
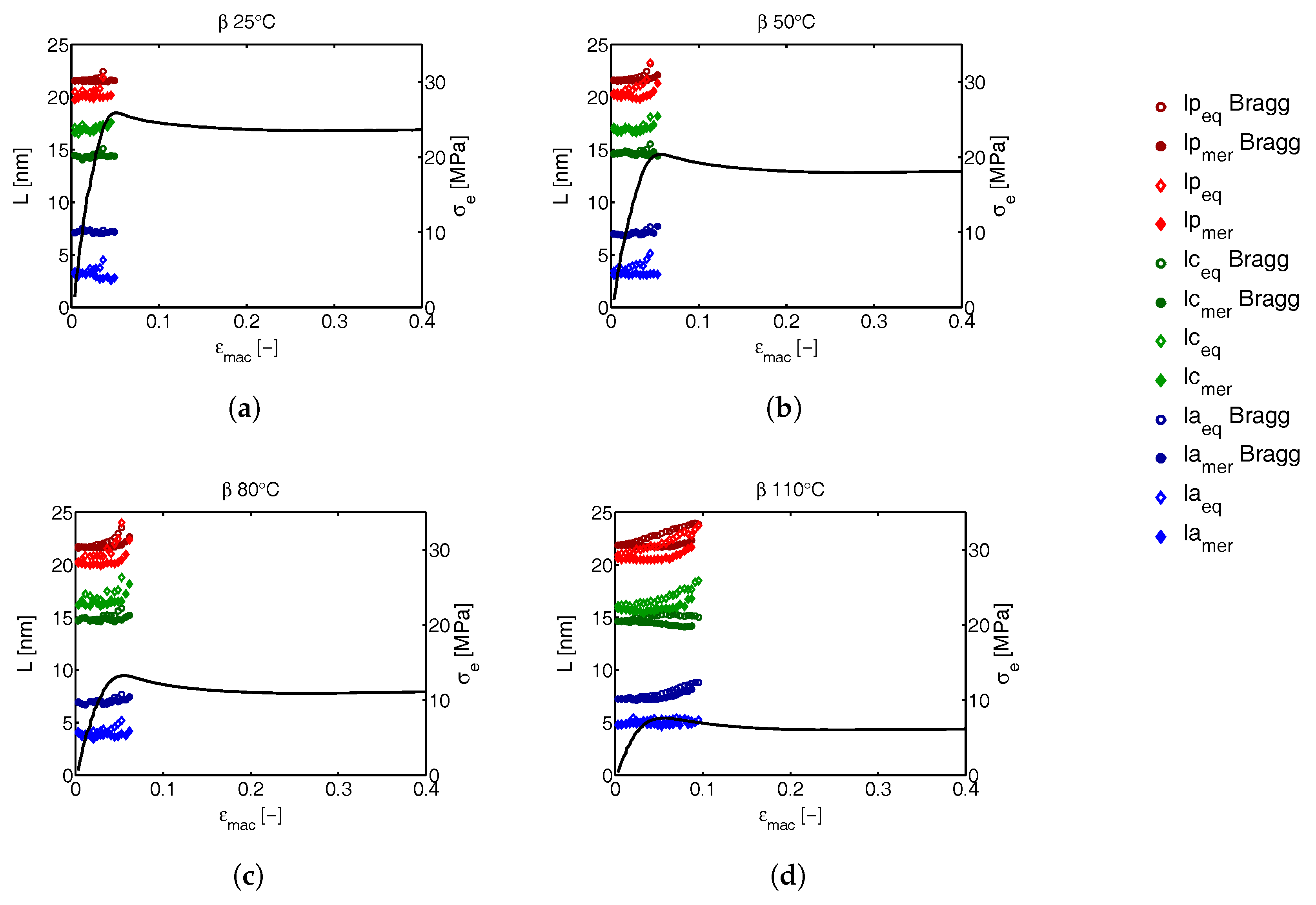
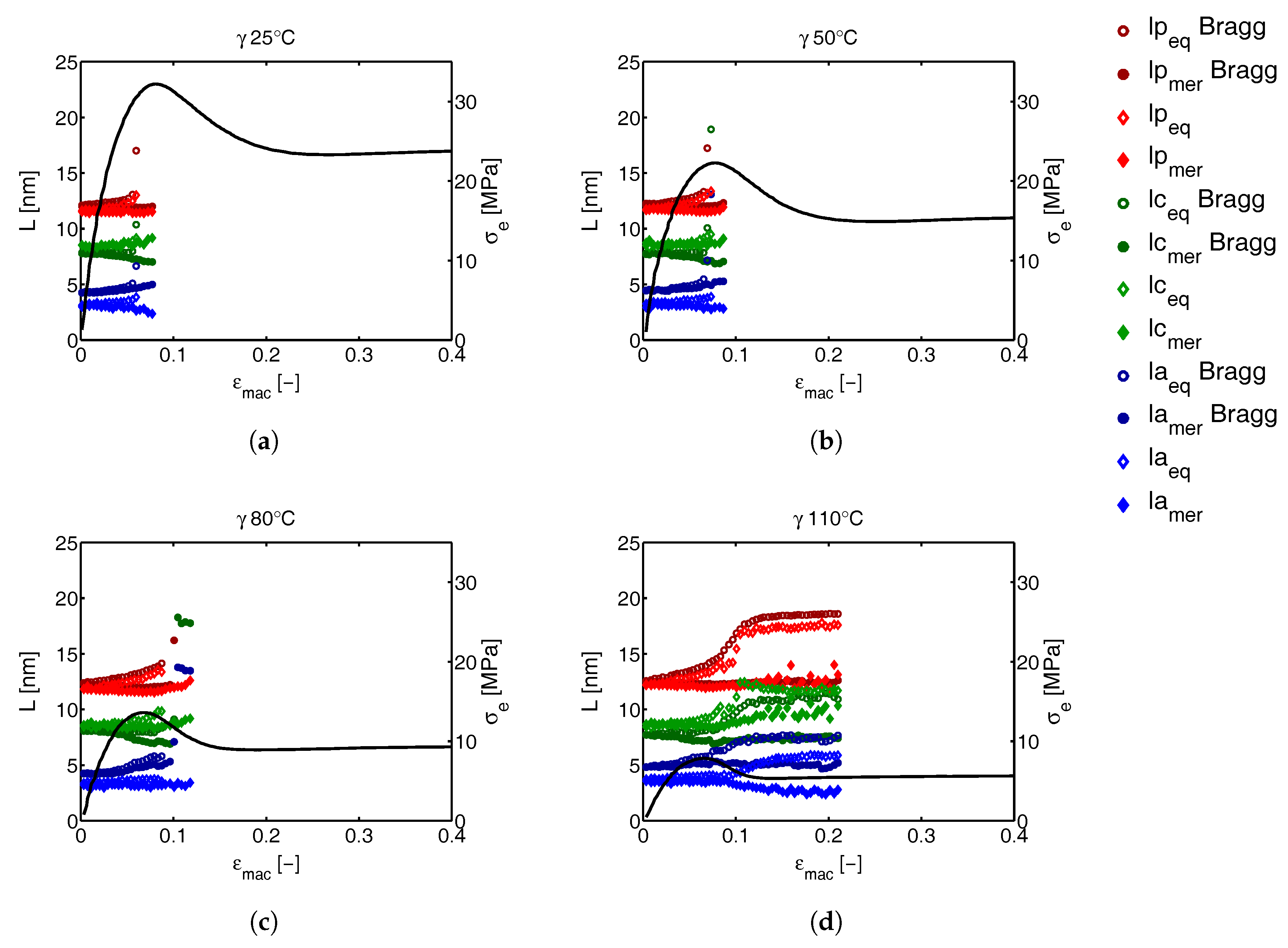
3.3.2. Lamellar Morphology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| iPP | isotactic polypropylene |
| 1D | one-dimensional |
| 2D | two-dimensional |
| SAXS | small angle X-ray scattering |
| WAXD | wide angle X-ray diffraction |
Appendix A




References
- Brückner, S.; Meille, S.; Petraccone, V.; Pirozzi, B. Polymorphism in isotactic polypropylene. Prog. Polym. Sci. 1991, 16, 361–404. [Google Scholar] [CrossRef]
- Lotz, B.; Wittmann, J.; Lovinger, A. Structure and morphology of poly(propylenes): A molecular analysis. Polymer 1996, 37, 4979–4992. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Di Capua, A.; Resconi, L.; Guidotti, S.; Camurati, I.; Nifant’ev, I.; Laishevtsev, I. Structure-property correlations in polypropylene from metallocene catalysts: Stereodefective, regioregular isotactic polypropylene. J. Am. Chem. Soc. 2004, 126, 17040–17049. [Google Scholar] [CrossRef] [PubMed]
- Varga, J. β-modification of isotactic polypropylene: Preparation, structure, processing, properties, and application. J. Macromol. Sci. Phys. 2002, 41, 1121–1171. [Google Scholar] [CrossRef]
- Roozemond, P.; Van Erp, T.; Peters, G. Flow-induced crystallization of isotactic polypropylene: Modeling formation of multiple crystal phases and morphologies. Polymer 2016, 89, 69–80. [Google Scholar] [CrossRef]
- Meille, S.; Ferro, D.; Brückner, S.; Lovinger, A.; Padden, F. Structure of β-isotactic polypropylene: A long-standing structural puzzle. Macromolecules 1994, 27, 2615–2622. [Google Scholar] [CrossRef]
- Varga, J.; Karger-Kocsis, J. Rules of supermolecular structure formation in sheared isotactic polypropylene melts. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 657–670. [Google Scholar] [CrossRef]
- Varga, J.; Mudra, I.; Ehrenstein, G. Highly active thermally stable β-nucleating agents for isotactic polypropylene. J. Appl. Polym. Sci. 1999, 74, 2357–2368. [Google Scholar] [CrossRef]
- Valdo Meille, S.; Brückner, S. Non-parallel chains in crystalline γ-isotactic polypropylene. Nature 1989, 340, 455–457. [Google Scholar] [CrossRef]
- Mezghani, K.; Phillips, P. The γ-phase of high molecular weight isotactic polypropylene: III. The equilibrium melting point and the phase diagram. Polymer 1998, 39, 3735–3744. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Di Girolamo, R.; De Ballesteros, O.; Pepe, M.; Tarallo, O.; Malafronte, A. Morphology and mechanical properties of the mesomorphic form of isotactic polypropylene in stereodefective polypropylene. Macromolecules 2013, 46, 5202–5214. [Google Scholar] [CrossRef]
- Caelers, H.; Parodi, E.; Cavallo, D.; Peters, G.; Govaert, L. Deformation and failure kinetics of iPP polymorphs. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 729–747. [Google Scholar] [CrossRef]
- Ma, Z.; Shao, C.; Wang, X.; Zhao, B.; Li, X.; An, H.; Yan, T.; Li, Z.; Li, L. Critical stress for drawing-induced α crystal-mesophase transition in isotactic polypropylene. Polymer 2009, 50, 2706–2715. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C. Stretching isotactic polypropylene: From “cross-β” to crosshatches, from γ form to α form. Macromolecules 2006, 39, 7635–7647. [Google Scholar] [CrossRef]
- Zuo, F.; Keum, J.; Chen, X.; Hsiao, B.; Chen, H.; Lai, S.Y.; Wevers, R.; Li, J. The role of interlamellar chain entanglement in deformation-induced structure changes during uniaxial stretching of isotactic polypropylene. Polymer 2007, 48, 6867–6880. [Google Scholar] [CrossRef]
- Xiong, B.; Lame, O.; Chenal, J.M.; Rochas, C.; Seguela, R.; Vigier, G. In-situ SAXS study of the mesoscale deformation of polyethylene in the pre-yield strain domain: Influence of microstructure and temperature. Polymer 2014, 55, 1223–1227. [Google Scholar] [CrossRef]
- Pawlak, A. Plastic Deformation and Cavitation in Semi-Crystalline Polymers; American Institute of Physics Inc.: Ischia, Italy, 2014; Volume 1599, pp. 118–121. [Google Scholar]
- Ran, S.; Zong, X.; Fang, D.; Hsiao, B.; Chu, B.; Phillips, R. Structural and morphological studies of isotactic polypropylene fibers during heat/draw deformation by in situ synchrotron SAXS/WAXD. Macromolecules 2001, 34, 2569–2578. [Google Scholar] [CrossRef]
- Na, B.; Lv, R.; Xu, W. Effect of network relaxation on void propagation and failure in isotactic polypropylene at large strain. J. Appl. Polym. Sci. 2009, 113, 4092–4099. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Chen, R.; Zhao, J.; Jiang, Z.; Men, Y. Cavitation in Isotactic Polypropylene at Large Strains during Tensile Deformation at Elevated Temperatures. Macromolecules 2015, 48, 5799–5806. [Google Scholar] [CrossRef]
- Lezak, E.; Bartczak, Z.; Galeski, A. Plastic deformation behavior of β-phase isotactic polypropylene in plane-strain compression at room temperature. Polymer 2006, 47, 8562–8574. [Google Scholar] [CrossRef]
- Lezak, E.; Bartczak, Z. Plastic deformation behavior of β-isotactic phase isotactic polypropylene in plane-strain compression at elevated temperatures. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 92–108. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, G.; Song, Y.; Zhao, Y.; Wang, D. Structural evolution of β-iPP during uniaxial stretching studied by in situ WAXS and SAXS. Polymer 2015, 55, 6915–6923. [Google Scholar] [CrossRef]
- Jia, C.; Liao, X.; Zhu, J.; An, Z.; Zhang, Q.; Yang, Q.; Li, G. Creep-resistant behavior of β-polypropylene with different crystalline morphologies. RSC Adv. 2016, 6, 30986–30997. [Google Scholar] [CrossRef]
- Lezak, E.; Bartczak, Z.; Galeski, A. Plastic deformation of the γ phase in isotactic polypropylene in plane-strain compression. Macromolecules 2006, 39, 4811–4819. [Google Scholar] [CrossRef]
- Humbert, S.; Lame, O.; Chenal, J.; Rochas, C.; Vigier, G. New insight on initiation of cavitation in semicrystalline polymers: In-situ SAXS measurements. Macromolecules 2010, 43, 7212–7221. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Z.; Fu, L.; Lu, Y.; Men, Y. Lamellar thickness and stretching temperature dependency of cavitation in semicrystalline polymers. PLoS ONE 2014, 9, e97234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Lame, O.; Chenal, J.M.; Rochas, C.; Seguela, R.; Vigier, G. Temperature-Microstructure Mapping of the Initiation of the Plastic Deformation Processes in Polyethylene via In Situ WAXS and SAXS. Macromolecules 2015, 48, 5267–5275. [Google Scholar] [CrossRef]
- Xiong, B.; Lame, O.; Chenal, J.; Rochas, C.; Seguela, R.; Vigier, G. In-situ SAXS study and modeling of the cavitation/crystal-shear competition in semi-crystalline polymers: Influence of temperature and microstructure in polyethylene. Polymer 2013, 54, 5408–5418. [Google Scholar] [CrossRef]
- Xiong, B.; Lame, O.; Chenal, J.M.; Rochas, C.; Seguela, R.; Vigier, G. Amorphous phase modulus and micro-macro scale relationship in polyethylene via in situ SAXS and WAXS. Macromolecules 2015, 48, 2149–2160. [Google Scholar] [CrossRef]
- Humbert, S.; Lame, O.; Vigier, G. Polyethylene yielding behaviour: What is behind the correlation between yield stress and crystallinity? Polymer 2009, 50, 3755–3761. [Google Scholar] [CrossRef]
- Schrauwen, B.; Janssen, R.; Govaert, L.; Meijer, H. Intrinsic deformation behavior of semicrystalline polymers. Macromolecules 2004, 37, 6069–6078. [Google Scholar] [CrossRef]
- Men, Y.; Rieger, J.; Strobl, G. Role of the Entangled Amorphous Network in Tensile Deformation of Semicrystalline Polymers. Phys. Rev. Lett. 2003, 91, 955021–955024. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Ushui, K.; Kondo, Y.; Hatada, K.; Gima, S. Effect of tie molecules on the craze strength of polypropylene. Polymer 1996, 37, 5375–5379. [Google Scholar] [CrossRef]
- Aboulfaraj, M.; G’Sell, C.; Ulrich, B.; Dahoun, A. In situ observation of the plastic deformation of polypropylene spherulites under uniaxial tension and simple shear in the scanning electron microscope. Polymer 1995, 36, 731–742. [Google Scholar] [CrossRef]
- Labour, T.; Gauthier, C.; Séguéla, R.; Vigier, G.; Bomal, Y.; Orange, G. Influence of the β crystalline phase on the mechanical properties of unfilled and CaCO3-filled polypropylene. I. Structural and mechanical characterisation. Polymer 2001, 42, 7127–7135. [Google Scholar] [CrossRef]
- Labour, T.; Vigier, G.; Séguéla, R.; Gauthier, C.; Orange, G.; Bomal, Y. Influence of the β-crystalline phase on the mechanical properties of unfilled and calcium carbonate-filled polypropylene: Ductile cracking and impact behavior. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 31–42. [Google Scholar] [CrossRef]
- Van Drongelen, M.; Van Erp, T.; Peters, G. Quantification of non-isothermal, multi-phase crystallization of isotactic polypropylene: The influence of cooling rate and pressure. Polymer 2012, 53, 4758–4769. [Google Scholar] [CrossRef]
- Haward, R. Strain hardening of thermoplastics. Macromolecules 1993, 26, 5860–5869. [Google Scholar] [CrossRef]
- Van Melick, H.; Govaert, L.; Meijer, H. Localisation phenomena in glassy polymers: Influence of thermal and mechanical history. Polymer 2003, 44, 3579–3591. [Google Scholar] [CrossRef]
- Bras, W.; Dolbnya, I.; Detollenaere, D.; Van Tol, R.; Malfois, M.; Greaves, G.; Ryan, A.; Heeley, E. Recent experiments on a combined small-angle/wide-angle X-ray scattering beam line at the ESRF. J. Appl. Crystallogr. 2003, 36, 791–794. [Google Scholar] [CrossRef]
- Zhang, H.; Scholz, A.; De Crevoisier, J.; Vion-Loisel, F.; Besnard, G.; Hexemer, A.; Brown, H.; Kramer, E.; Creton, C. Nanocavitation in carbon black filled styrene-butadiene rubber under tension detected by real time small angle X-ray scattering. Macromolecules 2012, 45, 1529–1543. [Google Scholar] [CrossRef]
- Vonk, C.; Kortleve, G. X-ray small-angle scattering of bulk polyethylene-II. Analyses of the scattering curve. Kolloid Z. Z. Polym. 1967, 220, 19–24. [Google Scholar] [CrossRef]
- Ruland, W. The evaluation of the small-angle scattering of lamellar two-phase systems by means of interface distribution functions. Colloid Polym. Sci. Kolloid Z. Z. Polym. 1977, 255, 417–427. [Google Scholar] [CrossRef]
- Debye, P.; Bueche, A. Scattering by an inhomogeneous solid. J. Appl. Phys. 1949, 20, 518–525. [Google Scholar] [CrossRef]
- Porod, G. Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen-I. Teil. Kolloid Z. 1951, 124, 83–114. [Google Scholar] [CrossRef]
- Koberstein, J.; Morra, B.; Stein, R. The determination of diffuse-boundary thicknesses of polymers by small-angle X-ray scattering. J. Appl. Crystallogr. 1980, 13, 34–45. [Google Scholar] [CrossRef]
- Jansen, B.; Rastogi, S.; Meijer, H.; Lemstra, P. Rubber-modified glassy amorphous polymers prepared via chemically induced phase separation. 2. Mode of microscopic deformation studied by in situ small-angle X-ray scattering during tensile deformation. Macromolecules 2001, 34, 4007–4018. [Google Scholar] [CrossRef]
- G’Sell, C.; Jonas, J. Yield and transient effects during the plastic deformation of solid polymers. J. Mater. Sci. 1981, 16, 1956–1974. [Google Scholar] [CrossRef]
- Smit, R.; Brekelmans, W.; Meijer, H. Prediction of the large-strain mechanical response of heterogeneous polymer systems: Local and global deformation behaviour of a representative volume element of voided polycarbonate. J. Mech. Phys. Solids 1999, 47, 201–221. [Google Scholar] [CrossRef]
- Caelers, H.; Govaert, L.; Peters, G. The prediction of mechanical performance of isotactic polypropylene on the basis of processing conditions. Polymer 2016, 83, 116–128. [Google Scholar] [CrossRef]
- Meijer, H.; Govaert, L. Mechanical performance of polymer systems: The relation between structure and properties. Prog. Polym. Sci. 2005, 30, 915–938. [Google Scholar] [CrossRef]
- Mezghani, K.; Phillips, P. The γ-phase of high molecular weight isotactic polypropylene. II: The morphology of the γ-form crystallized at 200 MPa. Polymer 1997, 38, 5725–5733. [Google Scholar] [CrossRef]
- Yamada, K.; Matsumoto, S.; Tagashira, K.; Hikosaka, M. Isotacticity dependence of spherulitic morphology of isotactic polypropylene. Polymer 1998, 39, 5327–5333. [Google Scholar] [CrossRef]
- Von Compostella, M.; Coen, A.; Bertinotti, F. Fasern und Filme aus isotaktischem Polypropylen. Angew. Chem. 1962, 74, 618–624. [Google Scholar] [CrossRef]
- Lotz, B.; Wittmann, J. The molecular origin of lamellar branching in the α (monoclinic) form of isotactic polypropylene. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 1541–1558. [Google Scholar] [CrossRef]
- Xu, W.; Martin, D.; Arruda, E. Finite strain response, microstructural evolution and β → α phase transformation of crystalline isotactic polypropylene. Polymer 2005, 46, 455–470. [Google Scholar] [CrossRef]
- Lezak, E.; Bartczak, Z. Plastic deformation of the γ phase isotactic polypropylene in plane-strain compression at elevated temperatures. Macromolecules 2007, 40, 4933–4941. [Google Scholar] [CrossRef]
- Plummer, C.; Kausch, H.H. Micronecking in thin films of isotactic polypropylene. Macromol. Chem. Phys. 1996, 197, 2047–2063. [Google Scholar] [CrossRef]
- Butler, M.; Donald, A.; Ryan, A. Time resolved simultaneous small- and wide-angle X-ray scattering during polyethylene deformation-II. Cold drawing of linear polyethylene. Polymer 1998, 39, 39–52. [Google Scholar] [CrossRef]
- Pawlak, A. Cavitation during tensile deformation of high-density polyethylene. Polymer 2007, 48, 1397–1409. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A. Plastic deformation of crystalline polymers: The role of cavitation and crystal plasticity. Macromolecules 2005, 38, 9688–9697. [Google Scholar] [CrossRef]
- Men, Y.; Rieger, J.; Homeyer, J. Synchrotron ultrasmall-angle X-ray scattering studies on tensile deformation of poly(1-butene). Macromolecules 2004, 37, 9481–9488. [Google Scholar] [CrossRef]
- Lode, U.; Pomper, T.; Karl, A.; Von Krosigk, G.; Cunis, S.; Wilke, W.; Gehrke, R. Development of crazes in polycarbonate, investigated by ultra small angle X-ray scattering of synchrotron radiation. Macromol. Rapid Commun. 1998, 19, 35–39. [Google Scholar] [CrossRef]
- Zafeiropoulos, N.; Davies, R.; Roth, S.; Burghammer, M.; Schneider, K.; Riekel, C.; Stamm, M. Microfocus X-ray scattering scanning microscopy for polymer applications. Macromol. Rapid Commun. 2005, 26, 1547–1551. [Google Scholar] [CrossRef]
- Ruland, W. Small- angle scattering studies on carbonized cellulose fibers. J. Polym. Sci. Polym. Symp. 1969, 28, 143–151. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (kg/m) | 946 | 921 | 938 | 880 | 850 |
| 25 °C | 50 °C | 80 °C | 110 °C | |
|---|---|---|---|---|
| of -iPP (MPa) | 32.0 | 22.3 | 12.6 | 8.4 |
| of -iPP (MPa) | 28.5 | 20.4 | 13.2 | 7.9 |
| of -iPP (MPa) | 32.3 | 22.8 | 13.6 | 7.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caelers, H.J.M.; Troisi, E.M.; Govaert, L.E.; Peters, G.W.M. Deformation-Induced Phase Transitions in iPP Polymorphs. Polymers 2017, 9, 547. https://doi.org/10.3390/polym9100547
Caelers HJM, Troisi EM, Govaert LE, Peters GWM. Deformation-Induced Phase Transitions in iPP Polymorphs. Polymers. 2017; 9(10):547. https://doi.org/10.3390/polym9100547
Chicago/Turabian StyleCaelers, Harm J. M., Enrico M. Troisi, Leon E. Govaert, and Gerrit W. M. Peters. 2017. "Deformation-Induced Phase Transitions in iPP Polymorphs" Polymers 9, no. 10: 547. https://doi.org/10.3390/polym9100547
APA StyleCaelers, H. J. M., Troisi, E. M., Govaert, L. E., & Peters, G. W. M. (2017). Deformation-Induced Phase Transitions in iPP Polymorphs. Polymers, 9(10), 547. https://doi.org/10.3390/polym9100547