One-Pot Synthesis of Charged Amphiphilic Diblock and Triblock Copolymers Via High-Throughput Cu(0)-Mediated Polymerization



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
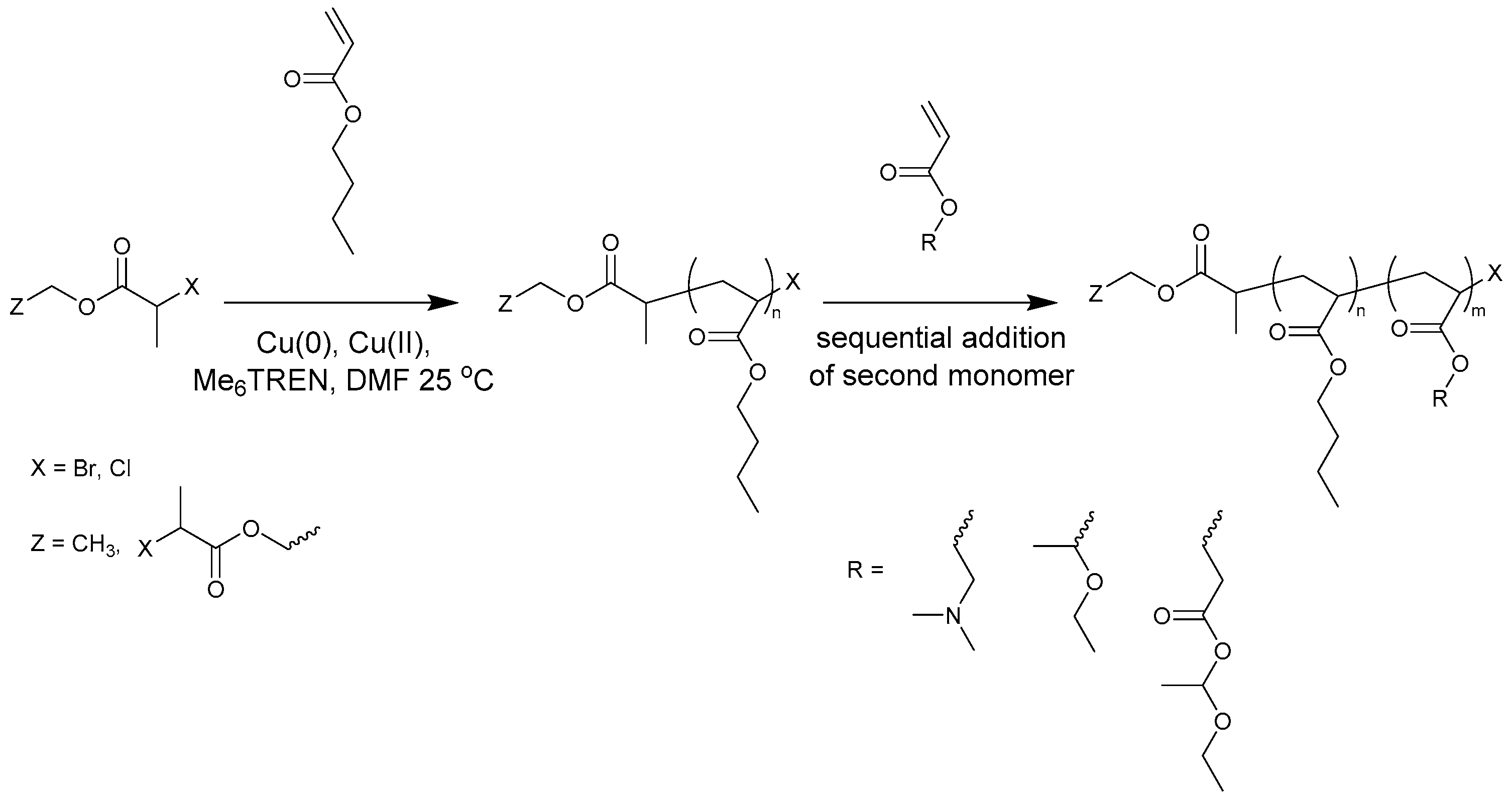
2.2. Automated Cu(0)-Mediated Polymerization
2.3. Gas Chromatography (GC)
2.4. Size Exclusion Chromatography (SEC)
2.5. Synthesis of Ethylene Glycol Bis(2-bromopropionyl) Ethane (BPE)
2.6. Synthesis of Ethylene Glycol Bis(2-chloropropionyl) Ethane (CPE)
2.7. Synthesis of 1-Ethoxyethyl Acrylate (EEA) and Protected 2-Carboxyethyl Acrylate (ProCEA)
3. Results and Discussion
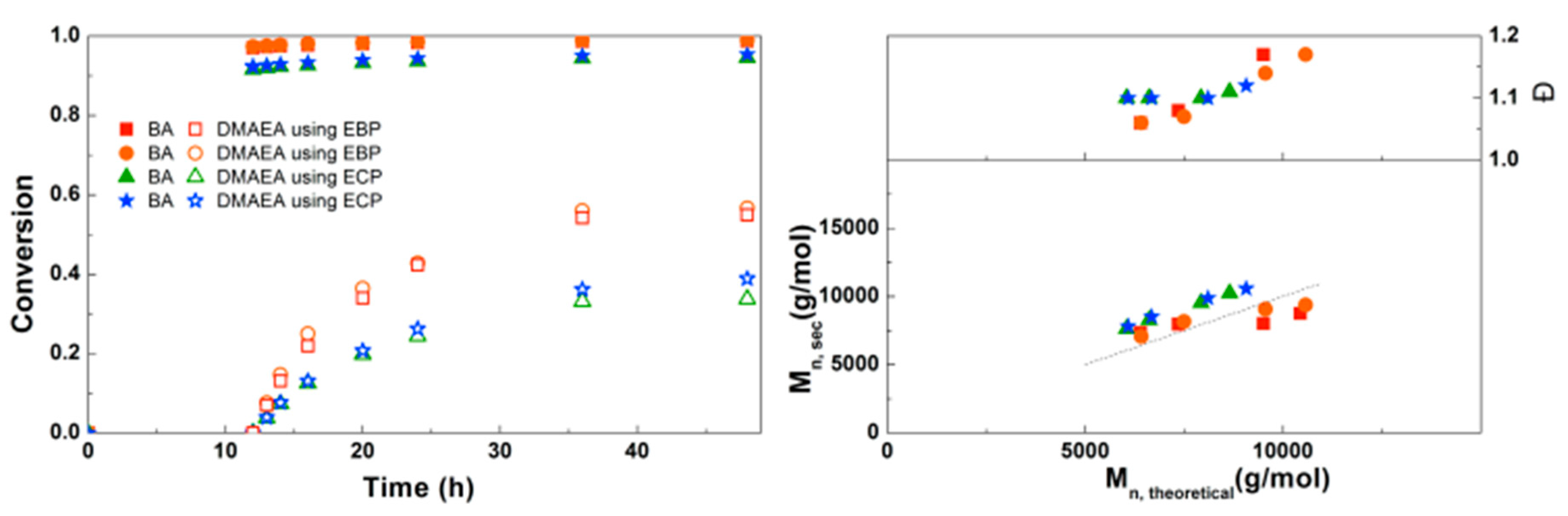
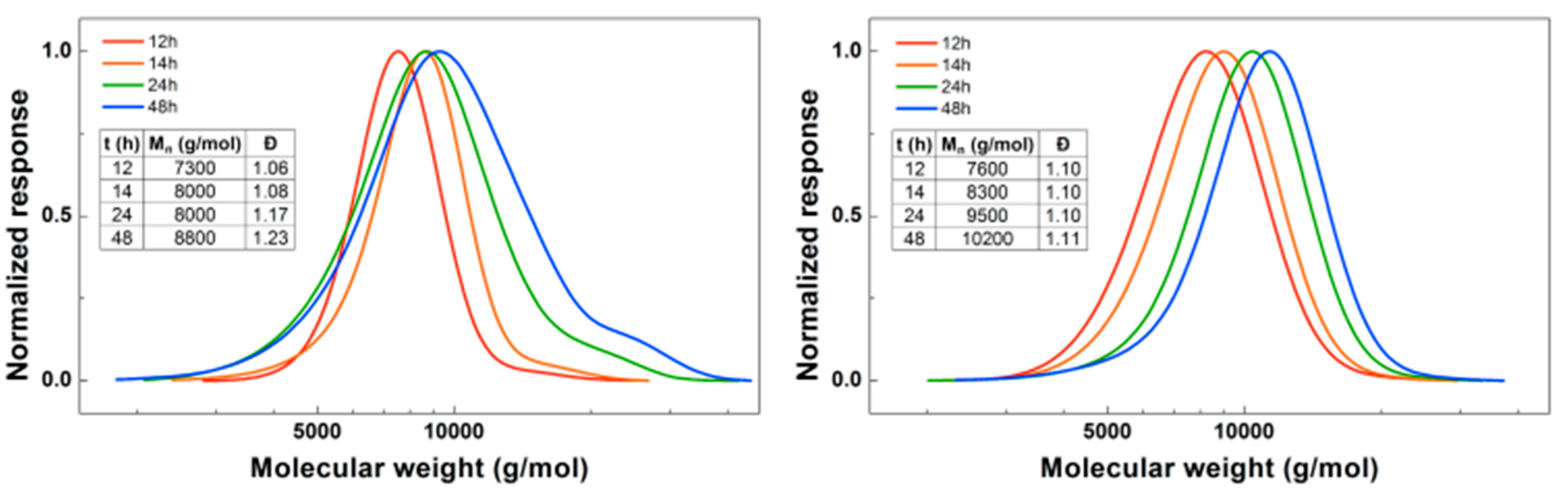
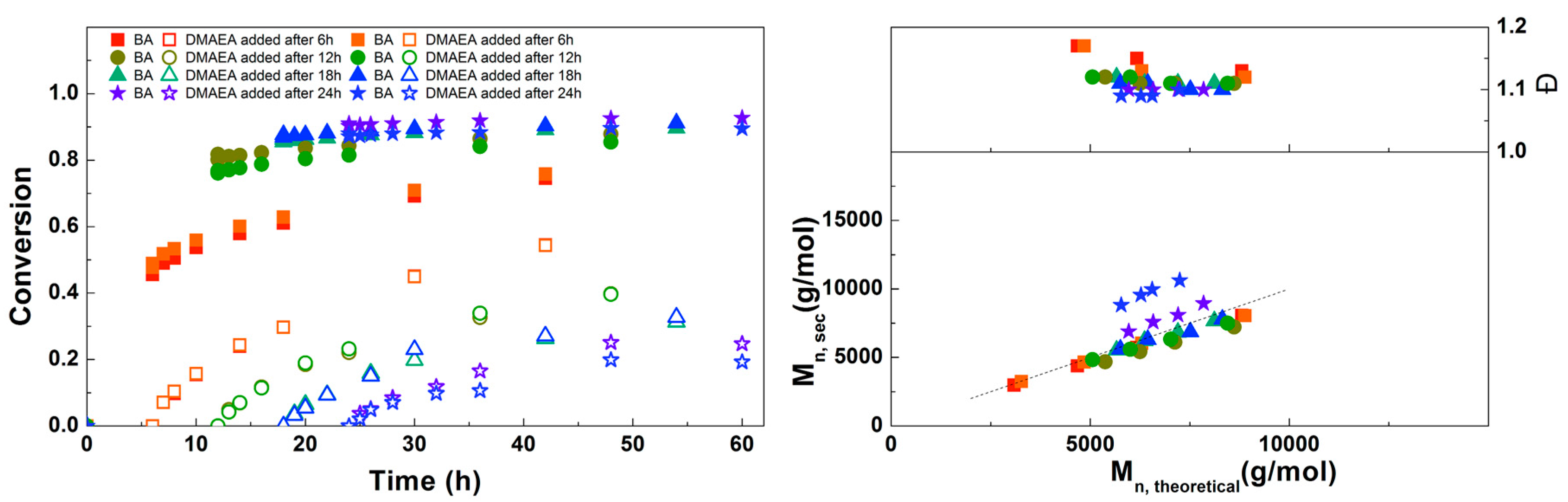
3.1. Synthesis of PBA-b-PDMAEA Diblock Copolymers
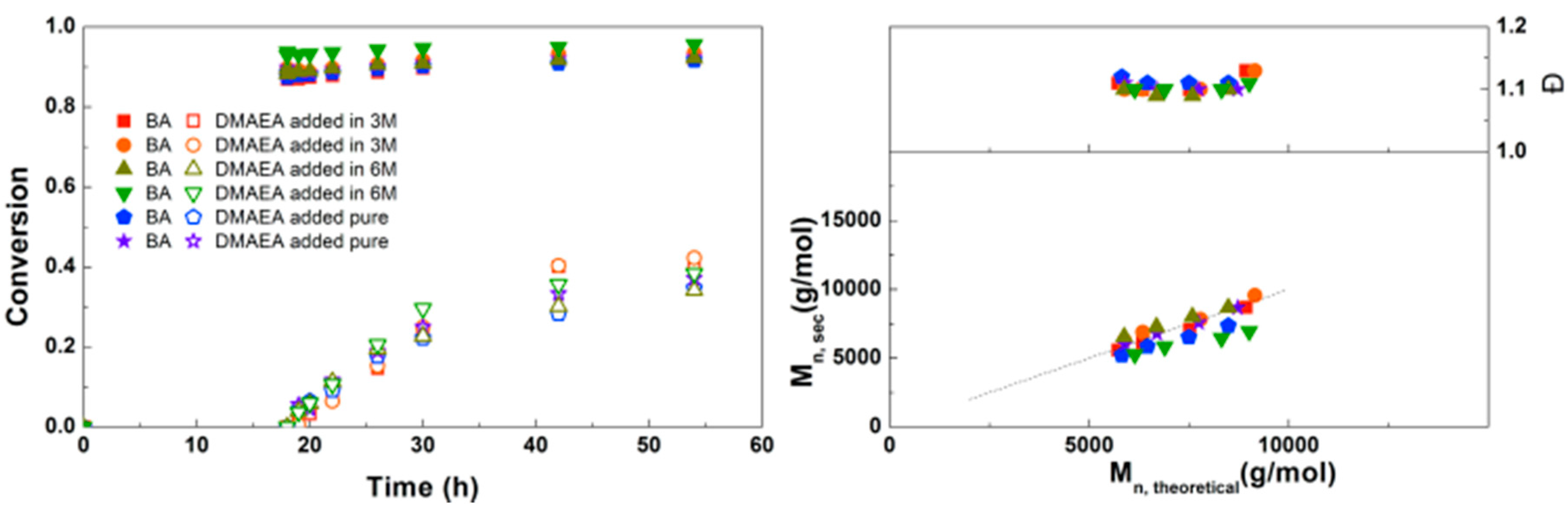
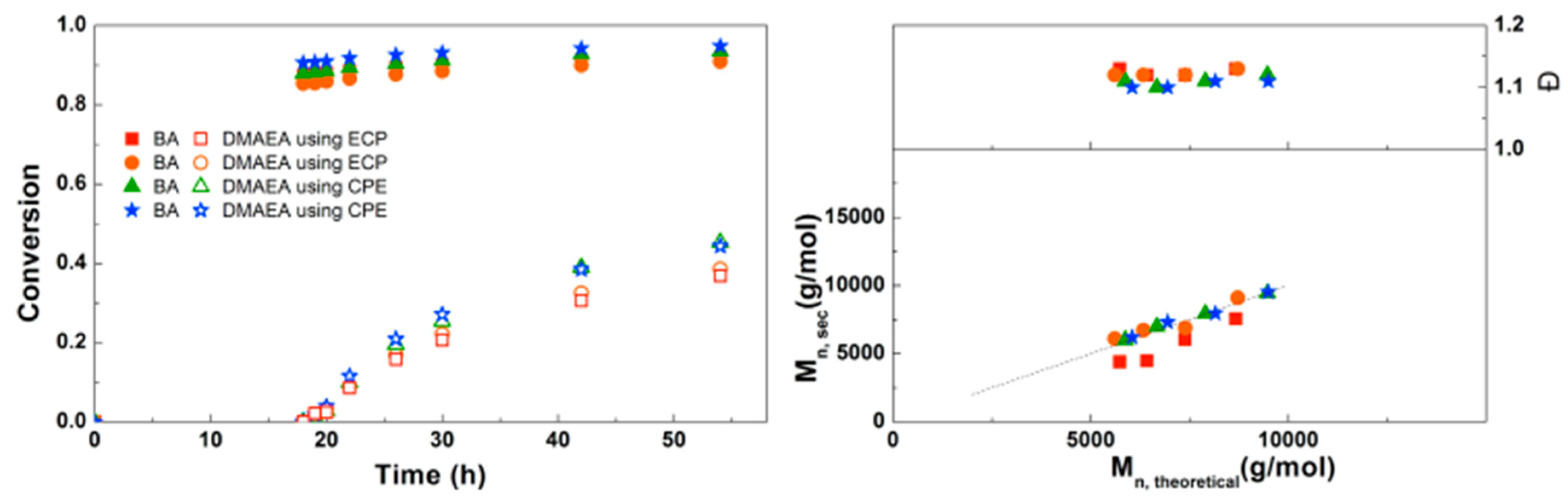
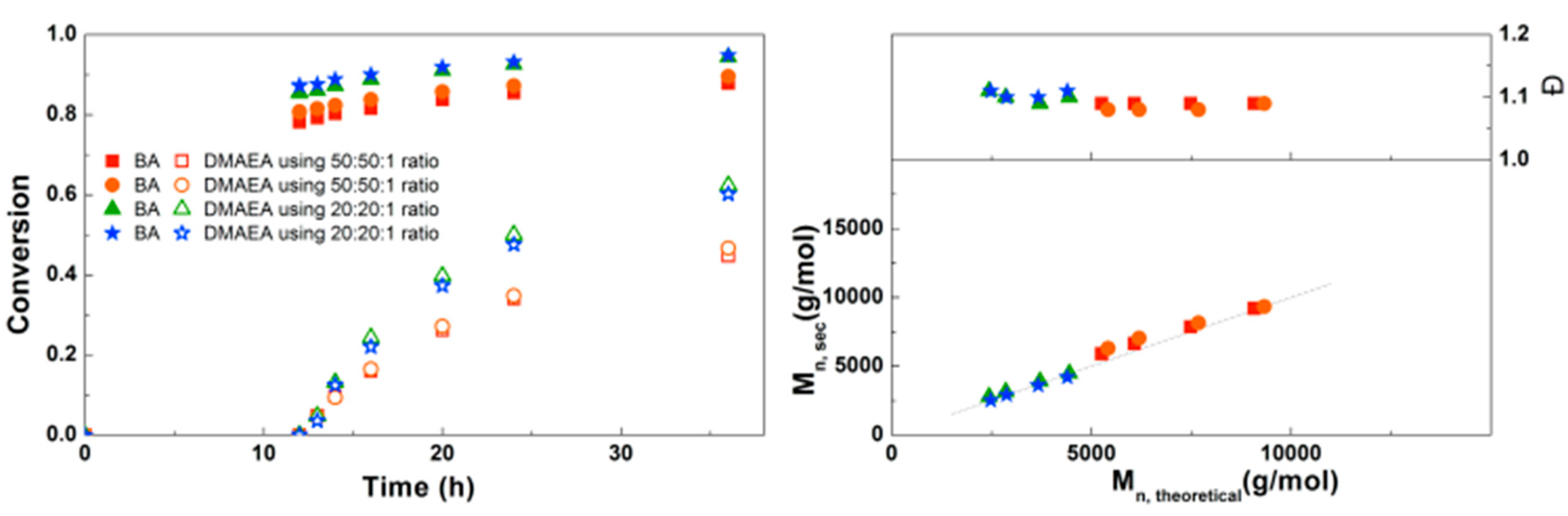
3.2. Synthesis of PDMAEA-b-PBA-b-PDMAEA Triblock Copolymers
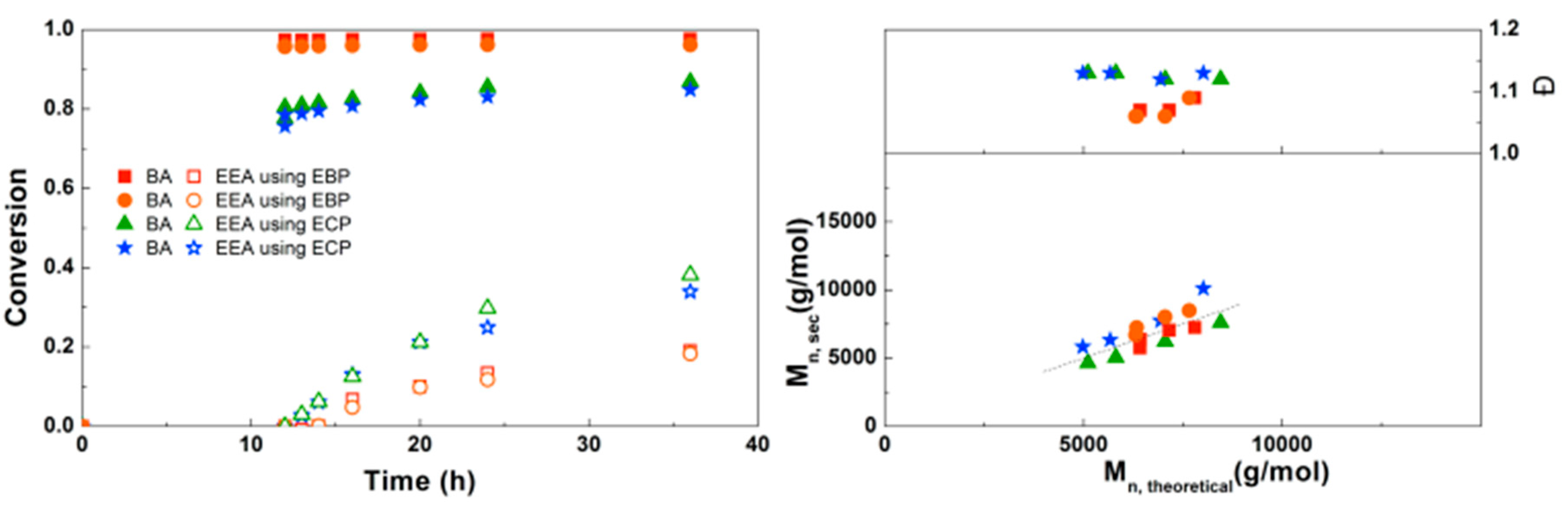
3.3. Synthesis of PBA-b-PEEA Diblock Copolymers
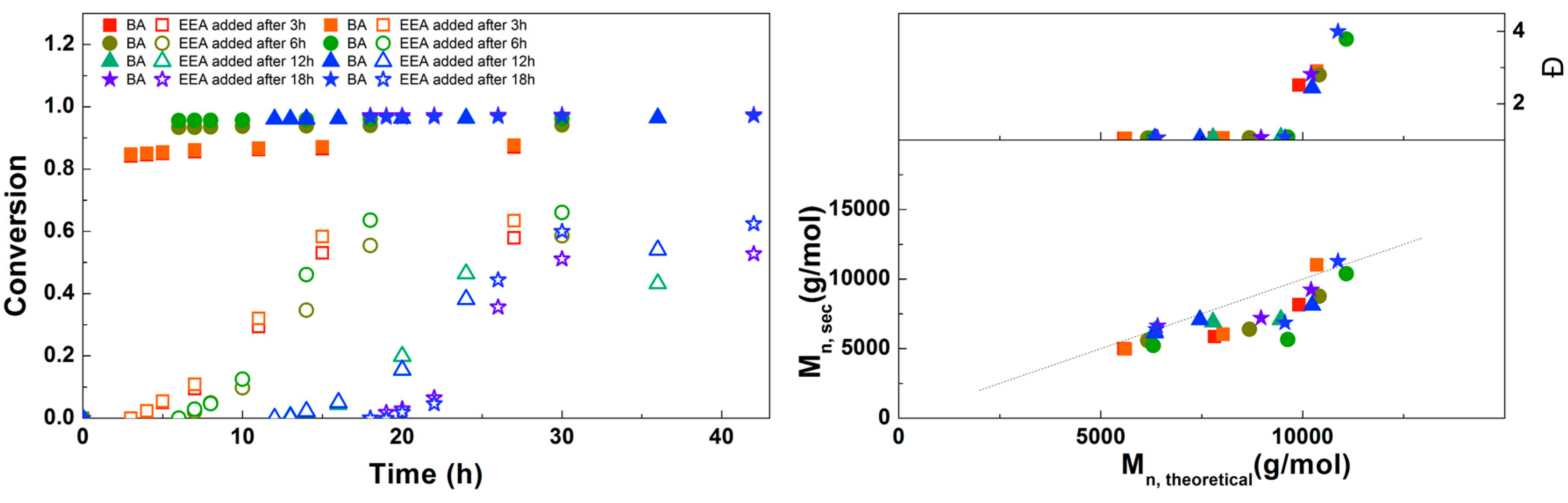
3.4. Synthesis of PEEA-b-PBA-b-PEEA Triblock Copolymers
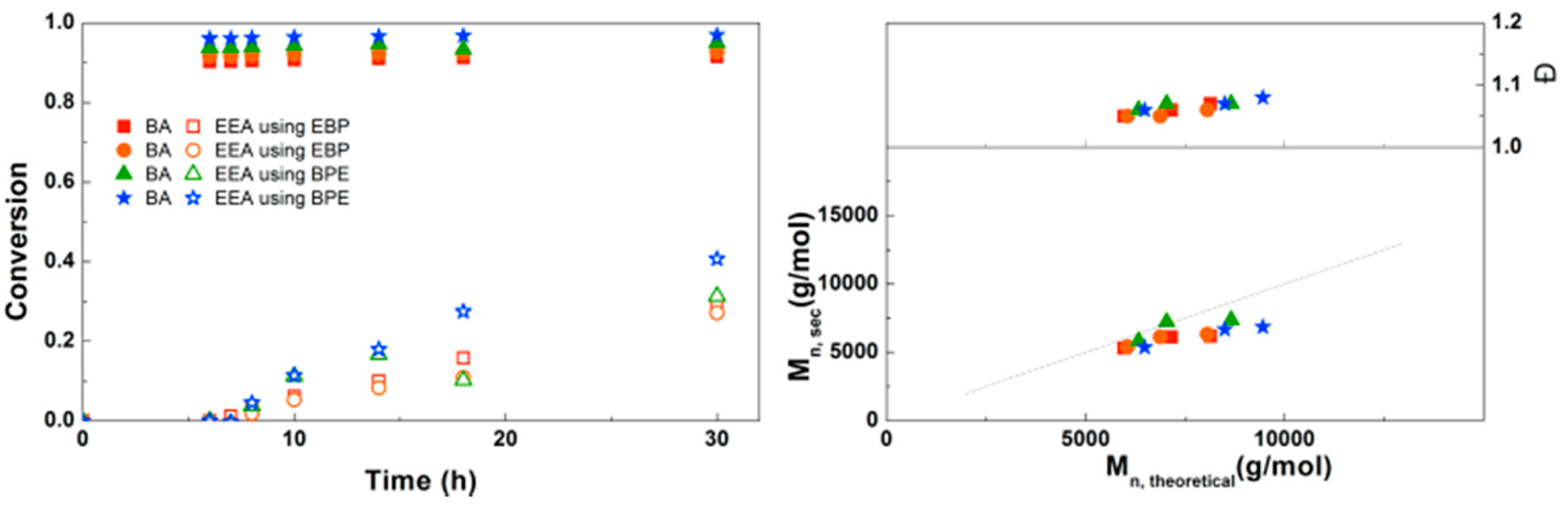
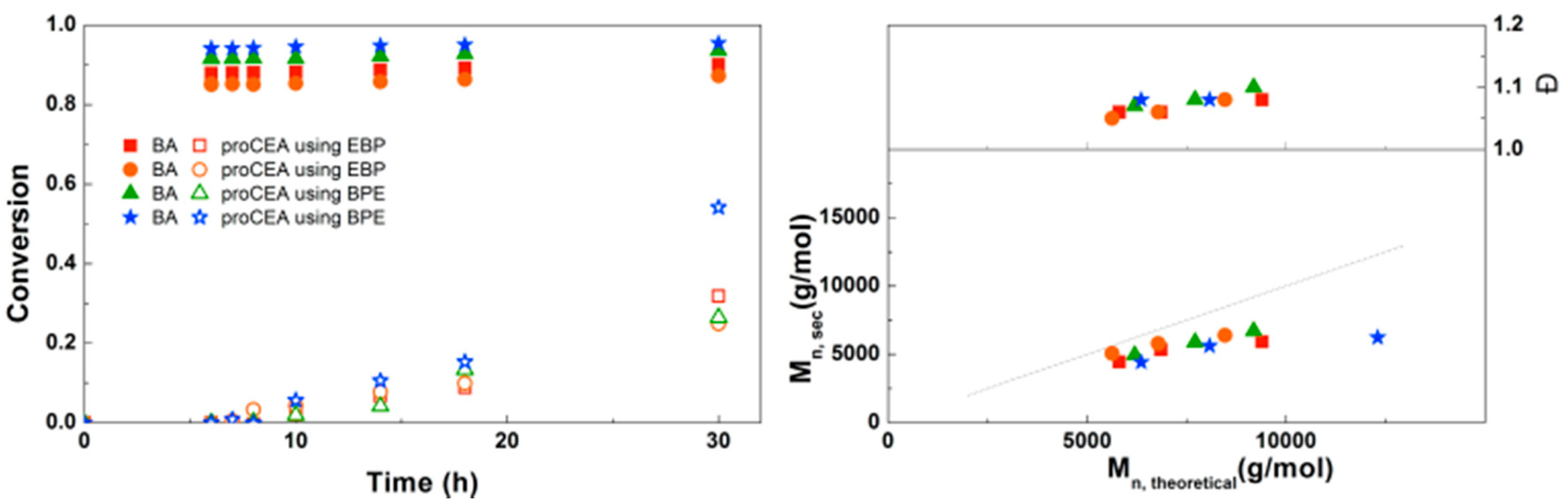
3.5. Synthesis of PBA-b-PproCEA Diblock and PproCEA-b-PBA-b-PproCEA Triblock Copolymers
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Voorhaar, L.; Wallyn, S.; Du Prez, F.E.; Hoogenboom, R. Cu(0)-mediated polymerization of hydrophobic acrylates using high-throughput experimentation. Polym. Chem. 2014, 5, 4268–4276. [Google Scholar] [CrossRef]
- Voorhaar, L.; Diaz, M.M.; Leroux, F.; Rogers, S.; Abakumov, A.M.; Van Tendeloo, G.; Van Assche, G.; Van Mele, B.; Hoogenboom, R. Supramolecular thermoplastics and thermoplastic elastomer materials with self-healing ability based on oligomeric charged triblock copolymers. NPG Asia Mater. 2017, 9, e385. [Google Scholar] [CrossRef]
- Boyer, C.; Zetterlund, P.B.; Whittaker, M.R. Synthesis of complex macromolecules using iterative copper(0)-mediated radical polymerization. J. Polym. Sci. A 2014, 52, 2083–2098. [Google Scholar] [CrossRef]
- Soeriyadi, A.H.; Boyer, C.; Nystrom, F.; Zetterlund, P.B.; Whittaker, M.R. High-Order Multiblock Copolymers via Iterative Cu(0)-Mediated Radical Polymerizations (SET-LRP): Toward Biological Precision. J. Am. Chem. Soc. 2011, 133, 11128–11131. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Soeriyadi, A.H.; Zetterlund, P.B.; Whittaker, M.R. Synthesis of Complex Multiblock Copolymers via a Simple Iterative Cu(0)-Mediated Radical Polymerization Approach. Macromolecules 2011, 44, 8028–8033. [Google Scholar] [CrossRef]
- Anastasaki, A.; Waldron, C.; Wilson, P.; Boyer, C.; Zetterlund, P.B.; Whittaker, M.R.; Haddleton, D. High Molecular Weight Block Copolymers by Sequential Monomer Addition via Cu(0)-Mediated Living Radical Polymerization (SET-LRP): An Optimized Approach. ACS Macro Lett. 2013, 2, 896–900. [Google Scholar] [CrossRef]
- Anastasaki, A.; Nikolaou, V.; Pappas, G.S.; Zhang, Q.; Wan, C.; Wilson, P.; Davis, T.P.; Whittaker, M.R.; Haddleton, D. Photoinduced sequence-control via one pot living radical polymerization of acrylates. Chem. Sci. 2014, 5, 3536–3542. [Google Scholar] [CrossRef]
- Chuang, Y.-M.; Ethirajan, A.; Junkers, T. Photoinduced Sequence-Controlled Copper-Mediated Polymerization: Synthesis of Decablock Copolymers. ACS Macro Lett. 2014, 3, 732–737. [Google Scholar] [CrossRef]
- Boyer, C.; Derveaux, A.; Zetterlund, P.B.; Whittaker, M.R. Synthesis of multi-block copolymer stars using a simple iterative Cu(0)-mediated radical polymerization technique. Polym. Chem. 2012, 3, 117–123. [Google Scholar] [CrossRef]
- Alsubaie, F.; Anastasaki, A.; Wilson, P.; Haddleton, D. Sequence-controlled multi-block copolymerization of acrylamides via aqueous SET-LRP at 0 °C. Polym. Chem. 2015, 6, 406–417. [Google Scholar] [CrossRef]
- Haddleton, D.; Simula, A.; Nikolaou, V.; Anastasaki, A.; Alsubaie, F.; Nurumbetov, G.; Wilson, P.; Kempe, K. Synthesis of well-defined α, ω -telechelic multiblock copolymers in aqueous medium: In situ generation of α, ω -diols. Polym. Chem. 2015, 6, 2226–2233. [Google Scholar]
- Zeng, F.; Shen, Y.; Zhu, S. Atom-Transfer Radical Polymerization of 2-(N,N-Dimethylamino)ethyl Acrylate. Macromol. Rapid Commun. 2002, 23, 1113–1117. [Google Scholar] [CrossRef]
- Anastasaki, A.; Waldron, C.; Wilson, P.; McHale, R.; Haddleton, D.M. The importance of ligand reactions in Cu(0)-mediated living radical polymerisation of acrylates. Polym. Chem. 2013, 4, 2672–2675. [Google Scholar] [CrossRef]
- Gu, W.; Jia, Z.; Truong, N.P.; Prasadam, I.; Xiao, Y.; Monteiro, M.J. Polymer Nanocarrier System for Endosome Escape and Timed Release of siRNA with Complete Gene Silencing and Cell Death in Cancer Cells. Biomacromolecules 2013, 14, 3386–3389. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, R.; Anastasaki, A.; Truong, N.P.; Wilson, P.; Kempe, K.; Burns, J.A.; Davis, T.P.; Haddleton, D.M. Well-Defined PDMAEA Stars via Cu(0)-Mediated Reversible Deactivation Radical Polymerization. Macromolecules 2016, 49, 8914–8924. [Google Scholar] [CrossRef]
- Nakane, Y.; Ishidoya, M.; Endo, T. Synthesis and thermal dissociation of polymers having hemiacetal ester moieties. J. Polym. Sci. A 1999, 37, 609–614. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Schubert, U.S.; Van Camp, W.; Du Prez, F.E. RAFT polymerization of 1-ethoxyethyl acrylate: A novel route toward near-monodisperse poly(acrylic acid) and derived block copolymer structures. Macromolecules 2005, 38, 7653–7659. [Google Scholar] [CrossRef]
- Van Camp, W.; Du Prez, F.E.; Bon, S.A.F. Atom Transfer Radical Polymerization of 1-Ethoxyethyl (Meth)acrylate: Facile Route toward Near-Monodisperse Poly((meth)acrylic acid). Macromolecules 2004, 37, 6673–6675. [Google Scholar] [CrossRef]
- Dervaux, B.; Van Camp, W.; Van Renterghem, L.; Du Prez, F.E. Synthesis of poly(isobornyl acrylate) containing copolymers by atom transfer radical polymerization. J. Polym. Sci. A 2008, 46, 1649–1661. [Google Scholar] [CrossRef]
- Davis, K.A.; Matyjaszewski, K. Atom Transfer Radical Polymerization of tert-Butyl Acrylate and Preparation of Block Copolymers. Macromolecules 2000, 33, 4039–4047. [Google Scholar] [CrossRef]
- Colombani, O.; Ruppel, M.; Schubert, F.; Zettl, H.; Pergushov, D.V.; Müller, A.H.E. Synthesis of Poly(n-butyl acrylate)-block-poly(acrylic acid) Diblock Copolymers by ATRP and Their Micellization in Water. Macromolecules 2007, 40, 4338–4350. [Google Scholar] [CrossRef]
- Ciampolini, M.; Nardi, N. Five-Coordinated High-Spin Complexes of Bivalent Cobalt, Nickel, andCopper with Tris(2-dimethylaminoethyl)amine. Inorg. Chem. 1966, 5, 41–44. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Levere, M.E.; Percec, V. TREN versus Me6-TREN as ligands in SET-LRP of methyl acrylate. J. Polym. Sci. A 2012, 50, 35–46. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Lu, S.; Fan, Q.-L.; Liu, S.-Y.; Chua, S.-J.; Huang, W. Synthesis of Conjugated−Acidic Block Copolymers by Atom Transfer Radical Polymerization. Macromolecules 2002, 35, 9875–9881. [Google Scholar] [CrossRef]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voorhaar, L.; Hoogenboom, R. One-Pot Synthesis of Charged Amphiphilic Diblock and Triblock Copolymers Via High-Throughput Cu(0)-Mediated Polymerization. Polymers 2017, 9, 320. https://doi.org/10.3390/polym9080320
Voorhaar L, Hoogenboom R. One-Pot Synthesis of Charged Amphiphilic Diblock and Triblock Copolymers Via High-Throughput Cu(0)-Mediated Polymerization. Polymers. 2017; 9(8):320. https://doi.org/10.3390/polym9080320
Chicago/Turabian StyleVoorhaar, Lenny, and Richard Hoogenboom. 2017. "One-Pot Synthesis of Charged Amphiphilic Diblock and Triblock Copolymers Via High-Throughput Cu(0)-Mediated Polymerization" Polymers 9, no. 8: 320. https://doi.org/10.3390/polym9080320
APA StyleVoorhaar, L., & Hoogenboom, R. (2017). One-Pot Synthesis of Charged Amphiphilic Diblock and Triblock Copolymers Via High-Throughput Cu(0)-Mediated Polymerization. Polymers, 9(8), 320. https://doi.org/10.3390/polym9080320