Propene Polymerization with C1-Symmetric Fluorenyl-Metallocene Catalysts

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
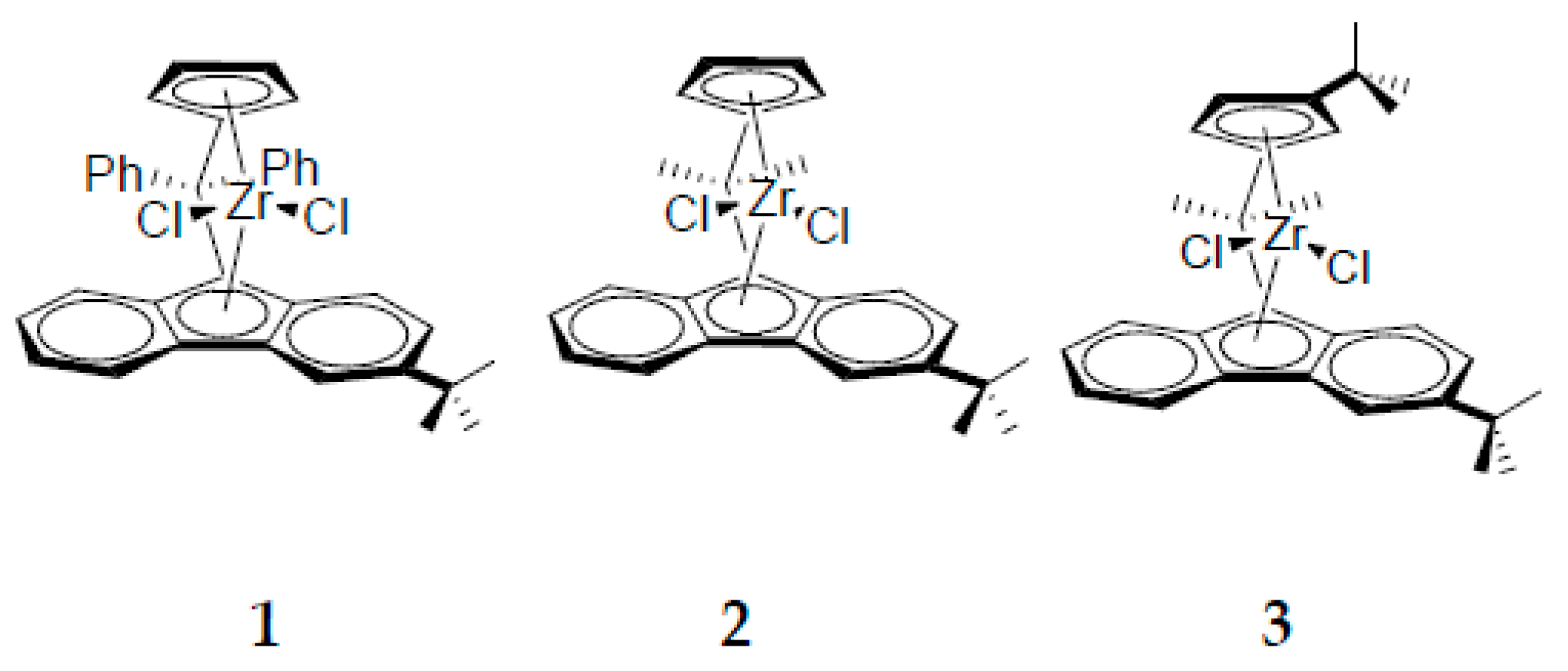
3. Results and Discussion
3.1. Polymer Synthesis and Analysis
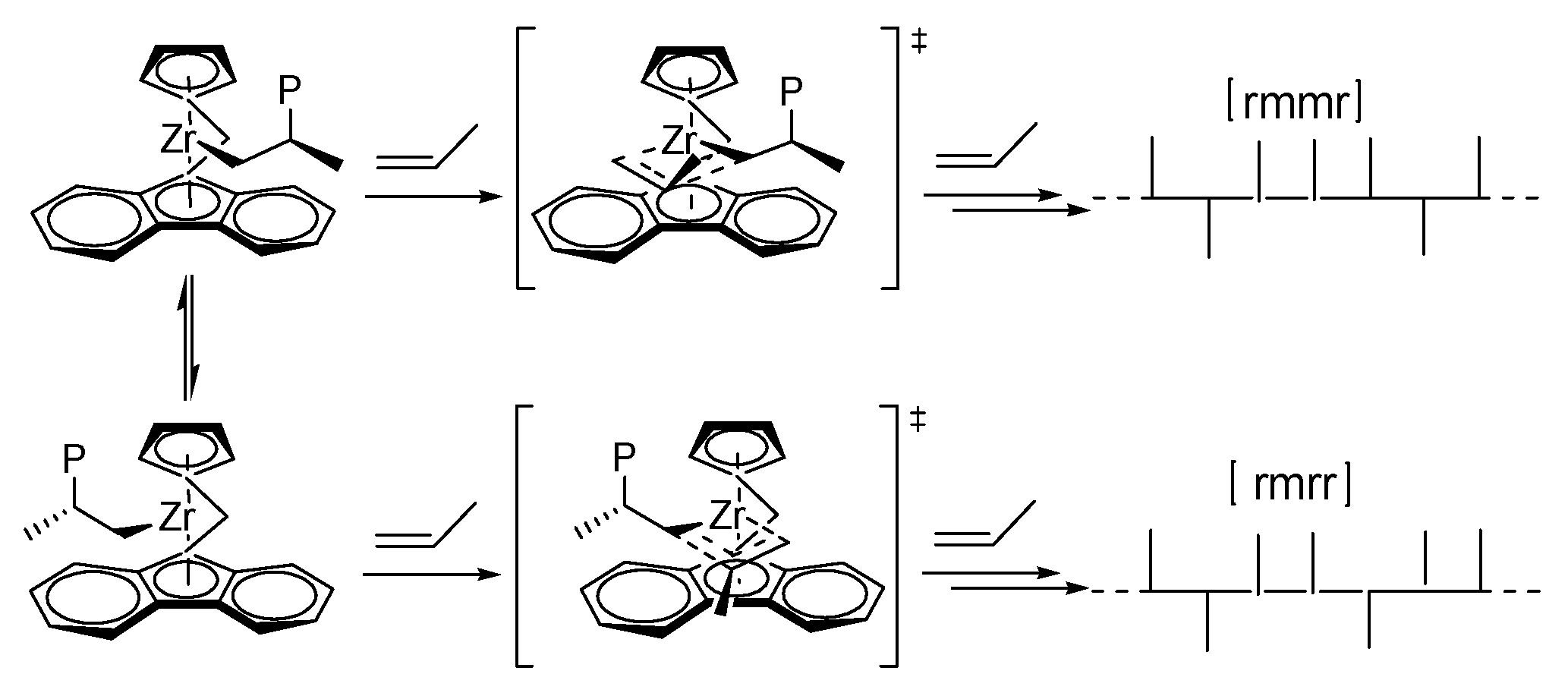
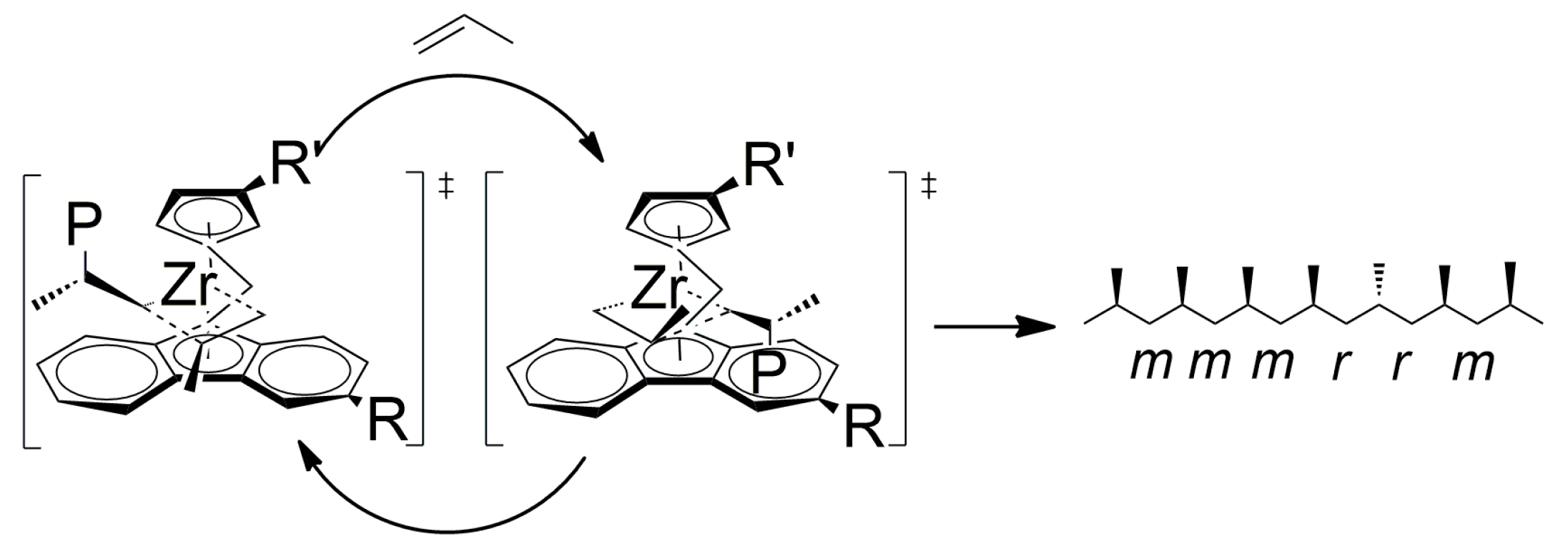
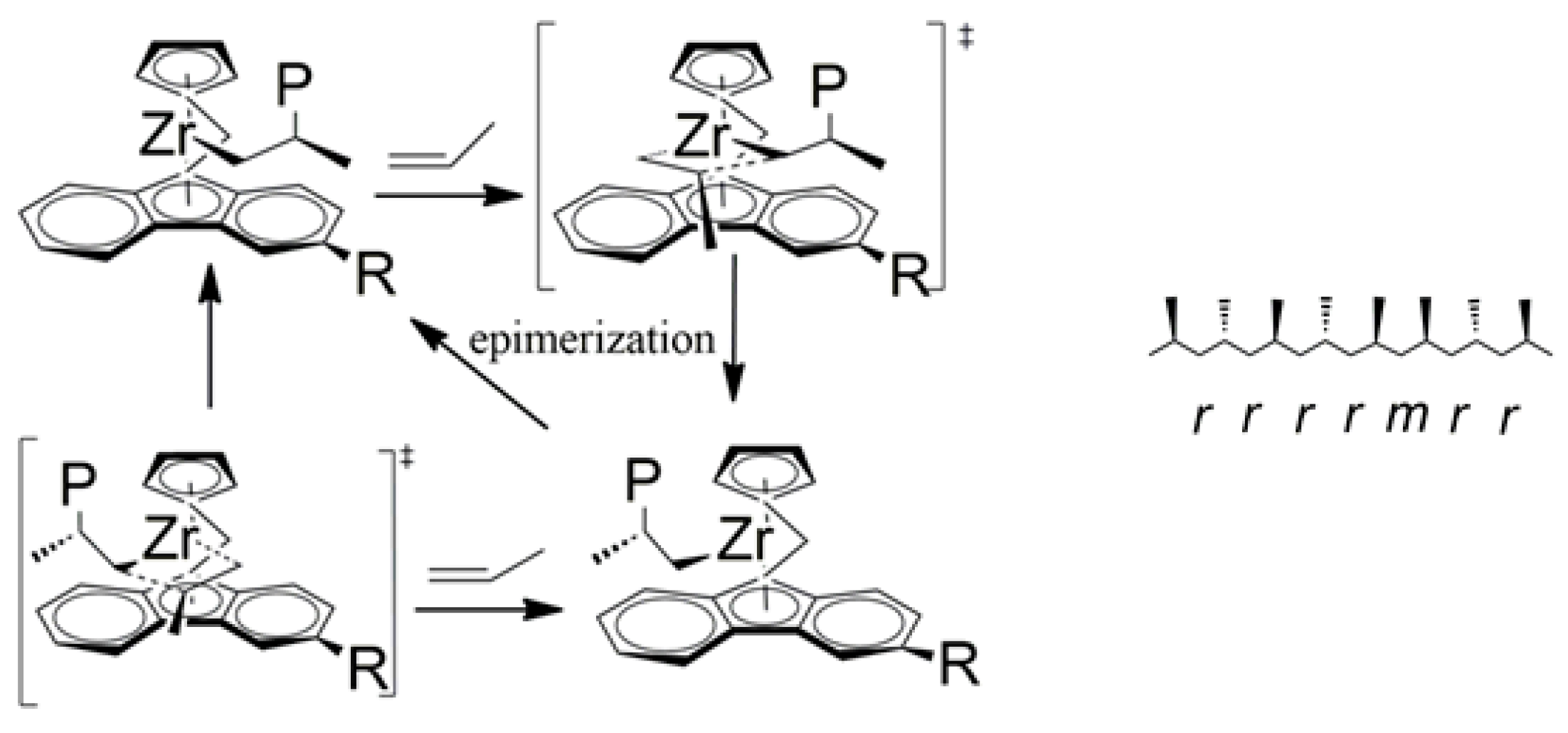
3.2. Polypropene Microstructure
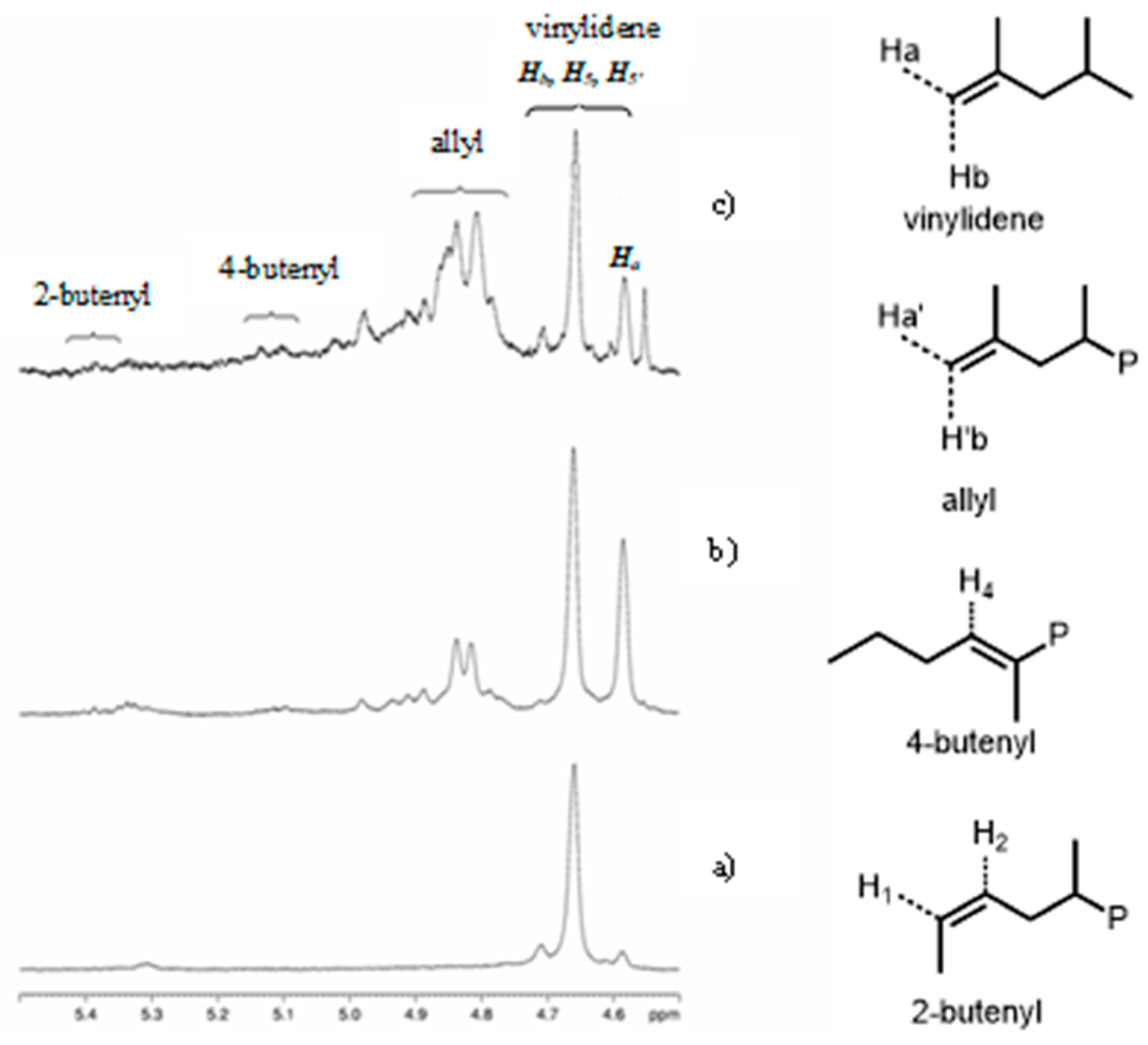
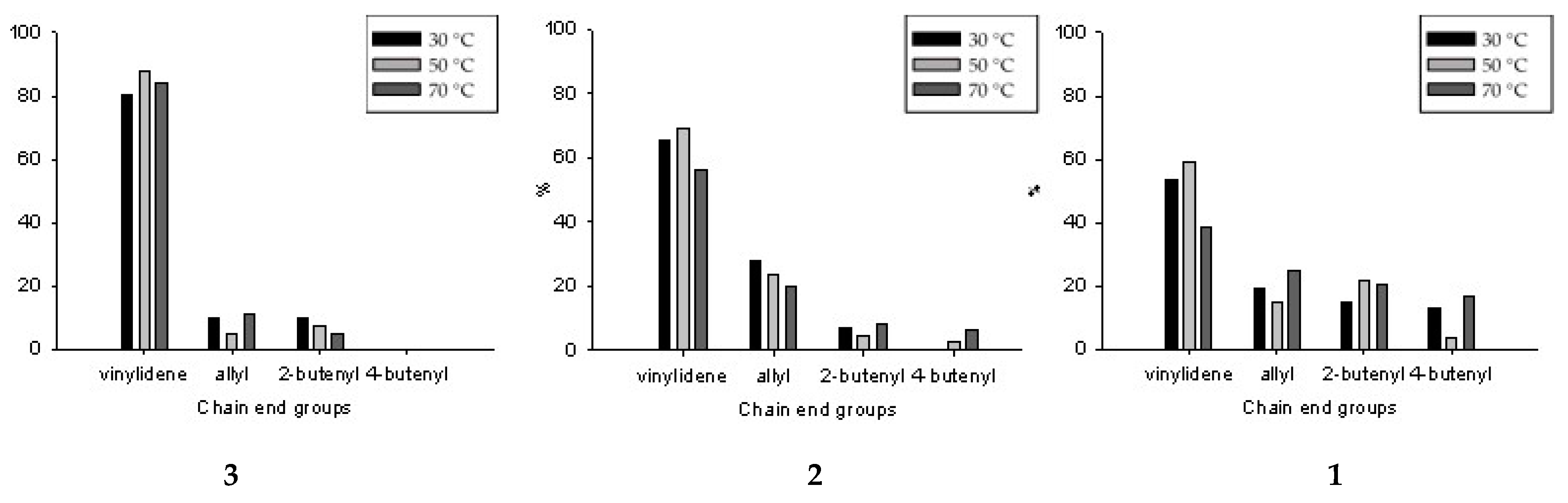
3.3. 1H NMR Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R. Stereospecific olefin polymerization with chiral metallocene catalysts. Angew. Chem. Int. Ed. Engl. 1995, 34, 1143–1170. [Google Scholar] [CrossRef]
- Resconi, L.; Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in propene polymerization with metallocene catalysts. Chem. Rev. 2000, 100, 1253–1345. [Google Scholar] [CrossRef] [PubMed]
- Razavi, A. Syndiotactic Polypropylene: Discovery, Development, and Industrialization via bridged Metallocene Catalysts. Adv. Polym. Sci. 2013, 258, 43–116. [Google Scholar] [CrossRef]
- Ewen, J.A.; Jones, R.L.; Razavi, A.; Ferrara, J.D. Syndiospecific propylene polymerizations with group-4 metallocenes. J. Am. Chem. Soc. 1988, 110, 6255–6256. [Google Scholar] [CrossRef] [PubMed]
- Spaleck, W.; Antberg, M.; Dolle, V.; Klein, R.; Rohrmann, J.; Winter, A. Stereorigid metallocenes: Correlations between structure and behavior in homopolymerizations of propylene. New J. Chem. 1990, 14, 499–503. [Google Scholar]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Haspeslagh, L.; Atwood, J.; Bott, S.G.; Robinson, K. Metallocene polypropylene structural relationships-implications on polymerization and stereochemical control mechanisms. Makromol. Chem. Rapid. Commun. 1991, 48/49, 253–295. [Google Scholar] [CrossRef]
- Razavi, A.; Atwood, J.L. Preparation and crystal-structures of the complexes (eta(5)-C(5)H(3)Me-CMe(2)-eta(5)-C13H8)MCl(2) (M=Zr or Hf)—Mechanistic aspects of the catalytic formation of a syndiotactic-isotactic stereoblock-type polypropylene. J. Organomet. Chem. 1995, 497, 105–111. [Google Scholar] [CrossRef]
- Razavi, A.; Atwood, J.L. Preparation and crystal-structures of the complexes (eta(5)-C5H4CPh2-eta(5)-C13H8)MCl2 (M = Zr, Hf) and the catalytic formation of high molecular weight high tacticity syndiotactic polypropylene. J. Organomet. Chem. 1996, 520, 115–120. [Google Scholar] [CrossRef]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Curtis, S.; Cheng, H.N. Syndiospecific Propylene Polymerization with iPr[CpFlu]ZrCl2. In Catalytic Olefin Polymerization, Studies in Surface Science and Catalysis; Keii, T., Soga, K., Eds.; Elsewier: New York, NY, USA, 1990; pp. 439–482. [Google Scholar]
- Farina, M.; Terragni, A. On the syndiotactic polymerization mechanism using metallocene catalysts. Makromol. Chem. Rapid Commun. 1993, 14, 791–798. [Google Scholar] [CrossRef]
- Razavi, A.; Peters, L.; Nafpliotis, L.; Vereecke, D.; Den Dauw, K. The geometry of the site and its relevance for chain migration and stereospecificity. Macromol. Symp. 1995, 89, 345–367. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Talarico, G.; Segre, A.L.; Caporaso, L. High-field C-13 NMR characterization of ethene-1-C-13/propene copolymers prepared with C-s-symmetric ansa-metallocene catalysts: A deeper insight into the regio- and stereoselectivity of syndiotactic propene polymerization. Macromolecules 1998, 31, 8720–8724. [Google Scholar] [CrossRef]
- Veghini, D.; Henling, L.M.; Burkhardt, T.J.; Bercaw, J.E. Mechanisms of stereocontrol for doubly silylene-bridged C-s- and C-1-symmetric zirconocene catalysts for propylene polymerization. Synthesis and molecular structure of Li-2[(1,2-Me2Si)(2){C5H2-4-(1R,2S,5R-menthyl)}{C5H-3,5-(CHMe2)(2)}].3THF and [(1,2-Me2Si)(2){eta(5)-C5H2-4-(1R,2S,5R-menthyl)}{eta(5)-C5H-3,5-(CHMe2)(2)}]ZrCl2. J. Am. Chem. Soc. 1999, 121, 564–573. [Google Scholar] [CrossRef]
- Yoder, J.C.; Bercaw, J.E. Chain epimerization during propylene polymerization with metallocene catalysts: Mechanistic studies using a doubly labeled molecular. J. Am. Chem. Soc. 2002, 124, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Roberts, J.A.S.; Marks, T.J. Marked counteranion effects on single-site olefin polymerization processes. Correlations of ion pair structure and dynamics with polymerization activity, chain transfer, and syndioselectivity. J. Am. Chem. Soc. 2004, 126, 4605–4625. [Google Scholar] [CrossRef] [PubMed]
- Cossee, P. On the reaction mechanism of the ethylene polymerization with heterogeneous Ziegler-Natta catalysts. Tetrahedron Lett. 1960, 38, 12–16. [Google Scholar] [CrossRef]
- Cossee, P. The formation of isotactic polypropylene under the influence of Ziegler-Natta catalysts. Tetrahedron Lett. 1960, 38, 17–21. [Google Scholar] [CrossRef]
- Angermund, K.; Fink, G.; Jensen, V.R.; Kleinschmidt, R. The role of intermediate chain migration in propene polymerization using substituted {Pr-i(CpFlu)}ZrCl2/MAO catalysts. Macromol. Rapid Commun. 2000, 21, 91–97. [Google Scholar] [CrossRef]
- Farina, M.; Di Silvestro, G.; Sozzani, P. Hemiisotactic polypropylene: A key point in the elucidation of the polymerization mechanism with metallocene catalysts. Macromolecules 1993, 26, 946–950. [Google Scholar] [CrossRef]
- Miller, S.A.; Bercaw, J.E. Mechanism of isotactic polypropylene formation with C-1-symmetric metallocene catalysts. Organometallics 2006, 25, 3576–3592. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Talarico, G.; Segre, A.L.; Chadwick, J.C. New evidence on the nature of the active sites in heterogeneous Ziegler-Natta catalysts for propene polymerization. Macromolecules 1997, 30, 4786–4790. [Google Scholar] [CrossRef]
- Leclerc, M.K.; Brintzinger, H.H. Zr-Alkyl isomerization in ansa-zirconocene-catalyzed olefin polymerizations. Contributions to stereoerror formation and chain termination. J. Am. Chem. Soc. 1996, 118, 9024–9032. [Google Scholar] [CrossRef]
- Busico, V.; Caporaso, L.; Cipullo, R.; Landriani, L.; Angelini, G.; Margonelli, A.; Segre, A.L. Propene polymerization promoted by C-2-symmetric metallocene catalysts: From atactic to isotactic polypropene in consequence of an isotope effect. J. Am. Chem. Soc. 1996, 118, 2105–2106. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Ferro, D.R. Alternating isotactic ethylene-norbornene copolymers by C-1-symmetric metallocenes: Determination of the copolymerization parameters and mechanistic considerations on the basis of pentad analysis. Macromolecules 2004, 37, 9681–9693. [Google Scholar] [CrossRef]
- Tomasi, S.; Razavi, A.; Ziegler, T. Stereoregularity, Regioselectivity, and Dormancy in Polymerizations Catalyzed by C-1-Symmetric Fluorenyl-Based Metallocenes. A Theoretical Study Based on Density Functional Theory. Organometallics 2009, 28, 2609–2618. [Google Scholar] [CrossRef]
- Razavi, A.; Bellia, V.; Baekelmans, D.; Slawinsky, M.; Sirol, S.; Peters, L.; Thewald, U. Chain “stationary” insertion mechanism and production of isotactic polypropylene with C-1 symmetric catalyst systems. Kinet. Catal. 2006, 47, 257–267. [Google Scholar] [CrossRef]
- Razavi, A.; Bellia, V.; De Brauwer, Y.; Hortmann, K.; Peters, L.; Sirol, S.; Van Belle, S.; Thewald, U. Syndiotactic- and isotactic specific bridged cyclopentadienyl-fluorenyl based metallocenes; Structural features, catalytic behavior. Macromol. Chem. Phys. 2004, 205, 347–356. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Zampa, C.; Ferro, D.R. Ethylene-norbornene copolymers by C-s-symmetric metallocenes: Determination of the copolymerization parameters and mechanistic considerations on the basis of tetrad analysis. Macromolecules 2005, 38, 9910–9919. [Google Scholar] [CrossRef]
- Carvill, A.; Zetta, L.; Zannoni, G.; Sacchi, M.C. Ansa-zirconocene-catalyzed solution polymerization of propene: Influence of polymerization conditions on the unsaturated chain-end groups. Macromolecules 1998, 31, 3783–3789. [Google Scholar] [CrossRef]
- Resconi, L.; Camurati, I.; Sudmeijer, O. Chain transfer reactions in propylene polymerization with zirconocene catalysts. Top. Catal. 1999, 7, 145–163. [Google Scholar] [CrossRef]
- Resconi, L. On the mechanisms of growing-chain-end isomerization and transfer reactions in propylene polymerization with isospecific, C-2-symmetric zirconocene catalysts. J. Mol. Catal. A 1999, 146, 167. [Google Scholar] [CrossRef]
- Kawahara, N.; Kojoh, S.; Toda, Y.; Mizuno, A.; Kashiwa, N. The detailed analysis of the vinylidene structure of metallocene-catalyzed polypropylene. Polymer 2004, 45, 355–357. [Google Scholar] [CrossRef]
- Quevedo-Sanchez, B.; Henson, M.A.; Coughlin, E.B. Origin of the formation of the 4-butenyl end group in zirconocene-catalyzed propylene polymerization. J. Polym. Sci. A 2006, 44, 3724–3728. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
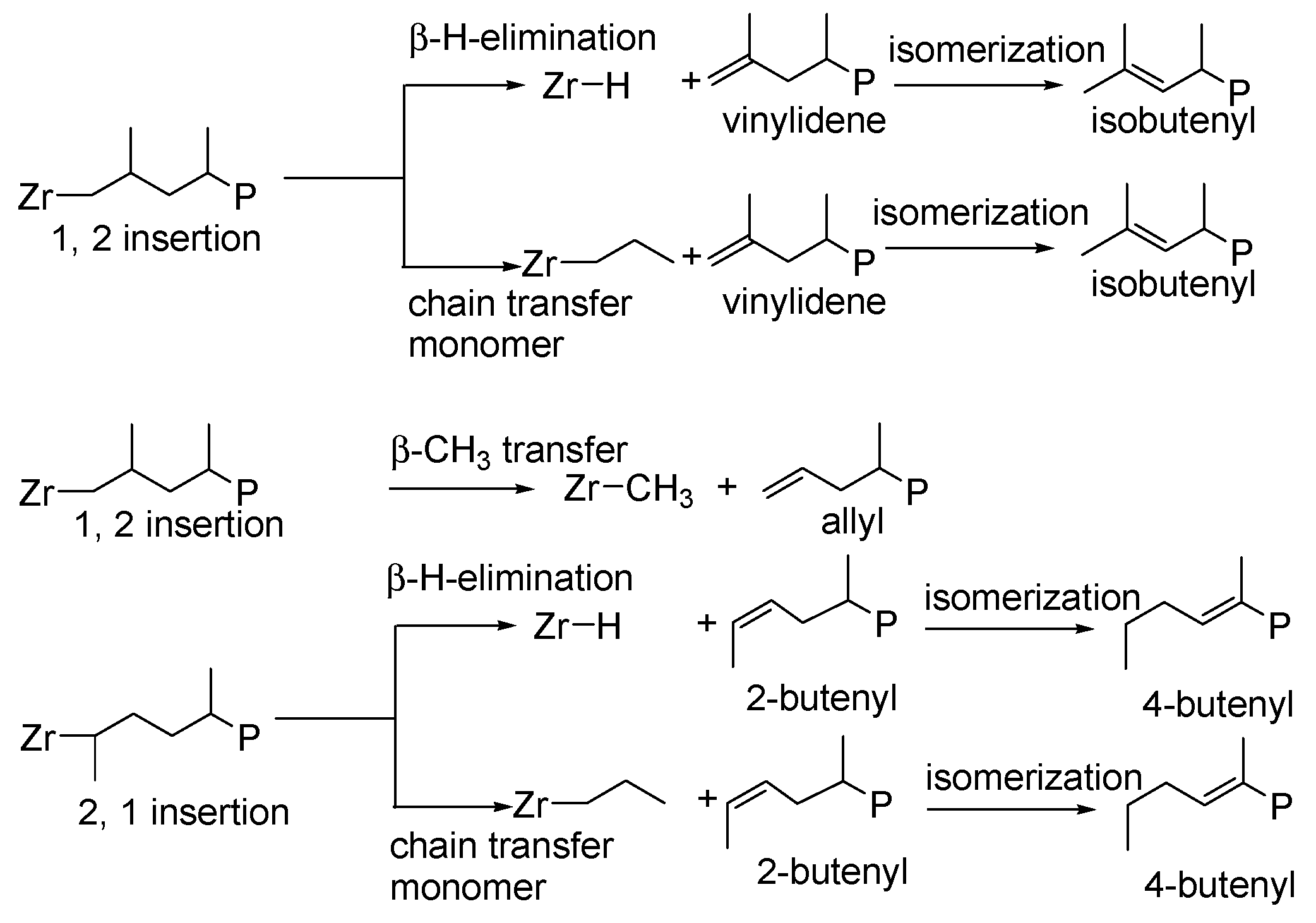{kind=link}
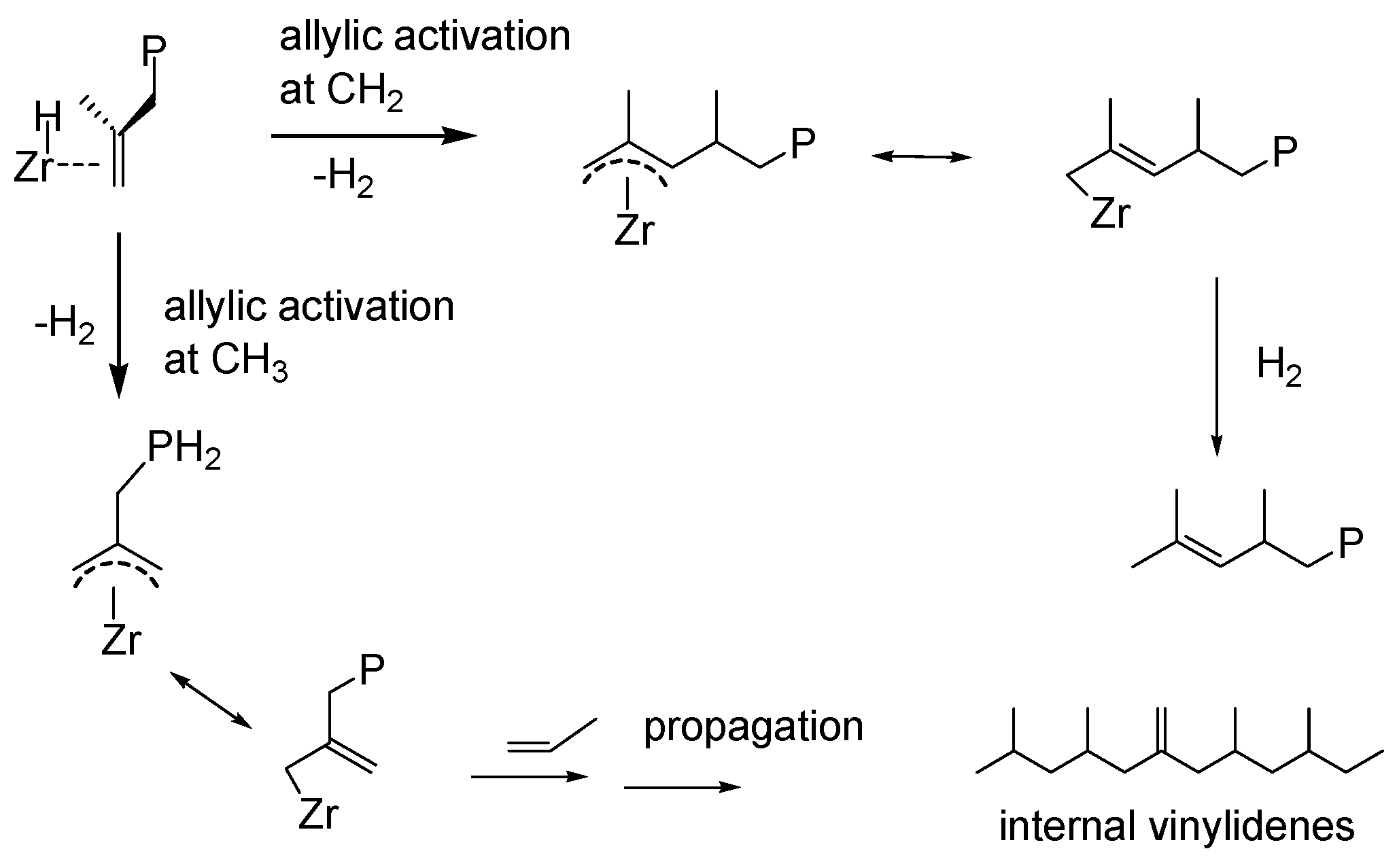{kind=link}
| Entry | Catalyst | T (°C) | Propene (molP/V) | Pressure (bar) | Activity 2 (KgPP/molZr·h·P) | Mw 3 (Kg/mol) | (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 30 | 5.3 | 4.7 | 2780 | 247 | 2.9 |
| 2 | 1 | 50 | 12.0 | 10.5 | 951 | 221 | 2.1 |
| 3 | 1 | 70 | 3.8 | 6.9 | 1026 | 129 | 2.1 |
| 4 | 2 | 30 | 12.5 | 7.7 | 1043 | 141 | 1.7 |
| 5 | 2 | 50 | 10.4 | 7.2 | - | 167 | 1.9 |
| 6 | 2 | 50 | 13.3 | 11.6 | 721 | 102 | 1.9 |
| 7 | 2 | 50 | 17.7 | 12.9 | 669 | 108 | 1.9 |
| 8 | 2 | 70 | 12.9 | 15.4 | 238 | 73 | 1.9 |
| 9 | 3 | 30 | 14.5 | 8.2 | 337 | 224 | 2.1 |
| 10 | 3 | 50 | 14.9 | 12.0 | 187 | 228 | 1.6 |
| 11 | 3 | 70 | 16.7 | 16.6 | 152 | 94 | 1.7 |
| Entry | Catalyst | T (°C) | Propene (molP/V) | Pressure (bar) | rrrr 1 (%) | mmmm 1 (%) | rmrr 1 (%) | rmmr 1 (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 30 | 5.3 | 4.7 | 69.8 | 0.8 | 10.5 | 2.1 |
| 2 | 1 | 50 | 12.0 | 10.5 | 54.1 | 1.2 | 17.4 | 2.9 |
| 3 | 1 | 70 | 3.8 | 6.9 | 58.9 | 0.8 | 15.5 | 2.6 |
| 4 | 2 | 30 | 12.5 | 7.7 | 77.4 | 0.8 | 5.8 | 2.1 |
| 5 | 2 | 50 | 10.4 | 7.2 | 75.8 | 1.9 | 7.5 | 1.8 |
| 6 | 2 | 50 | 13.3 | 11.6 | 77.6 | 0.7 | 6.0 | 2.0 |
| 7 | 2 | 50 | 17.7 | 12.9 | 69.4 | 1.9 | 9.1 | 2.4 |
| 8 | 2 | 70 | 12.9 | 15.4 | 68.0 | 1.2 | 8.9 | 2.6 |
| 9 | 3 | 30 | 14.5 | 8.2 | 0.56 | 86.9 | 0.7 | 0.9 |
| 10 | 3 | 50 | 14.9 | 12.0 | 1.25 | 87.3 | 1.2 | 0.0 |
| 11 | 3 | 70 | 16.7 | 16.6 | 1.01 | 84.0 | 1.9 | 1.0 |
 | 5 |  | 4 | ||||
|---|---|---|---|---|---|---|---|
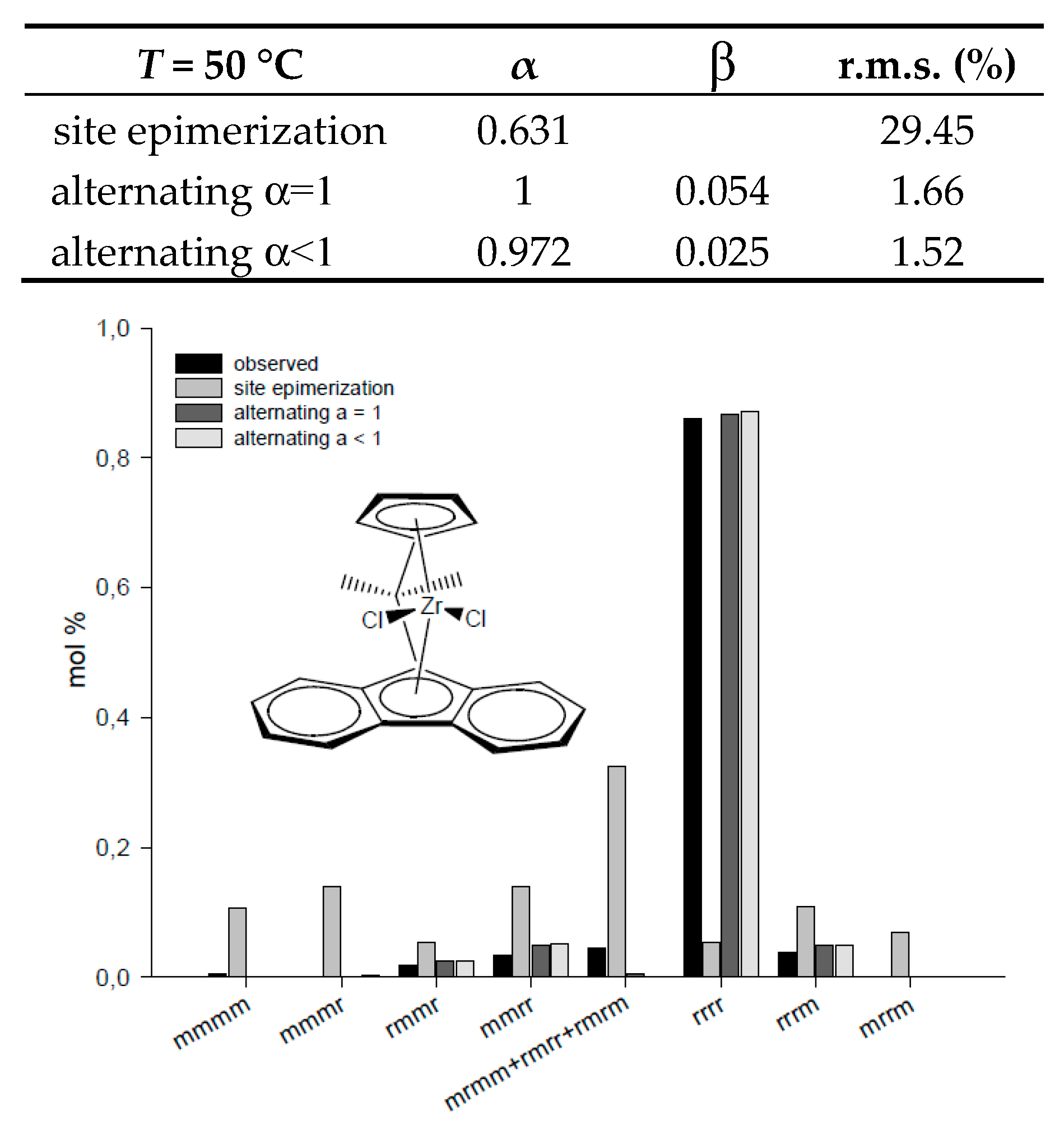
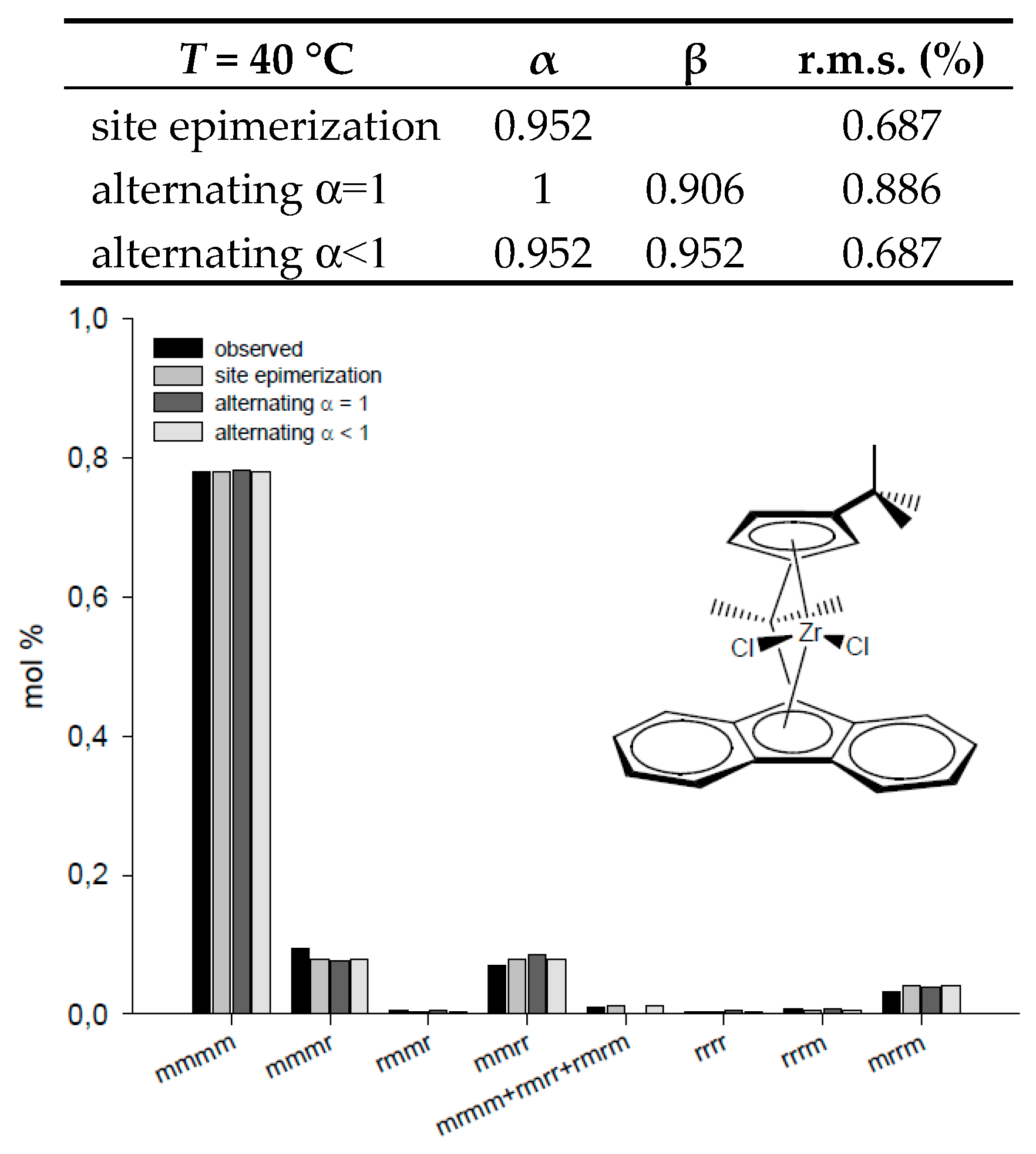
| T = 50 °C | α | β | r.m.s. (%) | T = 40 °C | α | β | r.m.s. (%) |
| site epimerization | 0.631 | 29.45 | site epimerization | 0.952 | 0.687 | ||
| alternating α = 1 | 1 | 0.054 | 1.66 | alternating α = 1 | 1 | 0.906 | 0.886 |
| alternating α < 1 | 0.972 | 0.025 | 1.52 | alternating α < 1 | 0.952 | 0.952 | 0.687 |
 | 2 | |||||
|---|---|---|---|---|---|---|
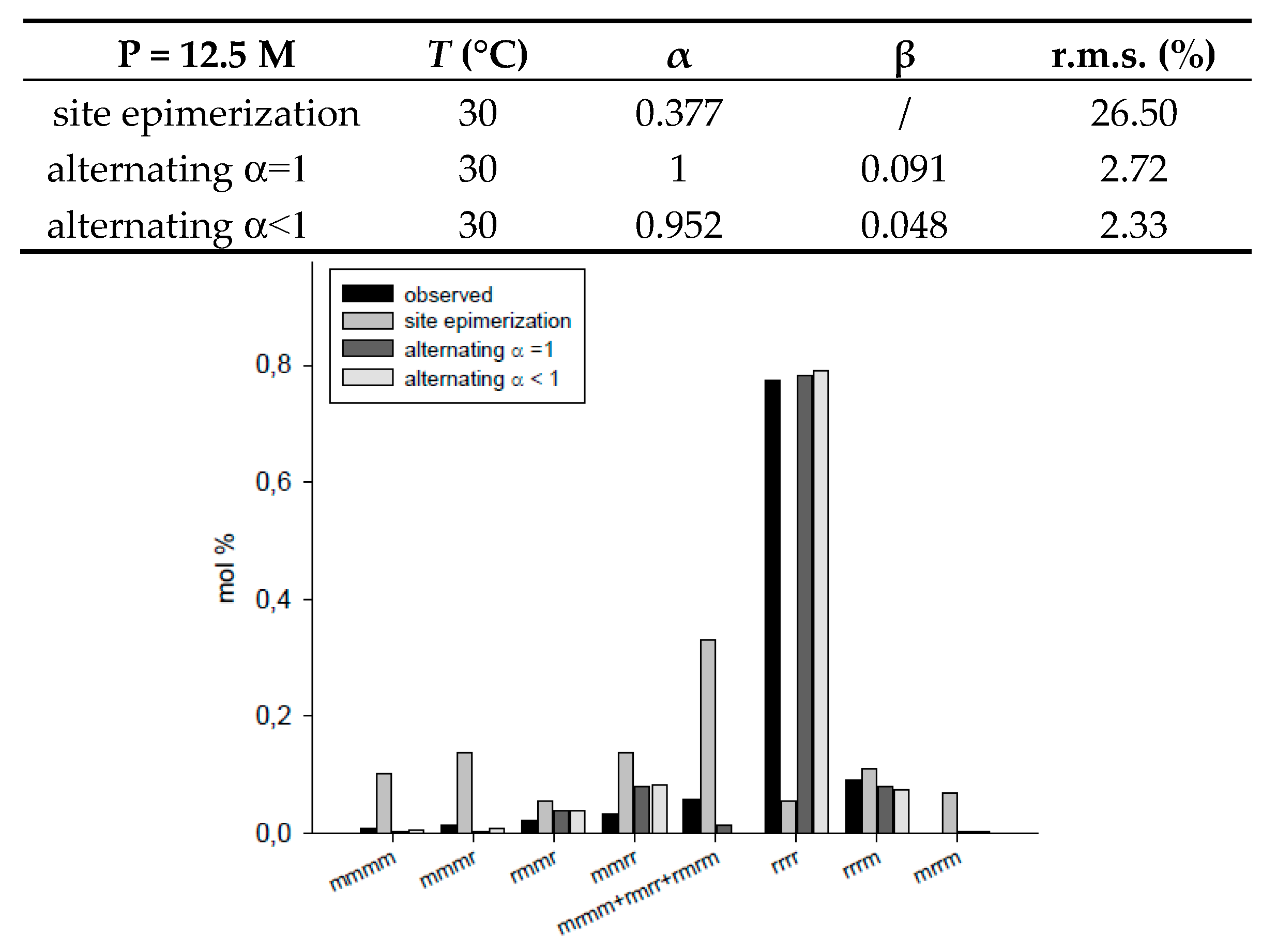
| P (mol/L) | T (°C) | α | β | r.m.s. (%) | ||
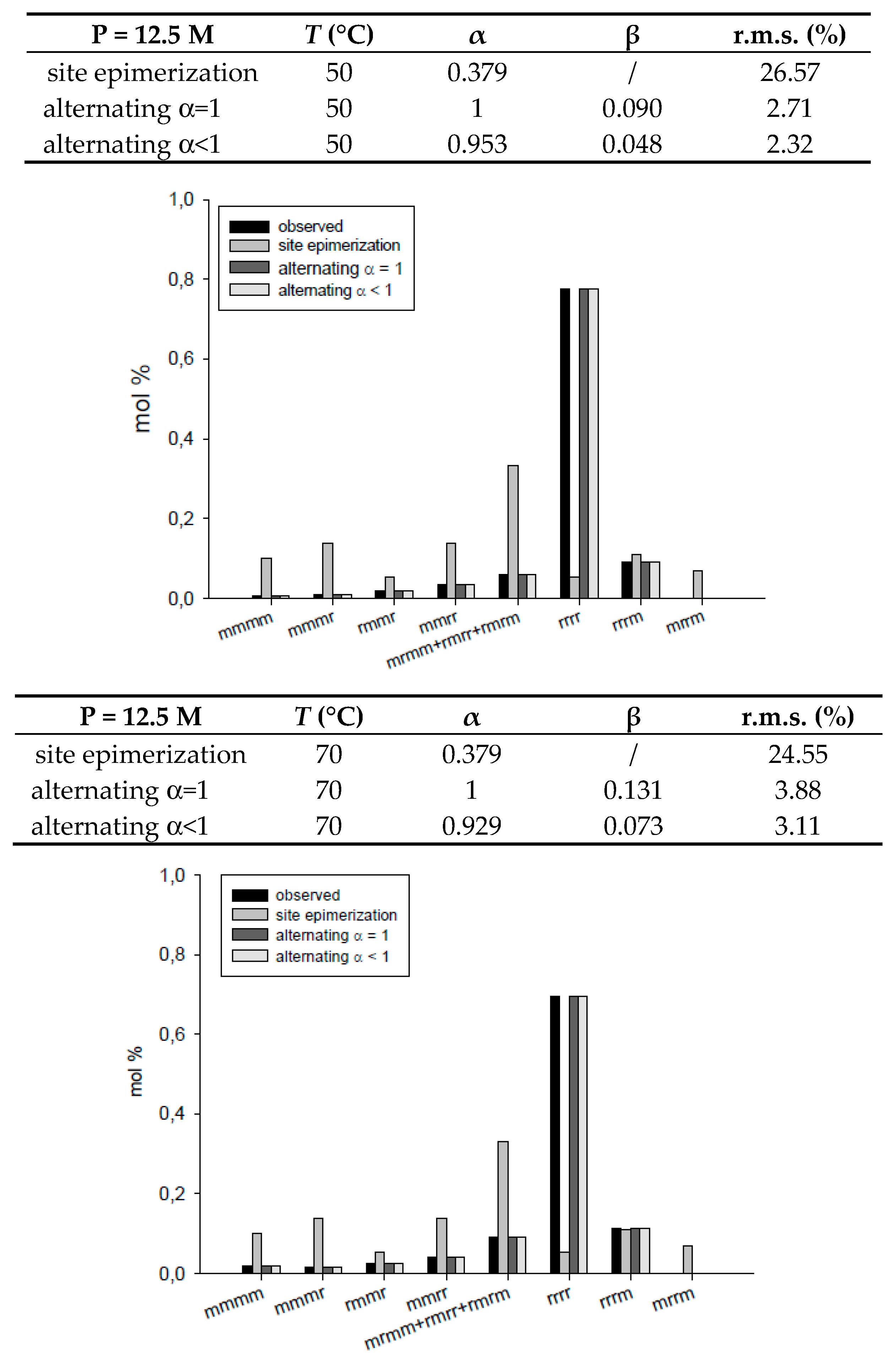
| site epimerization | 12.5 | 30 | 0.377 | / | 26.50 | |
| 13.3 | 50 | 0.379 | / | 26.57 | ||
| 12.9 | 70 | 0.379 | / | 24.55 | ||
| alternating α = 1 | 12.5 | 30 | 1 | 0.091 | 2.72 | |
| 13.3 | 50 | 1 | 0.090 | 2.71 | ||
| 12.9 | 70 | 1 | 0.131 | 3.88 | ||
| alternating α < 1 | 12.5 | 30 | 0.952 | 0.048 | 2.33 | |
| 13.3 | 50 | 0.953 | 0.048 | 2.32 | ||
| 12.9 | 70 | 0.929 | 0.073 | 3.11 | ||
| site epimerization | 13.3 | 50 | 0.379 | / | 26.57 | |
| site epimerization | 17.7 | 50 | 0.376 | / | 25.07 | |
| alternating α = 1 | 13.3 | 50 | 1 | 0.09 | 2.71 | |
| alternating α = 1 | 17.7 | 50 | 1 | 0.13 | 4.03 | |
| alternating α < 1 | 13.3 | 50 | 0.953 | 0.048 | 2.32 | |
| alternating α < 1 | 17.7 | 50 | 0.932 | 0.048 | 3.27 | |
 | 1 | |||
|---|---|---|---|---|
| α | β | r.m.s. (%) | ||
| T = 30 °C, P = 5.3 M | site epimerization | 0.5 | 23.65 | |
| alternating α = 1 | 1 | 0.121 | 4.30 | |
| alternating α < 1 | 0.933 | 0.067 | 3.57 | |
| T = 50 °C, P = 12 M | site epimerization | 0.5 | 17.86 | |
| alternating α = 1 | 1 | 0.198 | 6.88 | |
| alternating α < 1 | 0.881 | 0.121 | 4.87 | |
| T = 70 °C, P = 3.8 M | site epimerization | 0.5 | 20.89 | |
| alternating α = 1 | 1 | 0.102 | 6.33 | |
| alternating α < 1 | 0.898 | 0.067 | 4.75 | |
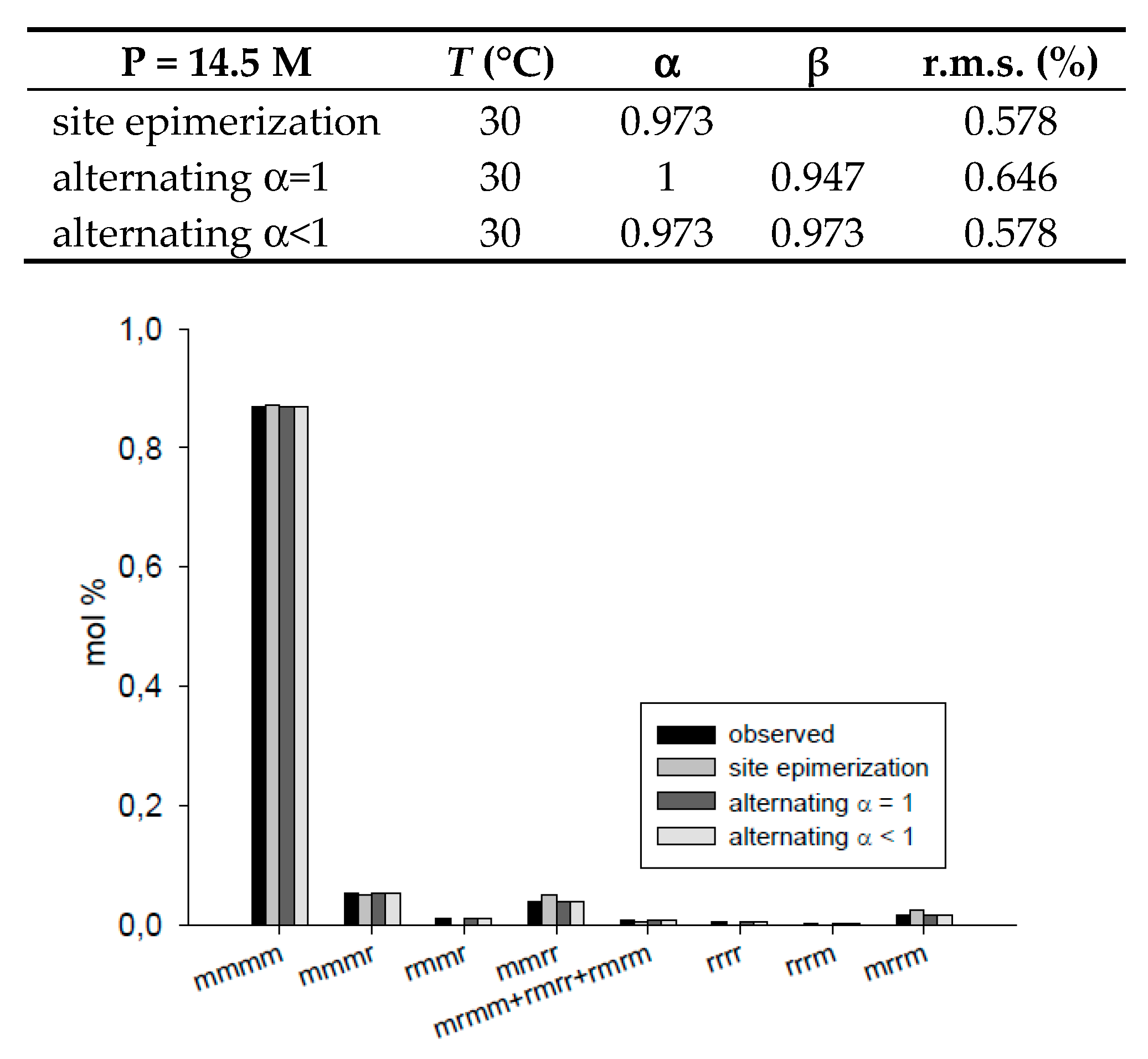
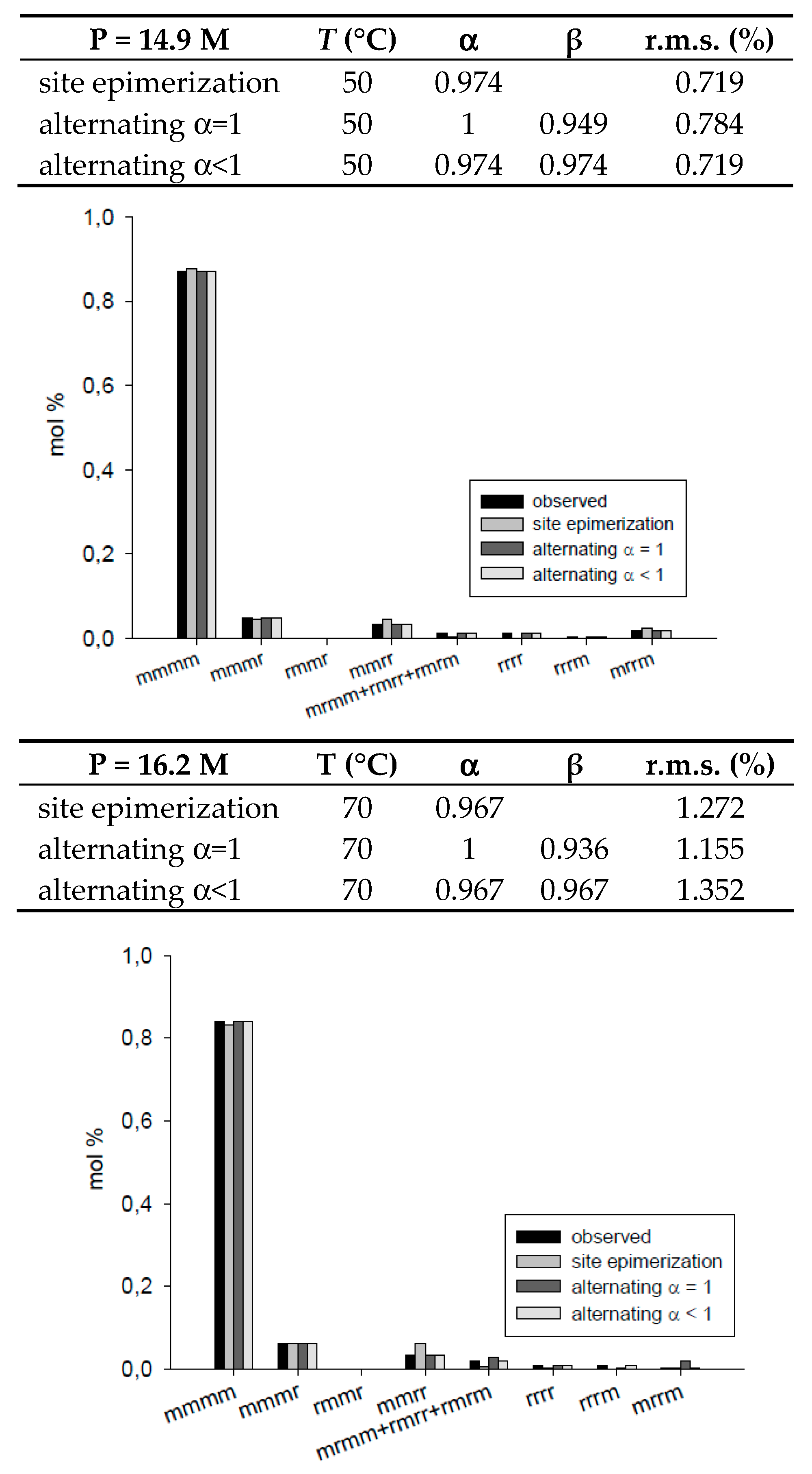
 | 3 | ||||
|---|---|---|---|---|---|
| α | β | r.m.s. (%) | |||
| T = 30 °C, P = 14.5 M | site epimerization | 0.973 | 0.578 | ||
| alternating α = 1 | 1 | 0.947 | 0.646 | ||
| alternating α < 1 | 0.973 | 0.973 | 0.578 | ||
| T = 50 °C, P = 14.9 M | site epimerization | 0.974 | 0.719 | ||
| alternating α = 1 | 1 | 0.949 | 0.784 | ||
| alternating α < 1 | 0.974 | 0.974 | 0.719 | ||
| T = 70 °C, P = 16.2 M | site epimerization | 0.967 | 1.272 | ||
| alternating α = 1 | 1 | 0.936 | 1.155 | ||
| alternating α < 1 | 0.967 | 0.967 | 1.352 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boggioni, L.; Cornelio, M.; Losio, S.; Razavi, A.; Tritto, I. Propene Polymerization with C1-Symmetric Fluorenyl-Metallocene Catalysts. Polymers 2017, 9, 581. https://doi.org/10.3390/polym9110581
Boggioni L, Cornelio M, Losio S, Razavi A, Tritto I. Propene Polymerization with C1-Symmetric Fluorenyl-Metallocene Catalysts. Polymers. 2017; 9(11):581. https://doi.org/10.3390/polym9110581
Chicago/Turabian StyleBoggioni, Laura, Massimiliano Cornelio, Simona Losio, Abbas Razavi, and Incoronata Tritto. 2017. "Propene Polymerization with C1-Symmetric Fluorenyl-Metallocene Catalysts" Polymers 9, no. 11: 581. https://doi.org/10.3390/polym9110581
APA StyleBoggioni, L., Cornelio, M., Losio, S., Razavi, A., & Tritto, I. (2017). Propene Polymerization with C1-Symmetric Fluorenyl-Metallocene Catalysts. Polymers, 9(11), 581. https://doi.org/10.3390/polym9110581