Genomic Prediction of Rust Resistance in Tetraploid Wheat under Field and Controlled Environment Conditions


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Seedling and Field Evaluations
2.3. Genotyping
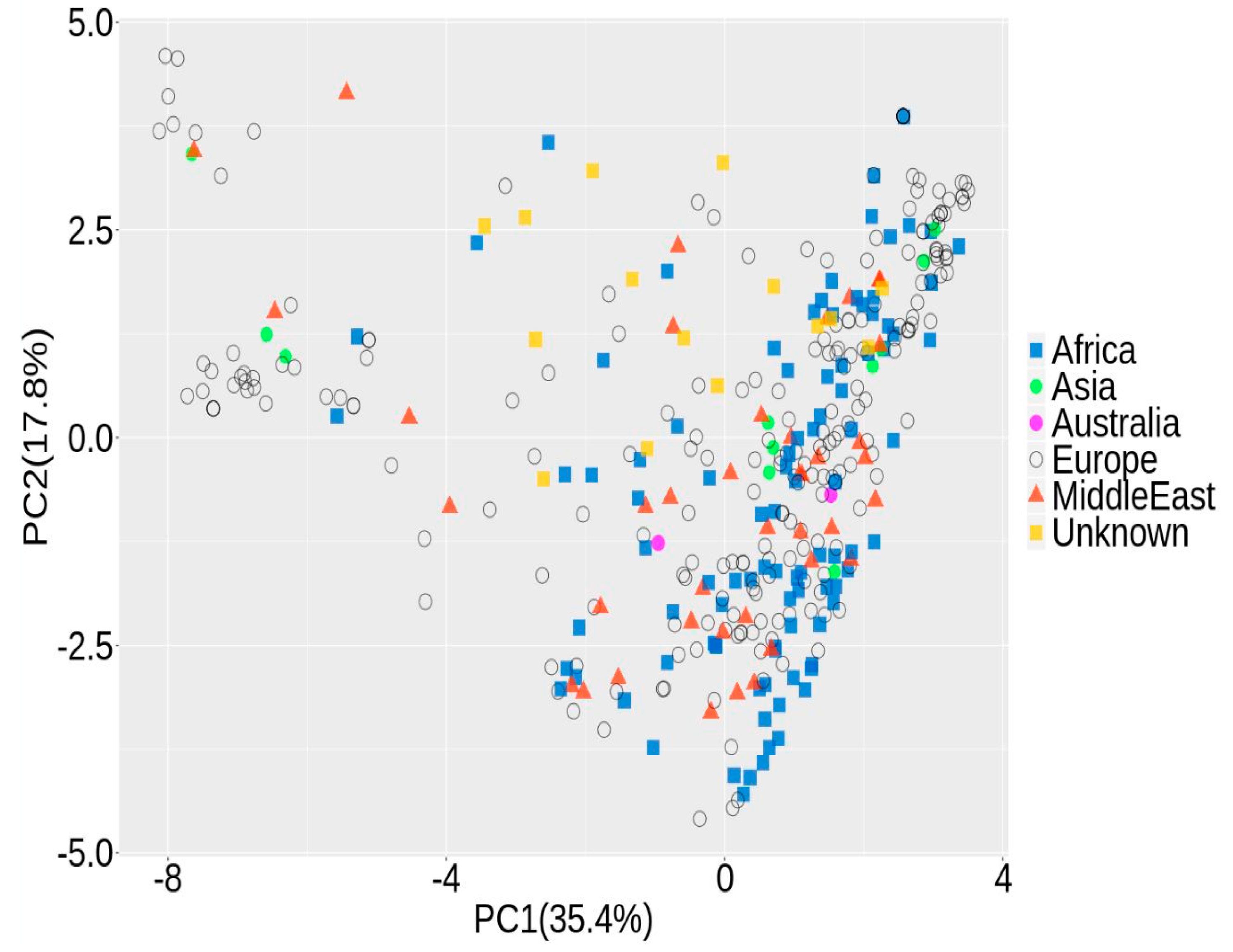
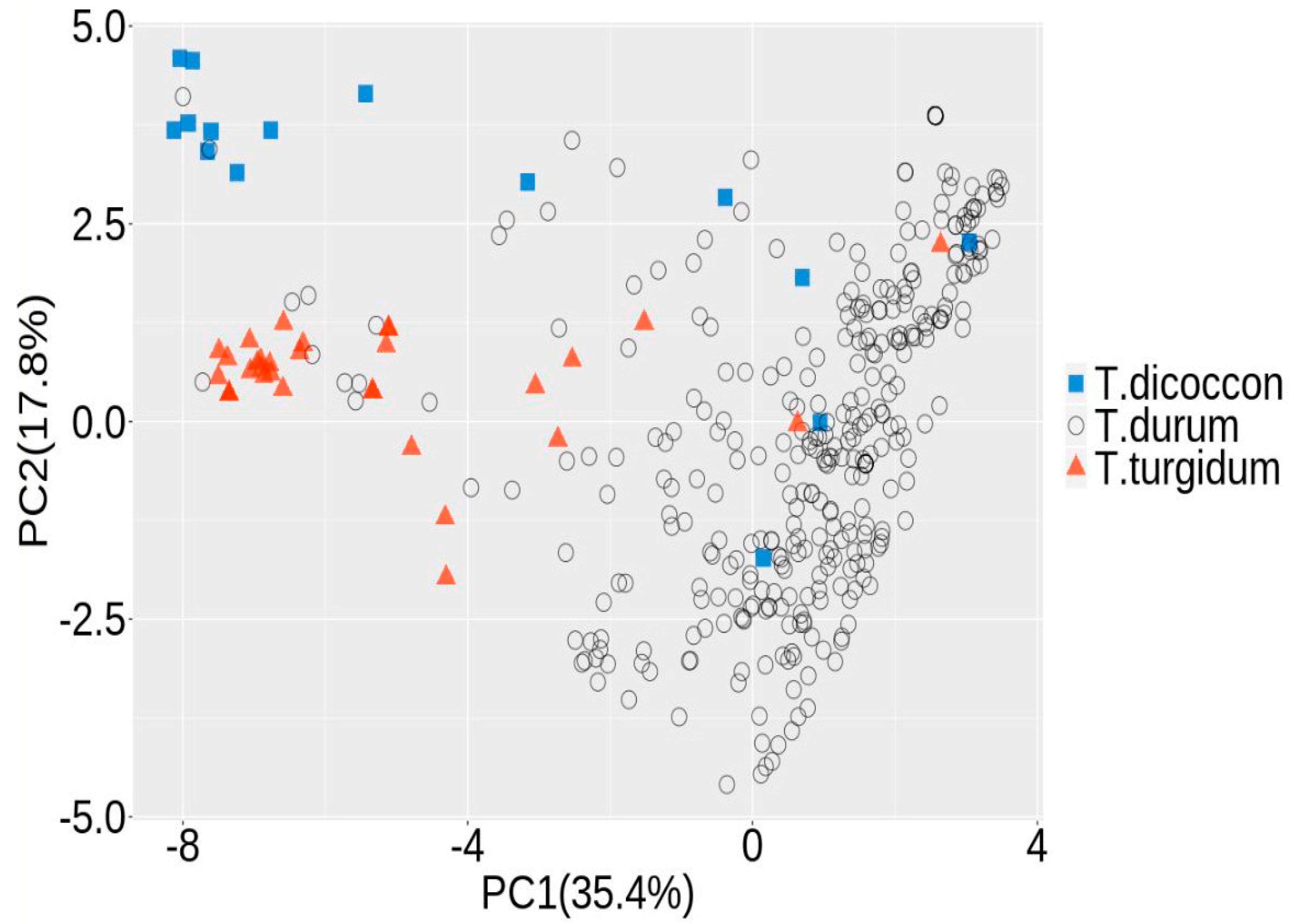
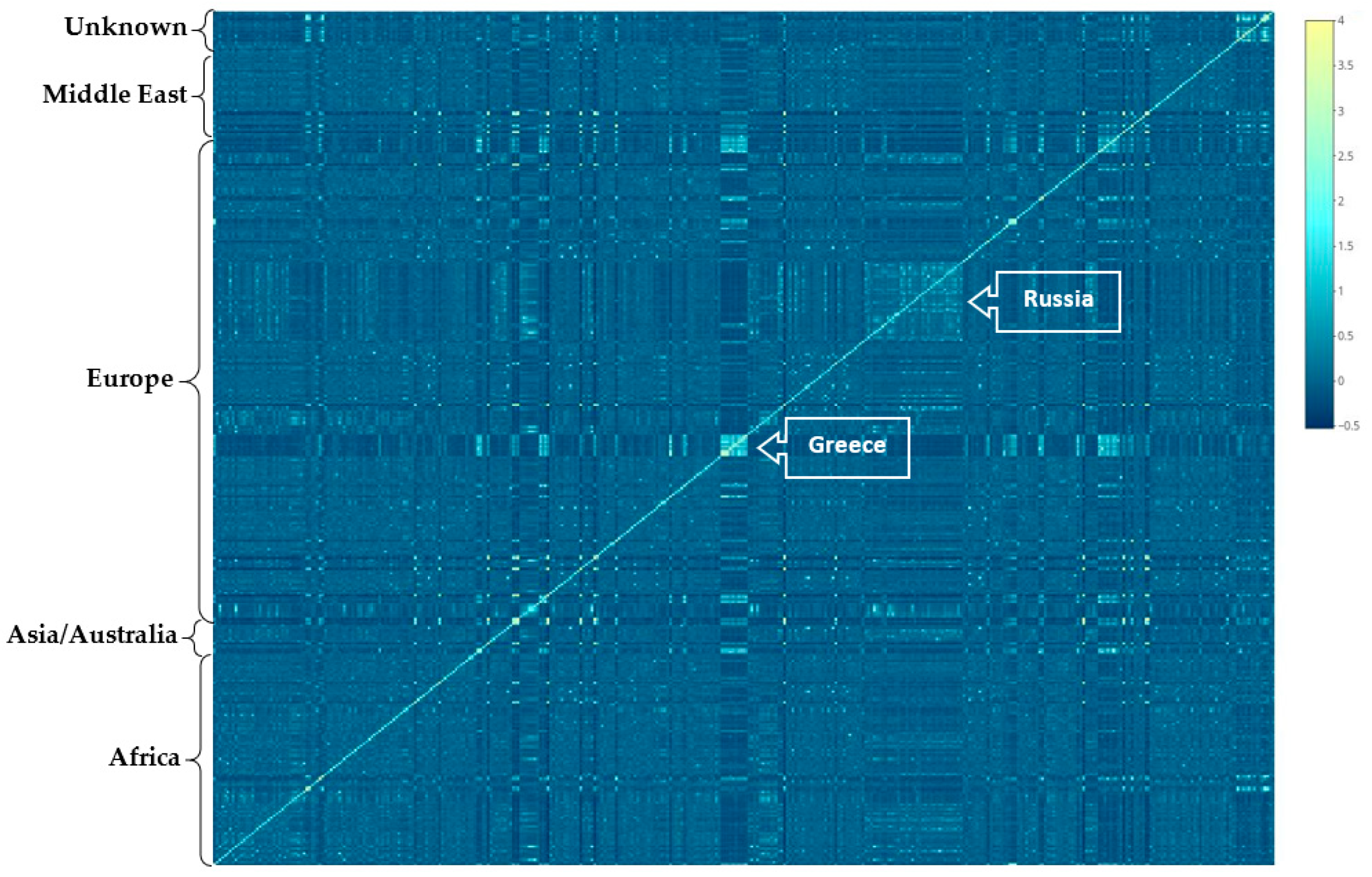
2.4. Population Structure
2.5. Genomic Prediction Models
2.5.1. Single Trait Genomic Model
2.5.2. Bi-Variate Genomic Model
2.5.3. Bayesian Models
BayesA
BayesB
BayesC
Bayes LASSO
Reproducing Kernel Hilbert Spaces
2.6. Heritability and Correlations
2.7. Evaluation of Genomic Prediction
3. Results
3.1. Population Structure
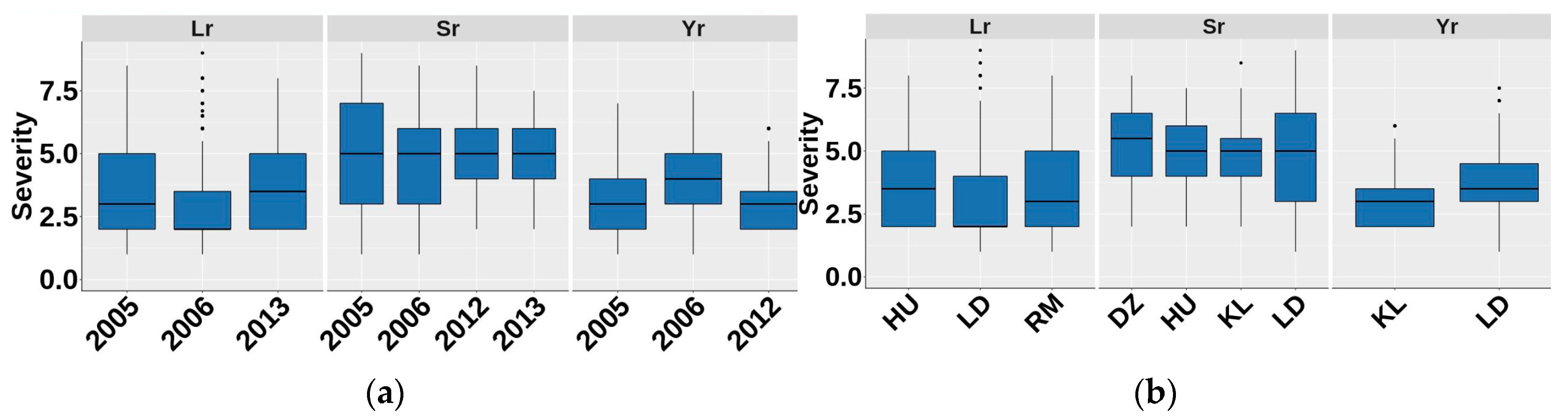
3.2. Phenotypic Variability and Heritability
3.3. Phenotypic and Genetic Correlations
3.4. Genomic Prediction
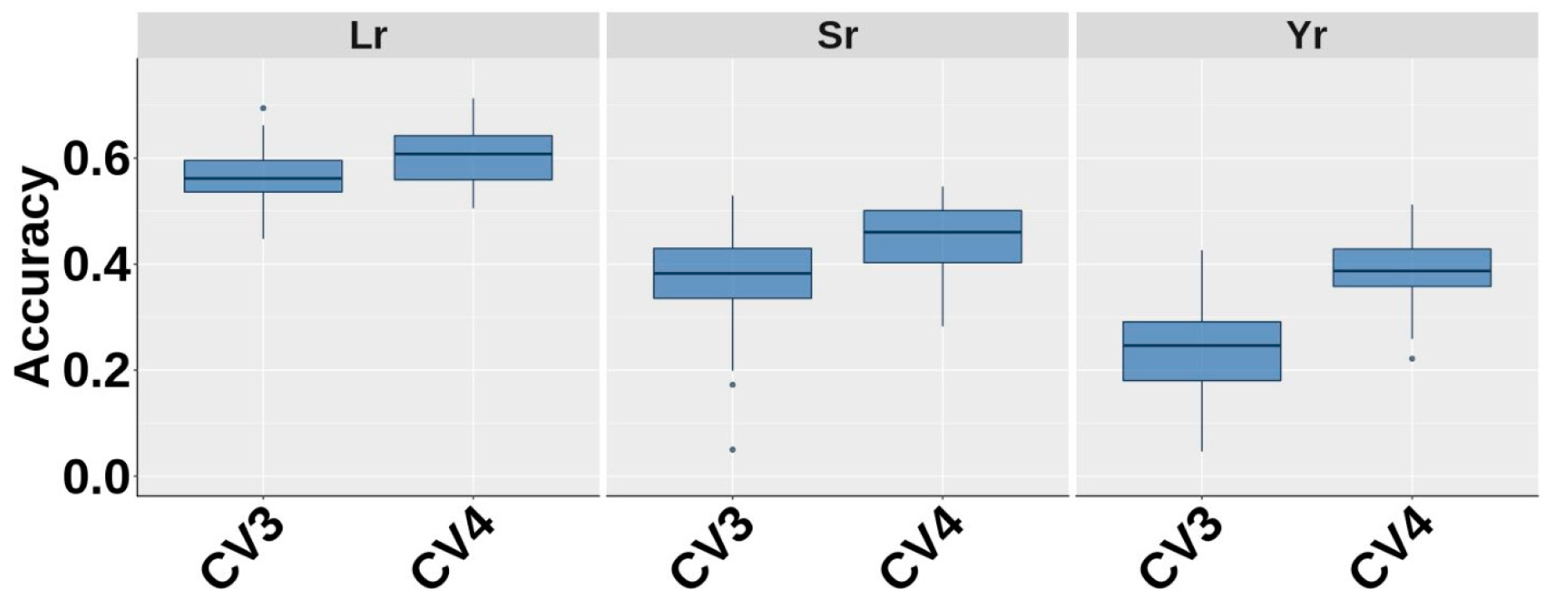
3.5. Genomic Prediction Across Years and Sites
3.6. Bivariate Genomic Prediction
4. Discussion
4.1. Predicting Across Years and Sites
4.2. Bivariate Prediction
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; Jakobsen, K.S.; Wulff, B.B.H.; Steuernagel, B.; Mayer, K.F.X.; Olsen, O.-A.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef]
- Figueroa, M.; Hammond-Kosack, K.E.; Solomon, P.S. A review of wheat diseases—A field perspective. Mol. Plant Pathol. 2018, 19, 1523–1536. [Google Scholar] [CrossRef]
- Bariana, H.S.; Bansal, U.K. Breeding for Disease Resistance. In Encyclopedia of Applied Plant Sciences; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 69–76. ISBN 9780123948083. [Google Scholar]
- Bariana, H.S. DISEASES|Breeding for Disease Resistance. In Encyclopedia of Applied Plant Sciences; Thomas, B., Ed.; Elsevier BV: Amsterdam, The Netherlands, 2003; pp. 244–253. ISBN 9780122270505. [Google Scholar]
- Ellis, J.G.; Lagudah, E.S.; Spielmeyer, W.; Dodds, P.N. The past, Present and future of breeding rust resistant wheat. Front. Plant Sci. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Goddard, M. Genomic selection: Prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Villanueva, B.; Woolliams, J.A. Accuracy of Predicting the Genetic Risk of Disease Using a Genome-Wide Approach. PLoS ONE 2008, 3, e3395. [Google Scholar] [CrossRef] [Green Version]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Pong-Wong, R.; Villanueva, B.; Woolliams, J.A. The Impact of Genetic Architecture on Genome-Wide Evaluation Methods. Genetics 2010, 185, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Pryce, J.; Chamberlain, A.J.; Bowman, P.J.; Goddard, M.E. Genetic Architecture of Complex Traits and Accuracy of Genomic Prediction: Coat Colour, Milk-Fat Percentage, and Type in Holstein Cattle as Contrasting Model Traits. PLoS Genet. 2010, 6, e1001139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.-L. Potential and Optimization of Genomic Selection for Fusarium Head Blight Resistance in Six-Row Barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Poland, J.; Rutkoski, J. Advances and Challenges in Genomic Selection for Disease Resistance. Annu. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef]
- Rutkoski, J.E.; Heffner, E.L.; Sorrells, M.E. Genomic selection for durable stem rust resistance in wheat. Euphytica 2011, 179, 161–173. [Google Scholar] [CrossRef]
- Ornella, L.; Singh, S.; Pérez-Rodríguez, P.; Burgueño, J.; Singh, R.; Tapia, E.; Bhavani, S.; Dreisigacker, S.; Braun, H.J.; Mathews, K.; et al. Genomic prediction of genetic values for resistance to wheat rusts. Plant Genome 2012, 5, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Bansal, U.K.; Bariana, H.S.; Hayden, M.J.; Hayes, B.J. Genomic prediction for rust resistance in diverse wheat landraces. Theor. Appl. Genet. 2014, 127, 1795–1803. [Google Scholar] [CrossRef]
- Rutkoski, J.E.; Poland, J.A.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.L.; Sorrells, M.E. Genomic selection for quantitative adult plant stem rust resistance in wheat. Plant Genome 2014, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Juliana, P.; Singh, R.P.; Singh, P.K.; Crossa, J.; Huerta-Espino, J.; Lan, C.; Bhavani, S.; Rutkoski, J.E.; Poland, J.A.; Bergstrom, G.C.; et al. Genomic and pedigree-based prediction for leaf, stem, and stripe rust resistance in wheat. Theor. Appl. Genet. 2017, 130, 1415–1430. [Google Scholar] [CrossRef] [Green Version]
- Muleta, K.T.; Bulli, P.; Zhang, Z.; Chen, X.; Pumphrey, M. Unlocking Diversity in Germplasm Collections via Genomic Selection: A Case Study Based on Quantitative Adult Plant Resistance to Stripe Rust in Spring Wheat. Plant Genome 2017, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Poland, J.; Jannink, J.L.; Sorrells, M.E. Efficient Use of Historical Data for Genomic Selection: A Case Study of Stem Rust Resistance in Wheat. Plant Genome 2015, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Poland, J.; Jannink, J.L.; Sorrells, M.E. Genetic Gain from Phenotypic and Genomic Selection for Quantitative Resistance to Stem Rust of Wheat. Plant Genome 2015, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.E.; Reader, S.M.; Devos, K.M.; Genetics, P.; Miller, T.E.; Reader, S.M.; Sciences, P.; Science, R. Items from The United Kingdom; John Innes Centre Norwich Research Park: Colney, Norwich, UK, 2000; The Watkins wheat collection; pp. 1–6. [Google Scholar]
- Olivera, P.D.; Jin, Y.; Rouse, M.; Badebo, A.; Fetch, T.; Singh, R.P.; Yahyaoui, A. Races of Puccinia graminis f. sp. tritici with Combined Virulence to Sr13 and Sr9e in a Field Stem Rust Screening Nursery in Ethiopia. Plant Dis. 2012, 96, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.F.; Campbell, A.B.; Hannah, A.E. A Diagrammatic Scale For Estimating Rust Intensity On Leaves And Stems Of Cereals. Can. J. Res. 1948, 26c, 496–500. [Google Scholar] [CrossRef]
- Roelfs, A.P.; Singh, R.P.; Saari, E.E. Rust Diseases of Wheat: Concepts and Methods of Disease Management; Cimmyt: El Batán, Mexico, 1992; ISBN 968612747X. [Google Scholar]
- Randhawa, M.; Bansal, U.; Lillemo, M.; Miah, H.; Bariana, H. Postulation of rust resistance genes in Nordic spring wheat genotypes and identification of widely effective sources of resistance against the Australian rust flora. J. Appl. Genet. 2016, 57, 453–465. [Google Scholar] [CrossRef]
- Long, D.L. A North American System of Nomenclature for Puccinia recondita f. sp. tritici. Phytopathology 1989, 79, 525–529. [Google Scholar] [CrossRef]
- McIntosh, R.A.; Wellings, C.R.; Park, R.F. Wheat Rusts: An Atlas of Resistance Genes. Australas. Plant Pathol. 1996, 25, 1–270. [Google Scholar] [CrossRef]
- Wellings, C.R.; McIntosh, R.A.; Walker, J. Puccinia striiformis f.sp. tritici in Eastern Australia possible means of entry and implications for plant quarantine. Plant Pathol. 1987, 36, 239–241. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.-Y.; Myles, S. LinkImpute: Fast and Accurate Genotype Imputation for Nonmodel Organisms. G3 Genes Genomes Genet. 2015, 5, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidro, J.; Jannink, J.L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias-Pazaran, G. Genome-Assisted Prediction of Quantitative Traits Using the R Package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: http://www.R-project.org/ (accessed on 10 October 2020).
- Tyner, S.; Briatte, F.; Hofmann, H. Network Visualization with ggplot2. R J. 2017, 9, 27–59. [Google Scholar] [CrossRef]
- Henderson, C.R. Best linear unbiased estimation and prediction under a selection model published by: International biometric society stable. Biometrics 1975, 31, 423–447. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.J.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tishbirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar]
- Gianola, D.; Fernando, R.L.; Stella, A. Genomic-Assisted Prediction of Genetic Value with Semiparametric Procedures. Genetics 2006, 173, 1761–1776. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–46. [Google Scholar] [CrossRef]
- Zhao, Y.; Zeng, J.; Fernando, R.; Reif, J.C. Genomic Prediction of Hybrid Wheat Performance. Crop Sci. 2013, 53, 802–810. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Todorovska, E.; Christov, N.; Slavov, S.; Christova, P.; Vassilev, D. Biotic Stress Resistance in Wheat—Breeding and Genomic Selection Implications. Biotechnol. Biotechnol. Equip. 2009, 23, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Arruda, M.P.; Brown, P.J.; Lipka, A.E.; Krill, A.M.; Thurber, C.; Kolb, F.L. Genomic Selection for Predicting Fusarium Head Blight Resistance in a Wheat Breeding Program. Plant Genome 2015, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, V.; He, S.; Zhao, Y.; Korzun, V.; Bothe, R.; Ebmeyer, E.; Reif, J.C.; Jiang, Y. Potential and limits of whole genome prediction of resistance to Fusarium head blight and Septoria tritici blotch in a vast Central European elite winter wheat population. Theor. Appl. Genet. 2015, 128, 2471–2481. [Google Scholar] [CrossRef]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Heffner, E.L.; Jannink, J.-L.; Sorrells, M.E. Genomic Selection Accuracy using Multifamily Prediction Models in a Wheat Breeding Program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Heslot, N.; Jannink, J.-L.; Sorrells, M.E. Perspectives for Genomic Selection Applications and Research in Plants. Crop Sci. 2015, 55, 1–12. [Google Scholar] [CrossRef]
- Charmet, G.; Storlie, E.; Oury, F.X.; Laurent, V.; Beghin, D.; Chevarin, L.; Lapierre, A.; Perretant, M.R.; Rolland, B.; Heumez, E.; et al. Genome-wide prediction of three important traits in bread wheat. Mol. Breed. 2014, 34, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; de los Campos, G.; Pérez, P.; Gianola, D.; Burgueño, J.; Araus, J.L.; Makumbi, D.; Singh, R.P.; Dreisigacker, S.; Yan, J.; et al. Prediction of Genetic Values of Quantitative Traits in Plant Breeding Using Pedigree and Molecular Markers. Genetics 2010, 186, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Lado, B.; Battenfield, S.; Guzmán, C.; Quincke, M.; Singh, R.P.; Dreisigacker, S.; Peña, R.J.; Fritz, A.; Silva, P.; Poland, J.; et al. Strategies for Selecting Crosses Using Genomic Prediction in Two Wheat Breeding Programs. Plant Genome 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haile, J.K.; N’Diaye, A.; Clarke, F.; Clarke, J.; Knox, R.; Rutkoski, J.; Bassi, F.M.; Pozniak, C.J. Genomic selection for grain yield and quality traits in durum wheat. Mol. Breed. 2018, 38, 75. [Google Scholar] [CrossRef]
- Howard, R.; Carriquiry, A.L.; Beavis, W.D. Parametric and Nonparametric Statistical Methods for Genomic Selection of Traits with Additive and Epistatic Genetic Architectures. G3 Genes Genomes Genet. 2014, 4, 1027–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.J.; Chao, S.; Asoro, F.G.; Heffner, E.L.; Hayashi, T.; Iwata, H.; Smith, K.P.; Sorrells, M.E.; Jannink, J.L. Genomic Selection in Plant Breeding. Knowledge and Prospects. Adv. Agron. 2011, 110, 77–123. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Ding, X. Advances in genomic selection in domestic animals. Chin. Sci. Bull. 2011, 56, 2655–2663. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Dekkers, J.C.M.; Fernando, R.L.; Jannink, J.-L. Factors Affecting Accuracy From Genomic Selection in Populations Derived from Multiple Inbred Lines: A Barley Case Study. Genetics 2009, 182, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Jarquin, D.; Crossa, J.; Reynolds, M. Genomic-enabled prediction accuracies increased by modeling genotype × environment interaction in Durum wheat. Plant Genome 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Jia, Y.; Jannink, J.-L. Multiple-Trait Genomic Selection Methods Increase Genetic Value Prediction Accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Bhatta, M.; Gutierrez, L.; Cammarota, L.; Cardozo, F.; Germán, S.; Gómez-Guerrero, B.; Pardo, M.F.; Lanaro, V.; Sayas, M.; Castro, A.J. Multi-trait Genomic Prediction Model Increased the Predictive Ability for Agronomic and Malting Quality Traits in Barley (Hordeum vulgare L.). G3 Amp Genes Genomes Genet. 2020, 10, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Khan, J.; Pradhan, S.; Shahi, D.; Khan, N.; Avci, M.; Mcbreen, J.; Harrison, S.; Brown-Guedira, G.; Murphy, J.P.; et al. Multi-Trait Genomic Prediction of Yield-Related Traits in US Soft Wheat under Variable Water Regimes. Genes 2020, 11, 1270. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Cericola, F.; Edriss, V.; Andersen, J.R.; Orabi, J.; Jensen, J.D.; Jahoor, A.; Janss, L.; Jensen, J. Use of multiple traits genomic prediction, genotype by environment interactions and spatial effect to improve prediction accuracy in yield data. PLoS ONE 2020, 15, e0232665. [Google Scholar] [CrossRef]
- Chen, Y.; Lübberstedt, T. Molecular basis of trait correlations. Trends Plant Sci. 2010, 15, 454–461. [Google Scholar] [CrossRef]
- Lozada, D.N.; Carter, A.H. Accuracy of Single and Multi-Trait Genomic Prediction Models for Grain Yield in US Pacific Northwest Winter Wheat. Crop Breed. Genet. Genom. 2019, 2, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Panozzo, J.; Walker, C.K.; Choy, A.L.; Kant, S.; Wong, D.; Tibbits, J.; Daetwyler, H.D.; Rochfort, S.; Hayden, M.J.; et al. Accelerating wheat breeding for end-use quality with multi-trait genomic predictions incorporating near infrared and nuclear magnetic resonance-derived phenotypes. Theor. Appl. Genet. 2017, 130, 2505–2519. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Year | Site | R | N | Mean Response | |
|---|---|---|---|---|---|---|
| Field | Lr | 2005 | Lansdowne | 1 | 376 | 3.45 ± 1.95 |
| 2005 | Richmond | 1 | 195 | 3.66 ± 1.87 | ||
| 2006 | Lansdowne | 2 | 388/355 | 2.73 ± 1.62 | ||
| 2013 | HorseUnit | 1 | 307 | 3.62 ± 1.52 | ||
| Sr | 2005 | Lansdowne | 1 | 388 | 4.90 ± 2.14 | |
| 2006 | Lansdowne | 1 | 383 | 4.84 ± 1.85 | ||
| 2012 | Karalee | 1 | 331 | 5.07 ± 1.71 | ||
| 2012 | Ethiopia | 1 | 350 | 4.59 ± 1.39 | ||
| 2013 | Horse Unit | 1 | 310 | 4.81 ± 1.42 | ||
| Yr | 2005 | Lansdowne | 1 | 389 | 3.68 ± 1.15 | |
| 2006 | Lansdowne | 2 | 390 | 3.89 ± 1.46 | ||
| 2012 | Karalee | 1 | 334 | 2.98 ± 1.03 | ||
| Greenhouse | Lr | 2012 | USA | 3 | 377 | 5.93 ± 2.32 |
| Sr | 2013 | AUS | 10 | 174 | 6.78 ± 1.69 | |
| Yr | 2013 | AUS | 10 | 391 | 6.09 ± 2.29 |
| Field | Greenhouse | |||||
|---|---|---|---|---|---|---|
| Lr | Sr | Yr | Lr | Sr | Yr | |
| Broad sense Heritability | 0.56 | 0.42 | 0.56 | 0.43 | 0.56 | 0.39 |
| Narrow sense Heritability | 0.33 | 0. 30 | 0.46 | 0.30 | 0.42 | 0.26 |
| GBLUP | 0.70 ± 0.06 | 0.49 ± 0.04 | 0.35 ± 0.03 | 0.53 ± 0.08 | 0.44 ± 0.16 | 0.51 ± 0.09 |
| BayesA | 0.71 ± 0.05 | 0.50 ± 0.08 | 0.35 ± 0.09 | 0.52 ± 0.1 | 0.45 ± 0.15 | 0.52 ± 0.08 |
| BayesB | 0.71 ± 0.07 | 0.50 ± 0.08 | 0.37 ± 0.09 | 0.52 ± 0.08 | 0.43 ± 0.19 | 0.53 ± 0.07 |
| BayesC | 0.71 ± 0.07 | 0.50 ± 0.08 | 0.36 ± 0.08 | 0.51 ± 0.09 | 0.45 ± 0.17 | 0.51 ± 0.07 |
| BayesLASSO | 0.71 ± 0.05 | 0.50 ± 0.08 | 0.36 ± 0.08 | 0.51 ± 0.07 | 0.46 ± 0.14 | 0.51 ± 0.09 |
| RKHS | 0.71 ± 0.06 | 0.50 ± 0.03 | 0.38 ± 0.05 | - | - | - |
| Yr | LD-05 | LD_06 | ||||
| LD_06 | 0.52 * | |||||
| KL-12 | 0.47 * | 0.51 * | ||||
| Sr | LD-05 | LD-06 | KL-12 | DZ-12 | ||
| LD-06 | 0.46 * | |||||
| KL-12 | 0.43 * | 0.63 * | ||||
| HU-13 | 0.43 * | 0.62 * | 0.73 * | |||
| DZ-12 | 0.28 ns | 0.24 ns | 0.22 ns | 0.29 ns | ||
| Lr | LD-05 | RM-05 | LD-06 | |||
| RM-05 | 0.43 * | |||||
| LD-06 | 0.67 * | 0.50 * | ||||
| HU-13 | 0.63 * | 0.41 * | 0.72 * |
| Lr | Sr | Yr | LrGH | SrGH | |
|---|---|---|---|---|---|
| Sr | 0.20 ± 0.09 | ||||
| Yr | 0.32 ± 0.10 | −0.07 ± 0.13 | |||
| LrGH 1 | 0.45 ± 0.10 | 0.22 ± 0.13 | 0.24 ± 0.15 | ||
| SrGH | −0.01 ± 0.13 | 0.65 ± 0.08 | −0.15 ± 0.16 | 0.10 ± 0.13 | |
| YrGH | −0.09 ± 0.11 | −0.25 ± 0.13 | 0.50 ± 0.12 | −0.09 ± 0.14 | −0.10 ± 0.20 |
| Lr | Sr | Yr | ||
|---|---|---|---|---|
| Site | Debre Zeit | - | 0.62 | - |
| Lansdowne | 0.71 | 0.59 | 0.51 | |
| Karalee | - | 0.29 | 0.51 | |
| Richmond | 0.56 | - | - | |
| Horse Unit | 0.66 | 0.65 | - | |
| Year | 2005 | 0.63 | 0.47 | 0.52 |
| 2006 | 0.69 | 0.57 | 0.56 | |
| 2012 | - | 0.58 | 0.51 | |
| 2013 | 0.66 | 0.65 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azizinia, S.; Bariana, H.; Kolmer, J.; Pasam, R.; Bhavani, S.; Chhetri, M.; Toor, A.; Miah, H.; Hayden, M.J.; Pino del Carpio, D.; et al. Genomic Prediction of Rust Resistance in Tetraploid Wheat under Field and Controlled Environment Conditions. Agronomy 2020, 10, 1843. https://doi.org/10.3390/agronomy10111843
Azizinia S, Bariana H, Kolmer J, Pasam R, Bhavani S, Chhetri M, Toor A, Miah H, Hayden MJ, Pino del Carpio D, et al. Genomic Prediction of Rust Resistance in Tetraploid Wheat under Field and Controlled Environment Conditions. Agronomy. 2020; 10(11):1843. https://doi.org/10.3390/agronomy10111843
Chicago/Turabian StyleAzizinia, Shiva, Harbans Bariana, James Kolmer, Raj Pasam, Sridhar Bhavani, Mumta Chhetri, Arvinder Toor, Hanif Miah, Matthew J. Hayden, Dunia Pino del Carpio, and et al. 2020. "Genomic Prediction of Rust Resistance in Tetraploid Wheat under Field and Controlled Environment Conditions" Agronomy 10, no. 11: 1843. https://doi.org/10.3390/agronomy10111843
APA StyleAzizinia, S., Bariana, H., Kolmer, J., Pasam, R., Bhavani, S., Chhetri, M., Toor, A., Miah, H., Hayden, M. J., Pino del Carpio, D., Bansal, U., & Daetwyler, H. D. (2020). Genomic Prediction of Rust Resistance in Tetraploid Wheat under Field and Controlled Environment Conditions. Agronomy, 10(11), 1843. https://doi.org/10.3390/agronomy10111843