The Influence of Calcium Glycerophosphate (GPCa) Modifier on Physicochemical, Mechanical, and Biological Performance of Polyurethanes Applicable as Biomaterials for Bone Tissue Scaffolds Fabrication


Abstract
:
1. Introduction
2. Experimental
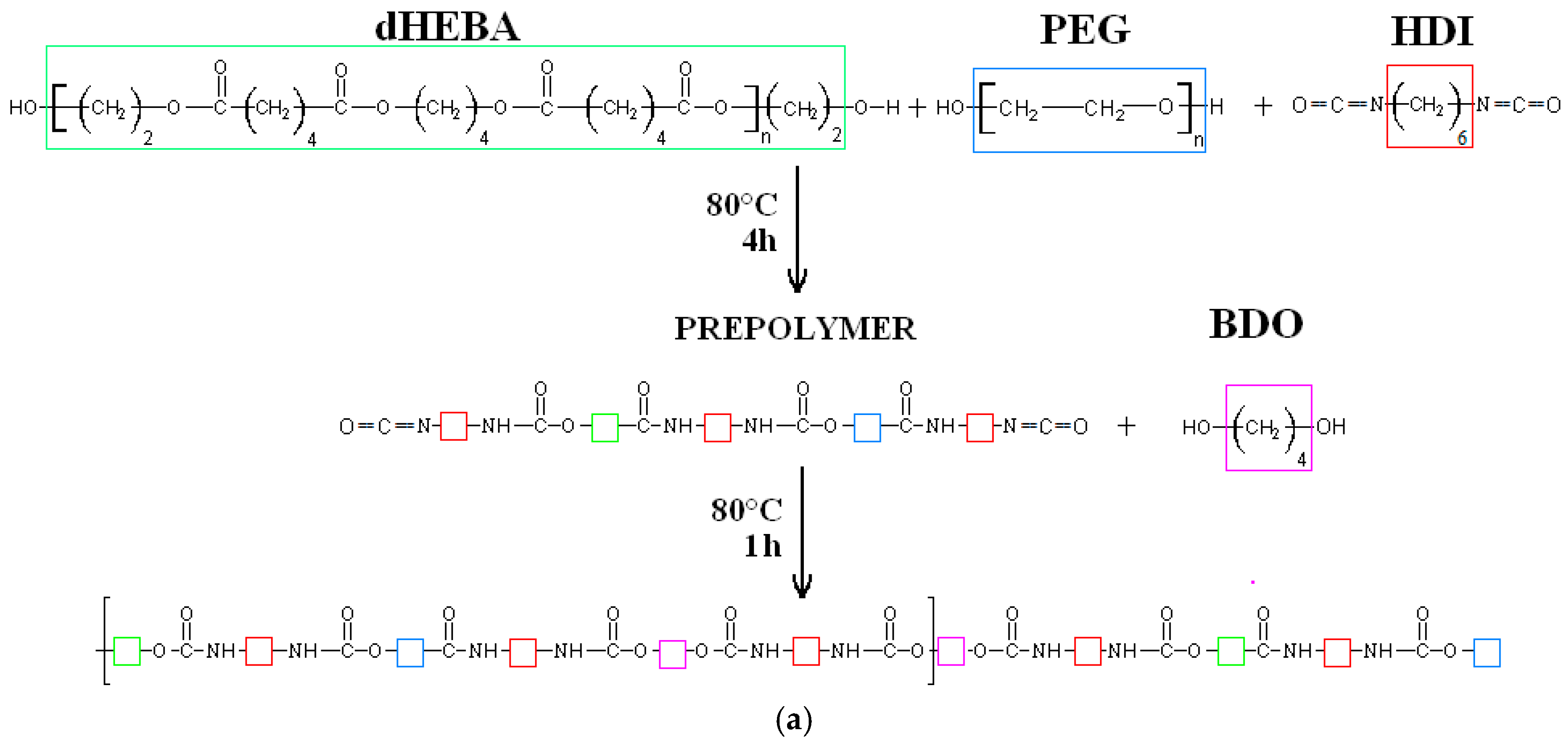
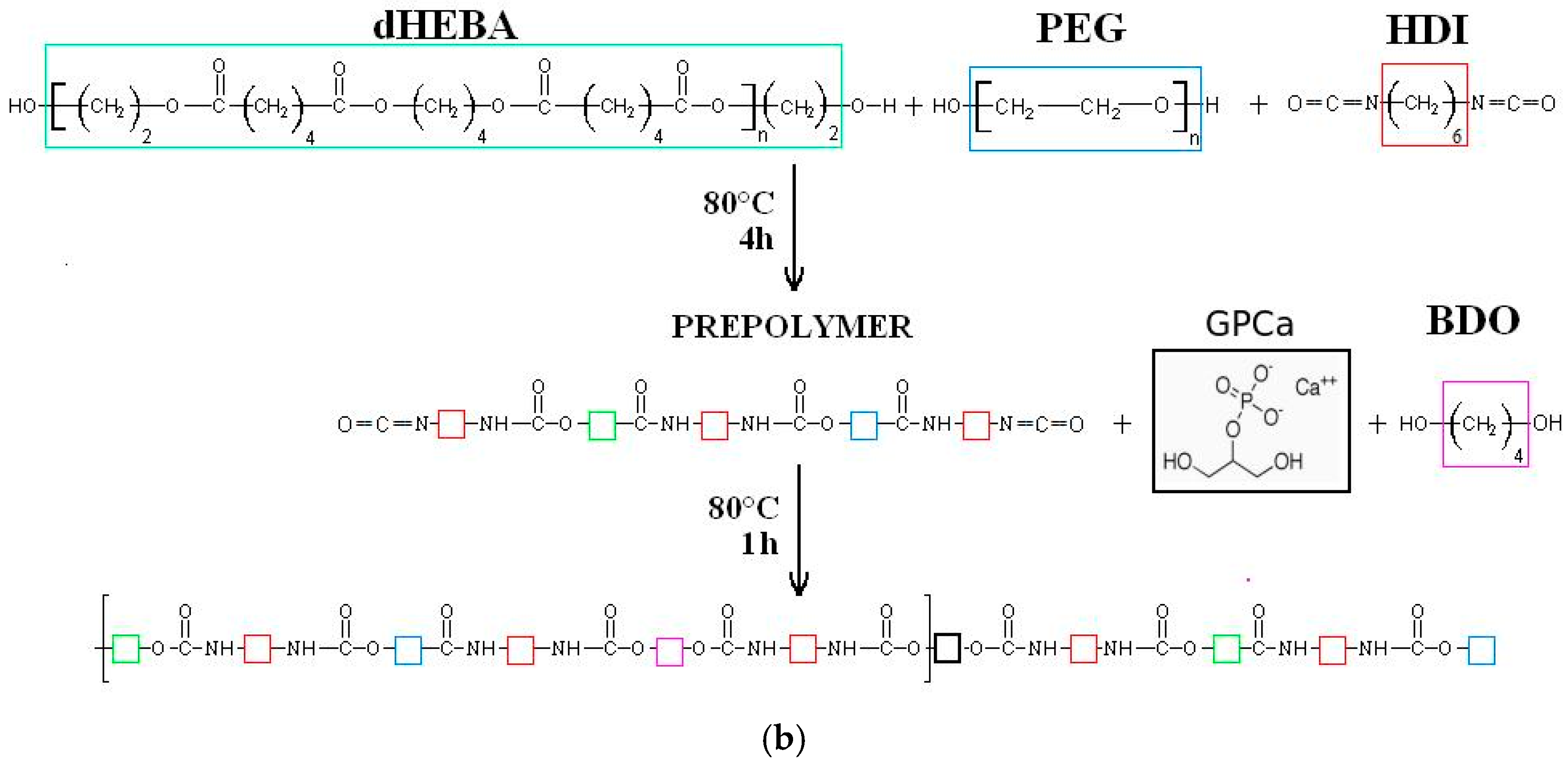
2.1. Poly(ester ether urethane)s Synthesis
2.2. Characterization Methods
2.2.1. Indications of Free Isocyanate Groups (FNCO) by the Acidimetric Method
2.2.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.2.3. Raman Spectroscopy
2.2.4. Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy (SEM/EDX)
2.2.5. Static Contact Angle Determination
2.2.6. Mechanical Properties
2.2.7. Short-Term Interactions Study Performed in Selected Environments
2.2.8. In Vitro Cytocompatibility
Cell Viability Assay
2.2.9. Calcification Study
2.2.10. Scaffold Fabrication
3. Results and Discussion
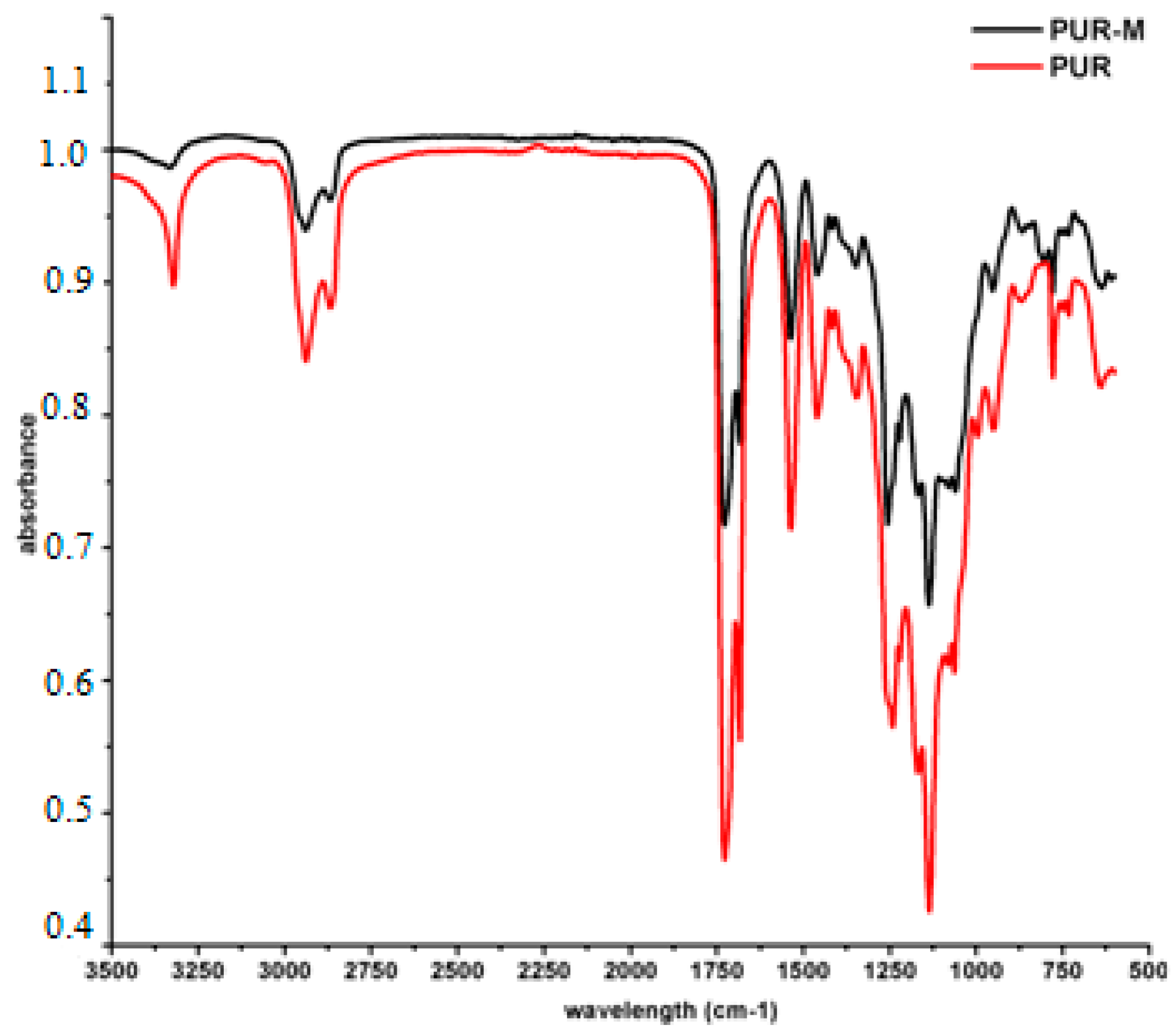
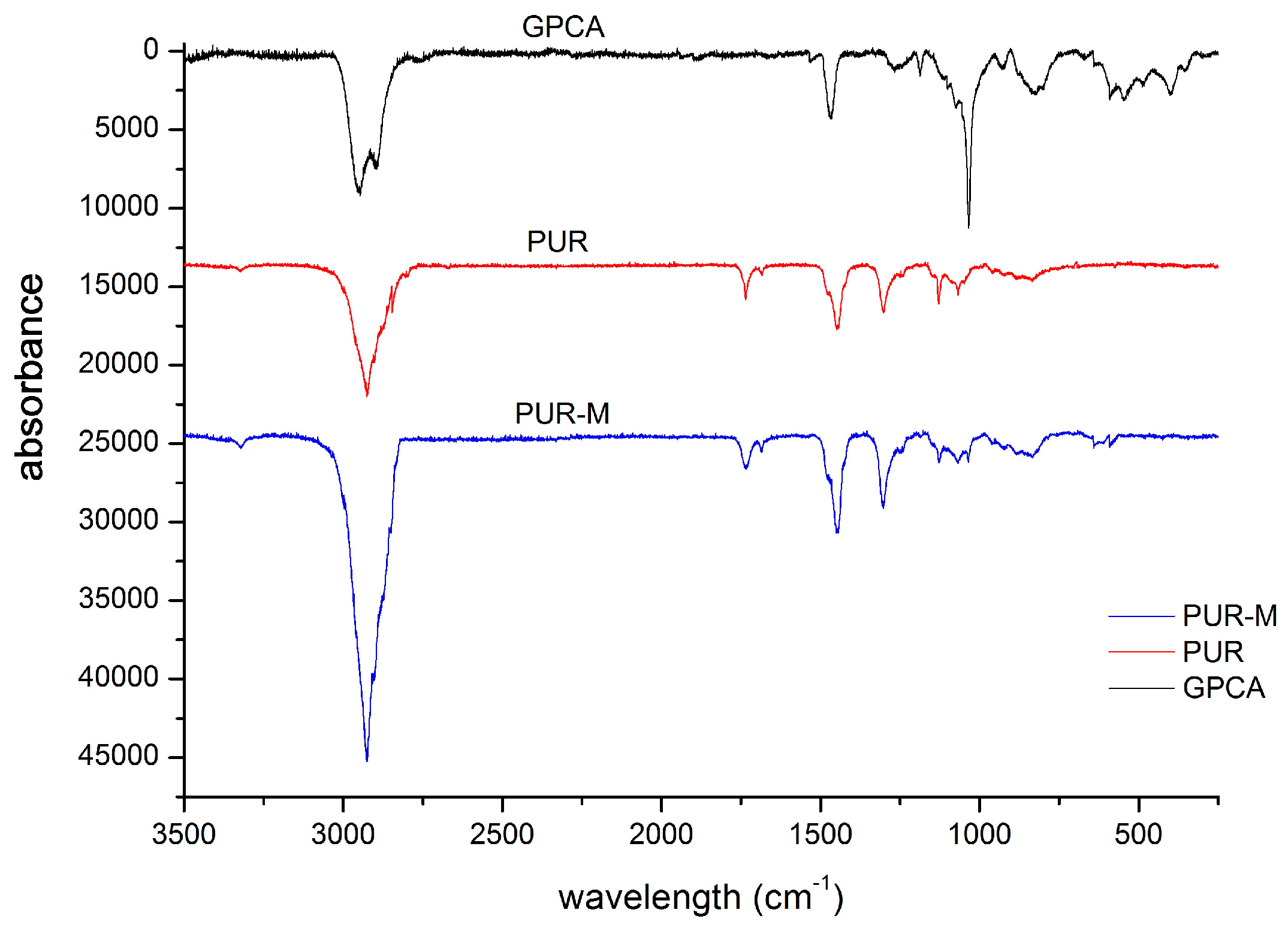
3.1. Fourier Transform Infrared Spectroscopy (FTIR)
3.2. Raman Spectroscopy
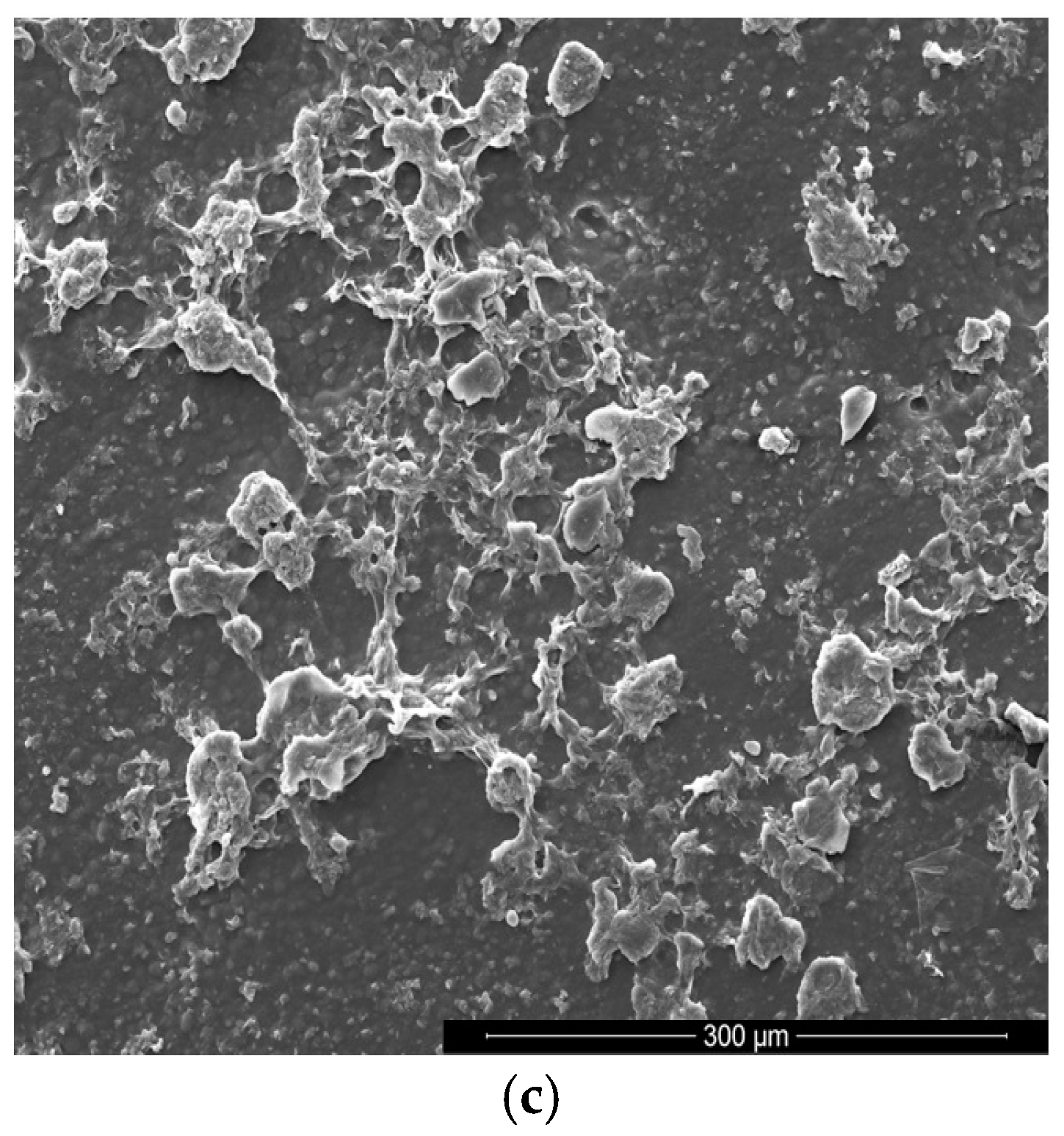
3.3. Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy(SEM/EDX)
3.4. Contact Angle
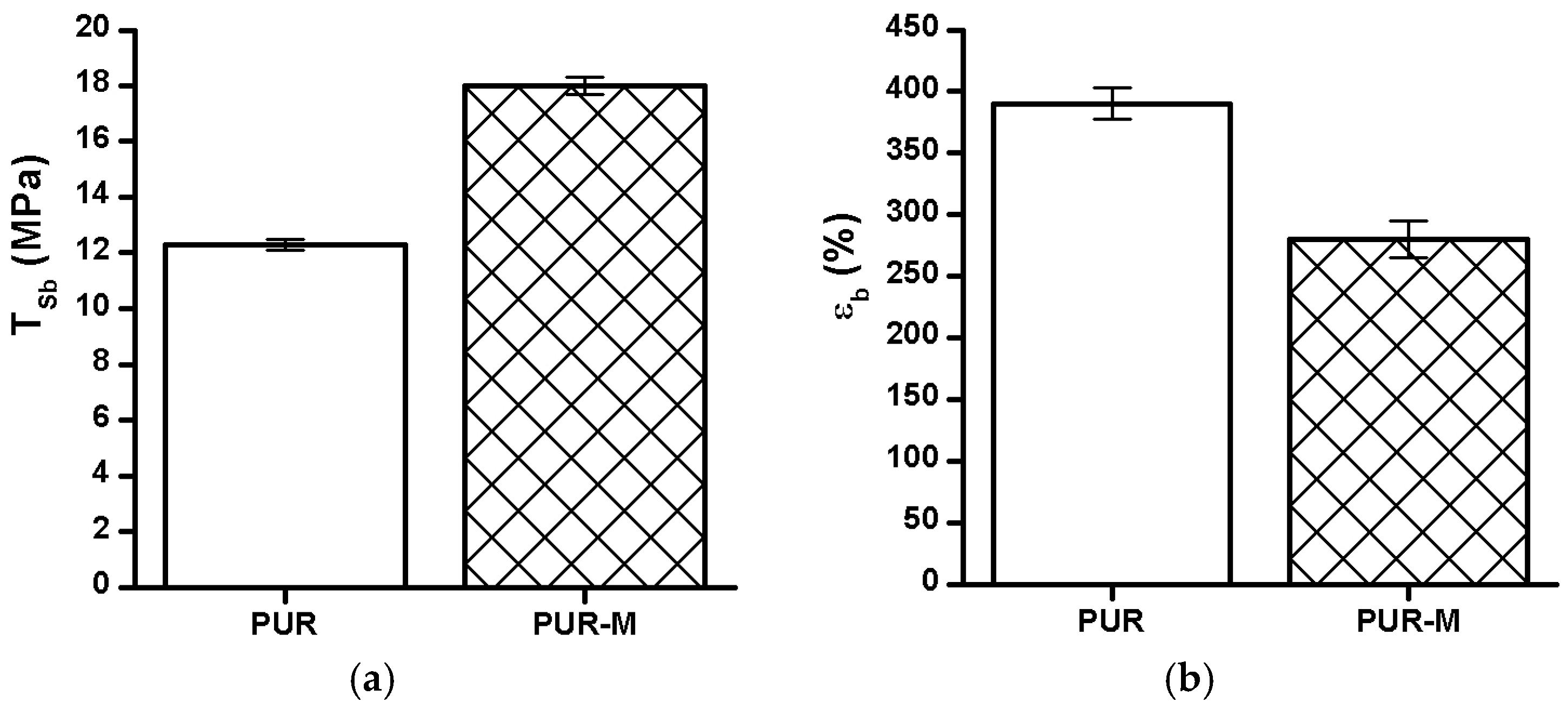
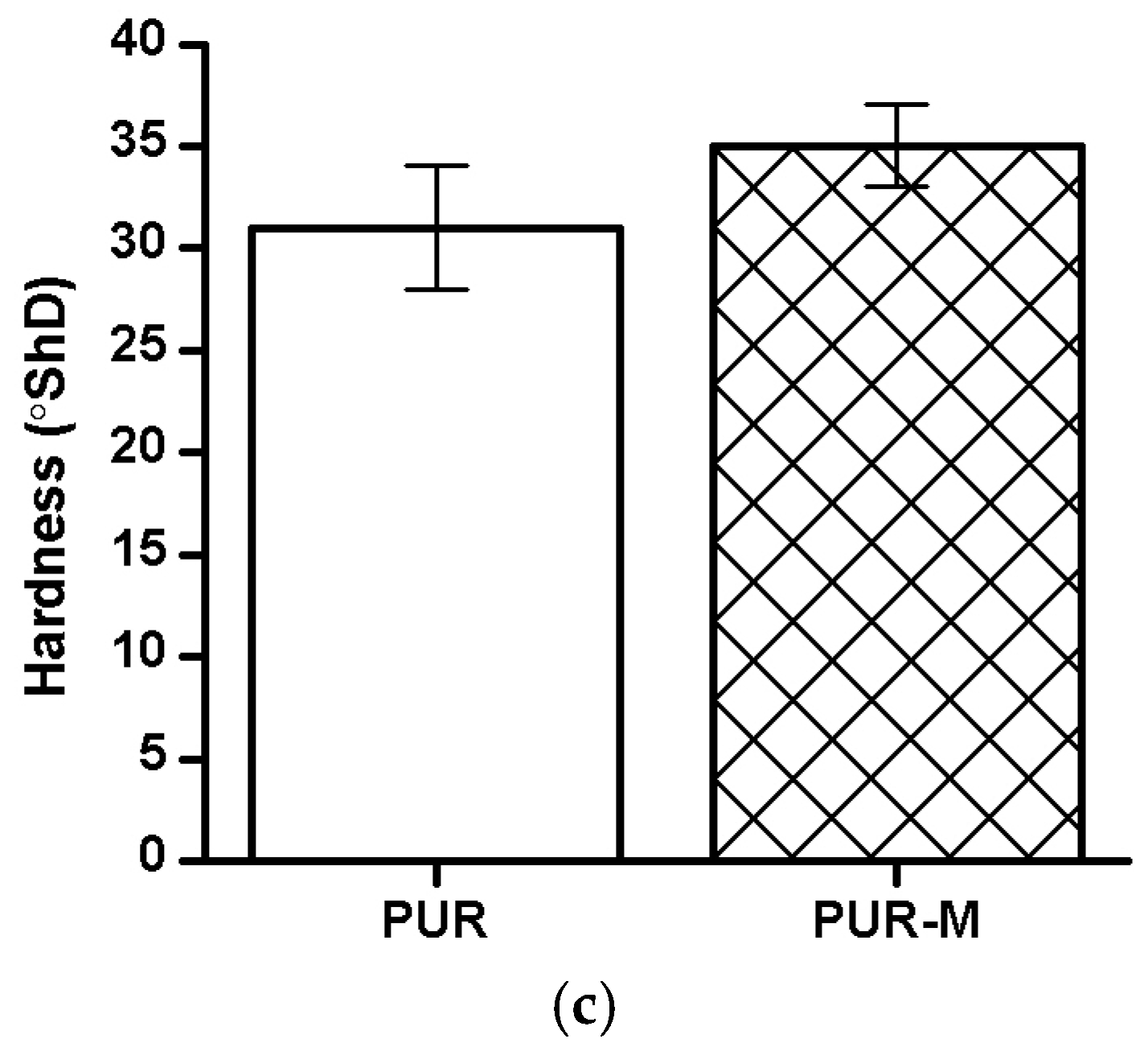
3.5. Mechanical Properties
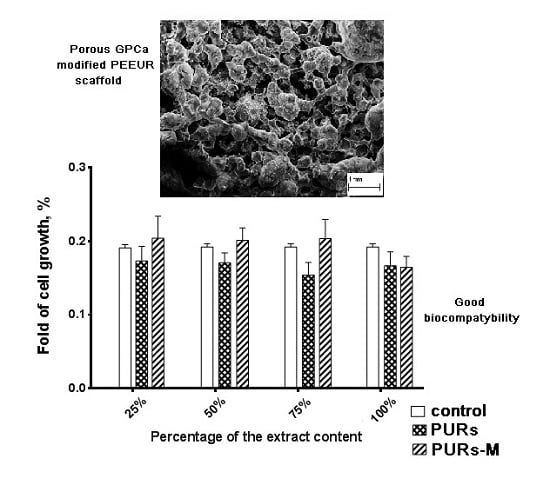
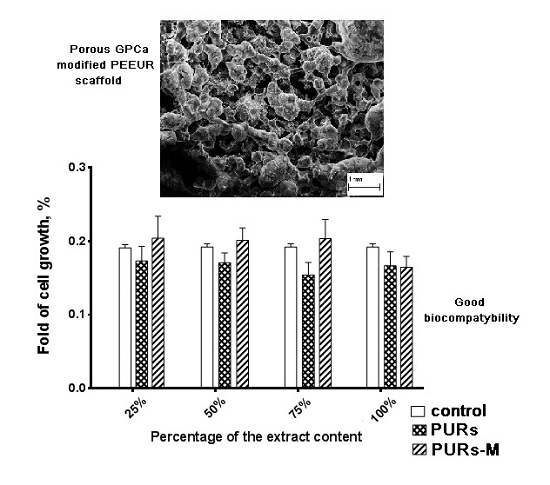
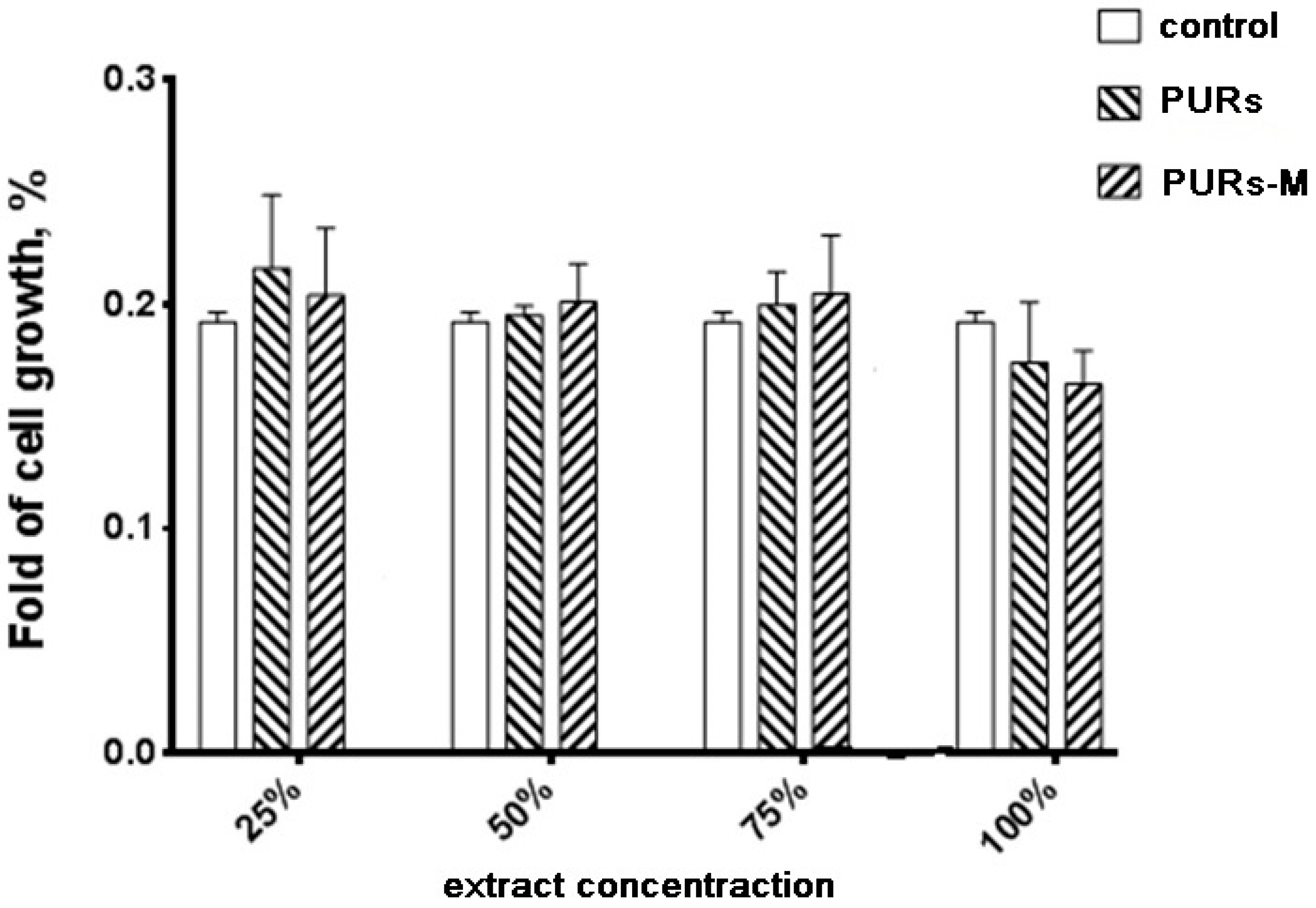
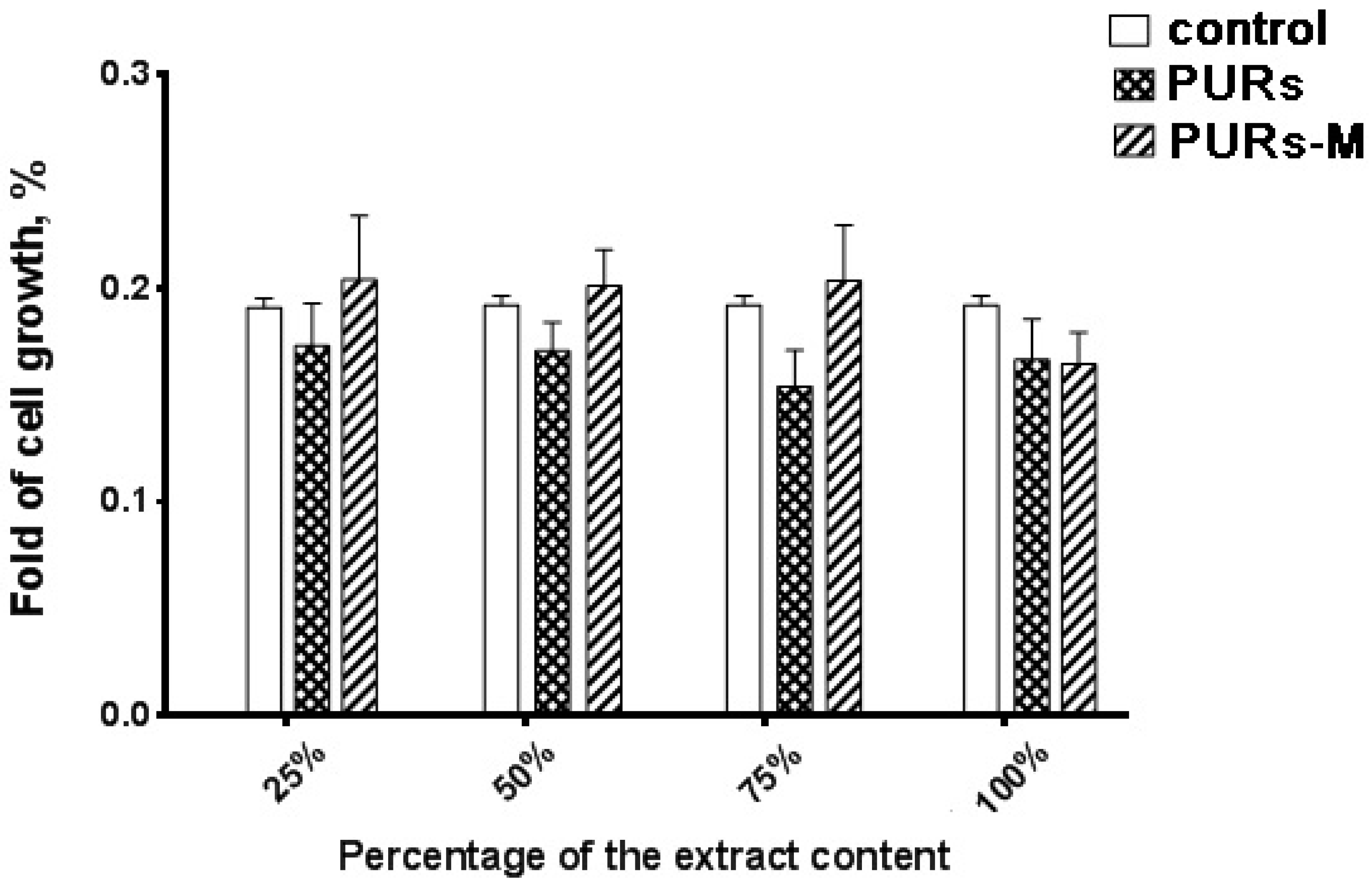
3.6. In Vitro Cytocompatibility
3.7. Short-Term Interactions Study Performed in Selected Environments
3.8. Calcification Study
3.9. Fabrication of PUR and PURs-M Scaffolds
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kucinska-Lipka, J.; Gubanska, I.; Strankowski, M.; Cieśliński, H.; Filipowicz, N.; Janik, H. Synthesis and characterization of cycloaliphatic hydrophilic polyurethanes, modified with L-ascorbic acid, as materials for soft tissue regeneration. Mater. Sci. Eng. C 2017, 75, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Kucinska-Lipka, J.; Gubanska, I.; Janik, H.; Sienkiewicz, M. Fabrication of polyurethane and polyurethane based composite fibres by the electrospinning technique for soft tissue engineering of cardiovascular system. Mater. Sci. Eng. C 2015, 46, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Woo, T.G.; Jeon, W.Y.; Park, H.H.; Lee, M.H.; Bae, T.S.; Keong, W.S. Surface characteristics of titanium anodized in the four different types of electrolyte. Electrochim. Acta 2007, 53, 863–870. [Google Scholar] [CrossRef]
- Du, J.; Zou, Q.; Zuo, Y.; Li, Y. Cytocompatibility and osteogenesis evaluation of HA/GCPU composite as scaffolds for bone tissue engineering. Int. J. Surg. 2014, 12, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Ryszkowska, J.L.; Auguścik, M.; Sheikh, A.; Boccaccini, A.R. Biodegradable polyurethane composite scaffolds containing Bioglass?? for bone tissue engineering. Compos. Sci. Technol. 2010, 70, 1894–1908. [Google Scholar] [CrossRef]
- Kucinska-Lipka, J.; Gubanska, I.; Janik, H.; Pokrywczynska, M.; Drewa, T. l-ascorbic acid modified poly(ester urethane)s as a suitable candidates for soft tissue engineering applications. React. Funct. Polym. 2015, 97, 105–115. [Google Scholar] [CrossRef]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Janik, H.; Marzec, M. A review: Fabrication of porous polyurethane scaffolds. Mater. Sci. Eng. C 2015, 48, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Kucinska-Lipka, J.; Gubanska, I.; Sienkiewicz, M. Thermal and mechanical properties of polyurethanes modified with L-ascorbic acid. J. Therm. Anal. Calorim. 2017, 127, 1631–1638. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Shi, P.; Zou, Q.; Zuo, Y.; Li, Y. Hydroxyapatite/polyurethane scaffold incorporated with drug-loaded ethyl cellulose microspheres for bone regeneration. J. Biomed. Mater. Res. B 2010, 95, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, G.; Khan, A.S.; Delaine-Smith, R.M.; Reilly, G.C.; Rehman, I.U. Electrospun polyurethane/hydroxyapatite bioactive Scaffolds for bone tissue engineering: The role of solvent and hydroxyapatite particles. J. Mech. Behav. Biomed. Mater. 2014, 39, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Li, Y.; Zou, Q. Degradation and biocompatibility of porous nano-hydroxyapatite/polyurethane composite scaffold for bone tissue engineering. Appl. Surf. Sci. 2009, 255, 6087–6091. [Google Scholar] [CrossRef]
- Laschke, M.W.; Strohe, A.; Menger, M.D.; Alini, M.; Eglin, D. In vitro and in vivo evaluation of a novel nanosize hydroxyapatite particles/poly(ester-urethane) composite scaffold for bone tissue engineering. Acta Biomater. 2010, 6, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Bonzani, I.C.; Adhikari, R.; Houshyar, S.; Mayadunne, R.; Gunatillake, P.; Stevens, M.M. Synthesis of two-component injectable polyurethanes for bone tissue engineering. Biomaterials 2007, 28, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.; Gunatillake, P.A.; Griffiths, I.; Tatai, L.; Wickramaratna, M.; Houshyar, S.; Moore, T.; Mayadunne, R.T.M.; Mc Gee, M.; Carbone, T. Biodegradable injectable polyurethanes: Synthesis and evaluation for orthopaedic applications. Biomaterials 2008, 29, 3762–3770. [Google Scholar] [CrossRef] [PubMed]
- Gorna, K.; Gogolewski, S. Preparation, degradation, and calcification of biodegradable polyurethane foams for bone graft substitutes. J. Biomed. Mater. Res. A 2003, 67, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Zawadzak, E.; Bil, M.; Ryszkowska, J.; Nazhat, S.N.; Cho, J.; Bretcanu, O.; Roethe, J.A.; Boccaccini, A.R. Polyurethane foams electrophoretically coated with carbon nanotubes for tissue engineering scaffolds. Biomed. Mater. 2009, 4, 15008. [Google Scholar] [CrossRef] [PubMed]
- Polish Ministry of Health. Polish Online System of Legal Acts. Available online: http://isap.sejm.gov.pl/ (accessed on 13 July 2016).
- American Food and Drug Administration. Available online: www.fda.gov (accessed on 13 July 2016).
- Zaze, A.C.S.F.; Dias, A.P.; Amaral, J.G.; Miyasaki, M.L.; Sassaki, K.T.; Delbem, A.C.B. In situ evaluation of low-fluoride toothpastes associated to calcium glycerophosphate on enamel remineralization. J. Dent. 2014, 42, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.S.; Bönecker, M.; Altenburger, M.J.; Buzalaf, M.A.R.; Sampaio, F.C.; Lussi, A. Fluoride varnishes containing calcium glycerophosphate: Fluoride uptake and the effect on in vitro enamel erosion. Clin. Oral Investig. 2015, 19, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.E.L.; Pilarek, M.; Kalaszczyńska, I.; Senderek, I.; Skwarczyńska, A.; Cuijpers, V.M.J.I.; Modrzejewska, Z.; Lewandowska-Szumieł, M.; Dubruel, P. Enrichment of chitosan hydrogels with perfluorodecalin promotes gelation and stem cell vitality. Mater. Lett. 2014, 128, 79–84. [Google Scholar] [CrossRef]
- Kavanaugh, T.E.; Clark, A.Y.; Chan-Chan, L.H.; Ramírez-Saldaña, M.; Vargas-Coronado, R.F.; Cervantes-Uc, J.M. Human mesenchymal stem cell behavior on segmented polyurethanes prepared with biologically active chain extenders. J. Mater. Sci. Mater. Med. 2016, 27, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.; Coelho, J.F.J.; Haack, J.; Rota, A.; Bruinink, A.; Gil, M.H. Surface modification and characterization of thermoplastic polyurethane. Eur. Polym. J. 2009, 45, 1412–1419. [Google Scholar] [CrossRef]
- Boloori Zadeh, P.; Corbett, S.C.; Nayeb-Hashemi, H. In-vitro calcification study of polyurethane heart valves. Mater. Sci. Eng. C 2014, 35, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, A.; Boffito, M.; Sartori, S.; Ciardelli, G. Biomimetic materials and scaffolds for myocardial tissue regeneration. Macromol. Biosci. 2013, 13, 984–1019. [Google Scholar] [CrossRef] [PubMed]
- Courtney, T.; Sacks, M.S.; Stankus, J.; Guan, J.; Wagner, W.R. Design and analysis of tissue engineering scaffolds that mimic soft tissue mechanical anisotropy. Biomaterials 2006, 27, 3631–3638. [Google Scholar] [CrossRef] [PubMed]
- Karchin, A.; Simonovsky, F.I.; Ratner, B.D.; Sanders, J.E. Melt electrospinning of biodegradable polyurethane scaffolds. Acta Biomater. 2011, 7, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Ihara, M.; Inoguchi, H.; Kwon, I.K.; Takamizawa, K.; Kidoaki, S. Mechano-active scaffold design of small-diameter artificial graft made of electrospun segmented polyurethane fabrics. J. Biomed. Mater. Res. A 2005, 73, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.M.; Lee, K.H.; Skrovanek, D.J.; Painter, P.C. Hydrogen bonding in polymers. 4. Infrared temperature studies of a simple polyurethane. Macromolecules 1986, 19, 2149–2157. [Google Scholar] [CrossRef]
- Coleman, M.M.; Skrovanek, D.J.; Hu, J.; Painter, P.C. Hydrogen bonding in polymer blends. 1. FTIR studies of urethane-ether blends. Macromolecules 1988, 21, 59–65. [Google Scholar] [CrossRef]
- Spirkova, M.; Poręba, R.; Pavlicevic, J.; Kobera, L.; Baldrian, J.; Pakarek, M. Aliphatic Polycarbonate-Based Polyurethane Elastomers and Nanocomposites. I. The Influence of Hard-Segment Content and Macrodiol-Constitution on Bottom-Up Self-Assembly. J. Appl. Polym. Sci. 2012, 126, 1016–1030. [Google Scholar] [CrossRef]
- Yilgor, I.; Yilgor, E.; Guler, I.G.; Ward, T.C.; Wilkes, G.L. FTIR investigation of the influence of diisocyanate symmetry on the morphology development in model segmented polyurethanes. Polymer 2006, 47, 4105–4114. [Google Scholar] [CrossRef]
- Yohannan Panicker, C.; Tresa Varghese, H.; Philip, D. FT-IR, FT-Raman and SERS spectra of Vitamin C. Spectrochim. Acta A 2006, 65, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Asefnejad, A.; Khorasani, M.T.; Behnamghader, A.; Farsadzadeh, B.; Bonakdar, S. Manufacturing of biodegradable polyurethane scaffolds based on polycaprolactone using a phase separation method: Physical properties and in vitro assay. Int. J. Nanomed. 2011, 6, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, T.; Peng, C.; Adegbite, S. Influence of acid and alkali pre-treatments on thermal degradation behaviour of polyisocyanurate foam and its carbon morphology. Polym. Degrad. Stab. 2017, 141, 104–118. [Google Scholar] [CrossRef]
- Cooke, S.L.; Whittington, A.R. Influence of therapeutic radiation on polycaprolactone and polyurethane biomaterials. Mater. Sci. Eng. C 2016, 60, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Strankowski, M.; Włodarczyk, D.; Piszczyk, Ł.; Strankowska, J. Polyurethane Nanocomposites Containing Reduced Graphene Oxide, FTIR, Raman, and XRD Studies. J. Spectrosc. 2016, 2016. [Google Scholar] [CrossRef]
- Gough, J.E.; Notingher, I.; Hench, L.L. Osteoblast attachment and mineralized nodule formation on rough and smooth 45S5 bioactive glass monoliths. J. Biomed. Mater. Res. 2004, 68, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Notingher, I.; Gough, J.E.; Hench, L.L. Study of osteoblasts mineralisation in-vitro by Raman micro-spectroscopy. Key Eng. Mater. 2004, 254–256, 769–772. [Google Scholar] [CrossRef]
- Silve, C.; Lopez, E.; Vidal, B.; Smith, D.C.; Camprasse, S.; Camprasse, G.; Couly, G. Nacre initiates biomineralization by human osteoblasts maintained In Vitro. Calcif. Tissue Int. 1992, 51, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Barrioni, B.R.; De Carvalho, S.M.; Oréfice, R.L.; De Oliveira, A.A.R.; Pereira, M.D.M. Synthesis and characterization of biodegradable polyurethane films based on HDI with hydrolyzable crosslinked bonds and a homogeneous structure for biomedical applications. Mater. Sci. Eng. C 2015, 52, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Gogolewski, S.; Gorna, K.; Turner, A.S. Regeneration of bicortical defects in the iliac crest of estrogen-deficient sheep, using new biodegradable polyurethane bone graft substitutes. J. Biomed. Mater. Res. 2006, 77, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone Tissue Engineering: Recent Advances and Challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Woodruff, M.A.; Epari, D.R.; Steck, R.; Glatt, V.; Dickinson, I.C.; Choong, P.F.; Schuetz, M.A.; Hutmacher, D.W. Bone Regeneration Based on Tissue Engineering Conceptions—A 21st Century Perspective. Bone Res. 2013, 1, 216–248. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Tang, X.; Gohil, S.V.; Laurencin, C.T. Biomaterials for Bone Regenerative Engineering. Adv. Healthc. Mater. 2015, 4, 1268–1285. [Google Scholar] [CrossRef] [PubMed]
- Bil, M.; Ryszkowska, J.; Woźniak, P.; Kurzydłowski, K.J.; Lewandowska-Szumieł, M. Optimization of the structure of polyurethanes for bone tissue engineering applications. Acta Biomater. 2010, 6, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Guelcher, S.A.; Srinivasan, A.; Dumas, J.E.; Didier, J.E.; McBride, S.; Hollinger, J.O. Synthesis, mechanical properties, biocompatibility, and biodegradation of polyurethane networks from lysine polyisocyanates. Biomaterials 2008, 29, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, M.K. Interfacial interaction between low energy surfaces. Mater. Sci. Eng. R Rep. 1996, 16, 97–159. [Google Scholar] [CrossRef]
- Król, P.; Król, B. Surface free energy of polyurethane coatings with improved hydrophobicity. Colloid Polym. Sci. 2012, 290, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable materials for bone repair and tissue engineering applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef]
- Teoh, S.H.; Tang, Z.G.; Hastings, G.W. Chapter 3 Thermoplastic Polymers In Biomedical Applications: Structures, Properties and Processing. In Handbook of Biomaterial Properties; Murphy, W., Black, J., Hastings, G., Eds.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Montini-Ballarin, F.; Caracciolo, P.C.; Rivero, G.; Abraham, G.A. In vitro degradation of electrospun poly(l-lactic acid)/segmented poly(ester urethane) blends. Polym. Degrad. Stab. 2016, 126, 159–169. [Google Scholar] [CrossRef]
- Gogolewski, S. Nonmetallic materials for bone substitutes. Eur. Cells Mater. 2001, 1 (Suppl. 2), 54–55. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time of prepolymeryzation (h) | Content of the free isocyanate groups in the unmodified prepolymer (NCO ) | Content of the free isocyanate groups in the modified prepolymer (NCO ) |
|---|---|---|
| PEEUR | PEEUR-M | |
| 0 | 10.14 ± 0.03 | 10.14 ± 0.03 |
| 1 | 9.04 ± 0.01 | 9.04 ± 0.01 |
| 2 | 8.83 ± 0.03 | 8.83 ± 0.03 |
| 3 | 8.54 ± 0.03 | 8.54 ± 0.03 |
| 4 | 8.54 ± 0.02 | 8.54 ± 0.02 |
| 5 | 8.54 ± 0.02 | 8.43 ± 0.01 * |
| 6 | 8.53 ± 0.03 | 8.33 ± 0.03 |
| 7 | 8.53 ± 0.01 | 8.14 ± 0.03 |
| 8 | 8.53 ± 0.02 | 8.12 ± 0.02 |
| 9 | 8.53 ± 0.01 | 8.12 ± 0.03 |
| PUR | PUR-M | Band | Description |
|---|---|---|---|
| Wavelength (cm−1) | |||
| 3392 3326w | 3380 3318w | νNH | stretching of NH groups, hydrogen bonded withthe C=O of the ester group present in macrodiol and in GCPa modifier |
| 2942m 2868m | 2942m 2868m | νCH2 | stretching of aliphatic asymmetric and symmetric CH2 groups present in the PUR chain and in GCPa modifier |
| 1728m | 1728s | νC=O | stretching of C=O in the ester and urethane groups, which were not bonded |
| 1680m | 1680s | νC=O | stretching of C=O groups which formed hydrogen bonds |
| 1535m | 1535s | νC–N | stretching between CN in urethane group |
| 1462w 1416w 1350w | 1459m 1415m 1347m | δCH2 | deformation vibrations of aliphatic CH2 groups present in the PUR and GPCa modifier: bending, wagging, scissoring in plane |
| 1260w 1214m | 1238s 1216s | νC–(C=O)–O | stretching vibrations of –C–(C=O)–O– of ester group, not hydrogen bonded |
| 1170m | 1170s | νNH–(C=O)–O | stretching vibrations of –NH–(C=O)–O– of urethane group |
| 1135m 1080m 1058m 947w | 1134s 1061s 993s 949s | νC–(C=O)–O νC–O | stretching vibration of hydrogen bonded –C–(C=O)–O–, |
| 868w 813w 775w 731w 636w | 867s 777s 730s 638s | δCH2, δNH δOH | out of the plane deformation of CH2 and CH3 groups as well as NH and OH groups |
| Wavelength (cm−1) | Assignments * |
|---|---|
| 3328 | stretching vibrations of N–H in urethane groups (as for II-ary amides) |
| 2925, 2890 | the strongest polarized stretching vibrations of asymmetric and symmetric CH2 groups present in PUR chains. |
| 1735, 1685 | stretching vibrations of carbonyl groups present in macrodiol Polios 55/20 and urethane groups (as for II-ary amides) respectively |
| 1480, 1450, 1443, 1424 | strong planar deforming vibrations (scissoring) of CH2 groups |
| 1302 | swinging and bending vibrations of CH2 groups outside of plane |
| 1249 | stretching vibrations of C–N in urethane groups |
| 1127, 1068 | stretching asymmetric and symmetric vibrations of C–O–C in ester groups respectively |
| 1035–1095 | stretching vibrations of saturated aliphatic chains C–C–C–C |
| 962, 935, 885, 832 | swaying vibrations of CH2 groups in different positions and deforming bending vibrations outside of the plane of N–H groups |
| 610 | deforming vibrations outside of the plane of ester groups and their fluctuations |
| Wavelength (cm−1) | Assignments * |
|---|---|
| 3315, 3326 | stretching vibrations of N–H in urethane groups (as for II amides), P–O–Ca stretch |
| 2925, 2883 | the strongest polarized stretching vibrations of asymmetric and symmetric CH2 groups present in PUR chains. Analogic CH2 in H2C–O–P–O stretch phonons included |
| 1733 | stretching vibrations of carbonyl groups present in macrodiol Polios 55/20 |
| 1684 | stretching vibrations of carbonyl groups C=O in urethane (HDI–BDO) |
| 1483, 1452, 1442, 1421 | deforming and scissoring vibrations of CH2 groups in both polymer and GPCa |
| 1302, 1261 | swinging and bending vibrations of CH2 out of the plane groups in polymer and filler with additional C–N stretch at the end |
| 1126, 1131 | stretching asymmetric vibrations of C–O–C in ester groups |
| 1040,1080 | complex CCCC stretch in branched alkanes with symmetric PO4 and C–O–P stretch |
| 959, 939, 882, 835 | swinging vibrations of CH2 groups in different positions and NH, CH bending deformation vibrations outside of the plane of urethane groups |
| 580,614 | deforming vibrations outside of the plane of ester groups with eventual phosphoester and PO4 fluctuations |
| Symbol | Contact angle (°) | Surface energy (mN/m) | ||||
|---|---|---|---|---|---|---|
| Formamide | Ethylene glycol | Water | Acid-part | Base-part | Total surface free energy | |
| PUR | 63.6 ± 2 | 68.2 ± 1 | 72.1 ± 2 | 0.04 | 20.83 | 31.82 |
| PUR-M | 35.8 ± 4 | 47.5 ± 3 | 57 ± 3 | 14.51 | 21.72 | 59.09 |
| Sample | Extracted mass (%) | ||
|---|---|---|---|
| 5 M NaOH | 2 N HCl | 0.1 M CoCl2/20% H2O2 | |
| PUR | 50.9 ± 0.2 | 38.62 ± 0.14 | 4.25 ± 0.08 |
| PUR -M | 54.7 ± 0.1 | 35.81 ± 0.11 | 2.03 ± 0.12 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucińska-Lipka, J.; Gubanska, I.; Korchynskyi, O.; Malysheva, K.; Kostrzewa, M.; Włodarczyk, D.; Karczewski, J.; Janik, H. The Influence of Calcium Glycerophosphate (GPCa) Modifier on Physicochemical, Mechanical, and Biological Performance of Polyurethanes Applicable as Biomaterials for Bone Tissue Scaffolds Fabrication. Polymers 2017, 9, 329. https://doi.org/10.3390/polym9080329
Kucińska-Lipka J, Gubanska I, Korchynskyi O, Malysheva K, Kostrzewa M, Włodarczyk D, Karczewski J, Janik H. The Influence of Calcium Glycerophosphate (GPCa) Modifier on Physicochemical, Mechanical, and Biological Performance of Polyurethanes Applicable as Biomaterials for Bone Tissue Scaffolds Fabrication. Polymers. 2017; 9(8):329. https://doi.org/10.3390/polym9080329
Chicago/Turabian StyleKucińska-Lipka, Justyna, Iga Gubanska, Olexandr Korchynskyi, Khrystyna Malysheva, Marcin Kostrzewa, Damian Włodarczyk, Jakub Karczewski, and Helena Janik. 2017. "The Influence of Calcium Glycerophosphate (GPCa) Modifier on Physicochemical, Mechanical, and Biological Performance of Polyurethanes Applicable as Biomaterials for Bone Tissue Scaffolds Fabrication" Polymers 9, no. 8: 329. https://doi.org/10.3390/polym9080329
APA StyleKucińska-Lipka, J., Gubanska, I., Korchynskyi, O., Malysheva, K., Kostrzewa, M., Włodarczyk, D., Karczewski, J., & Janik, H. (2017). The Influence of Calcium Glycerophosphate (GPCa) Modifier on Physicochemical, Mechanical, and Biological Performance of Polyurethanes Applicable as Biomaterials for Bone Tissue Scaffolds Fabrication. Polymers, 9(8), 329. https://doi.org/10.3390/polym9080329