Nuclear Magnetic Resonance with Fast Field-Cycling Setup: A Valid Tool for Soil Quality Investigation



Abstract
:1. Introduction
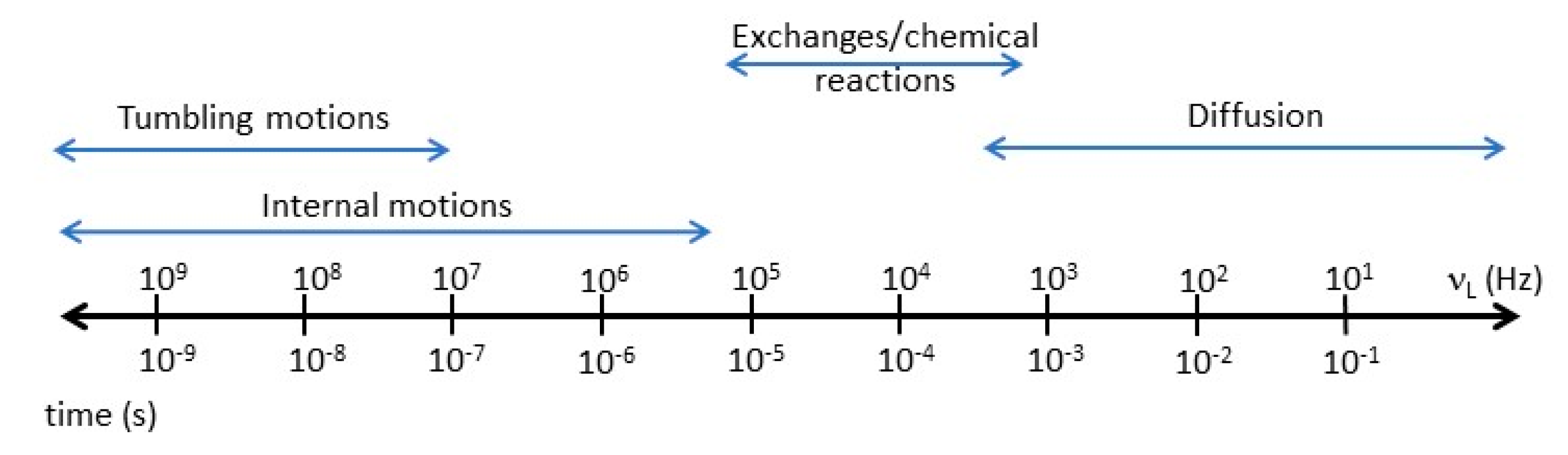
2. The Meaning of the T1 Value
3. The Fast Field-Cycling NMR Experiment and the Evaluation of the T1 Value
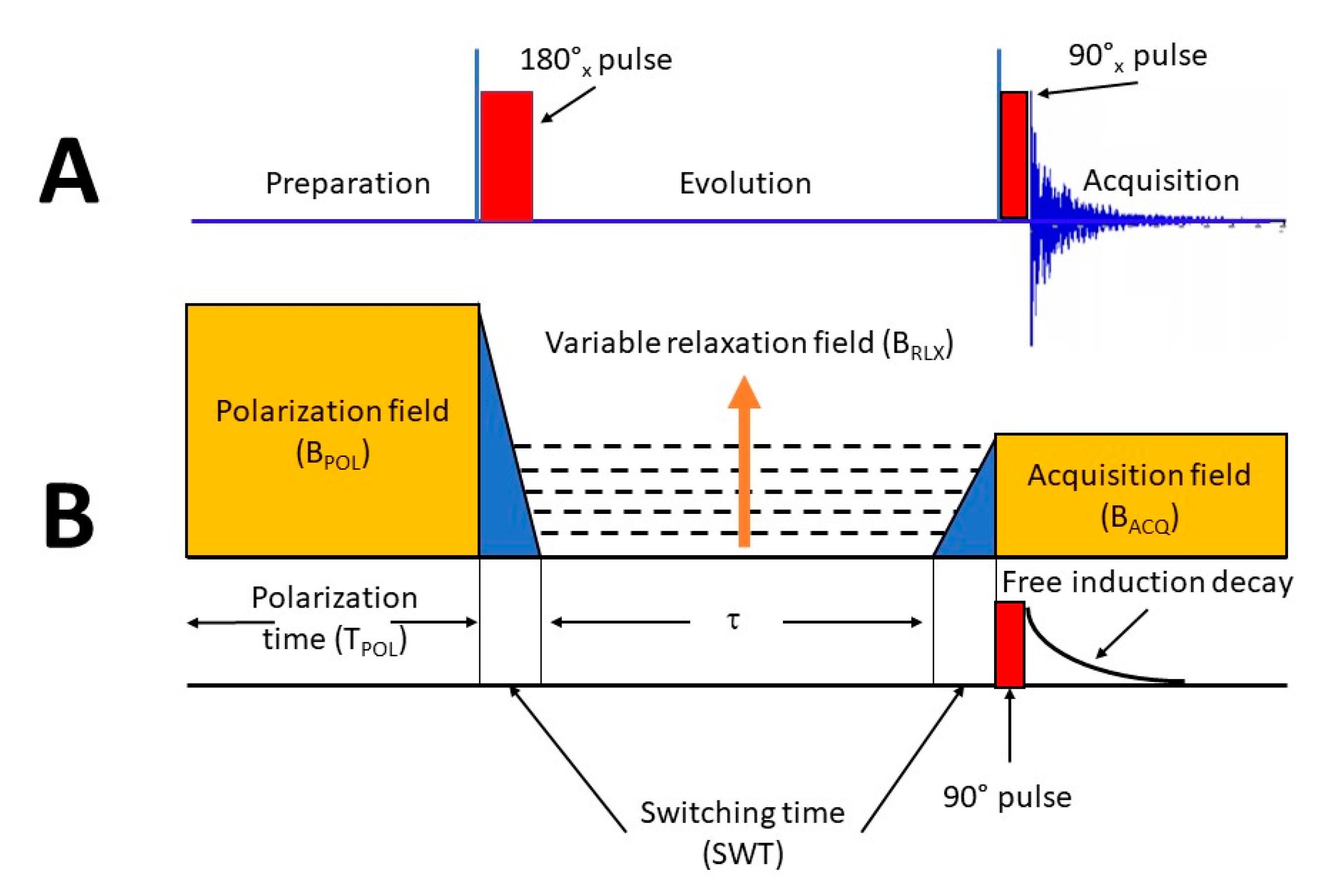
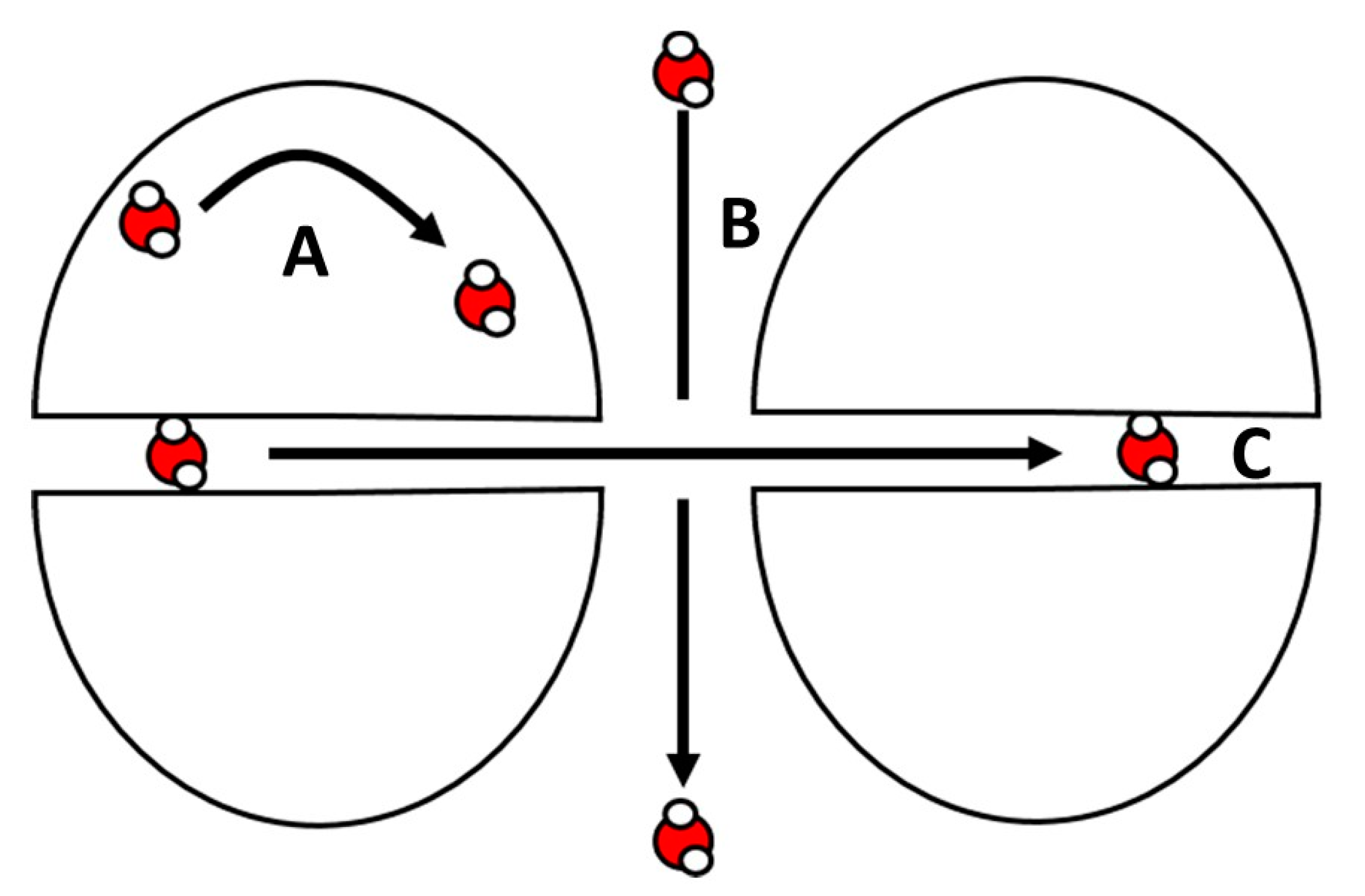
3.1. The Inversion Recovery Sequence and the Basic FFC NMR Experiment
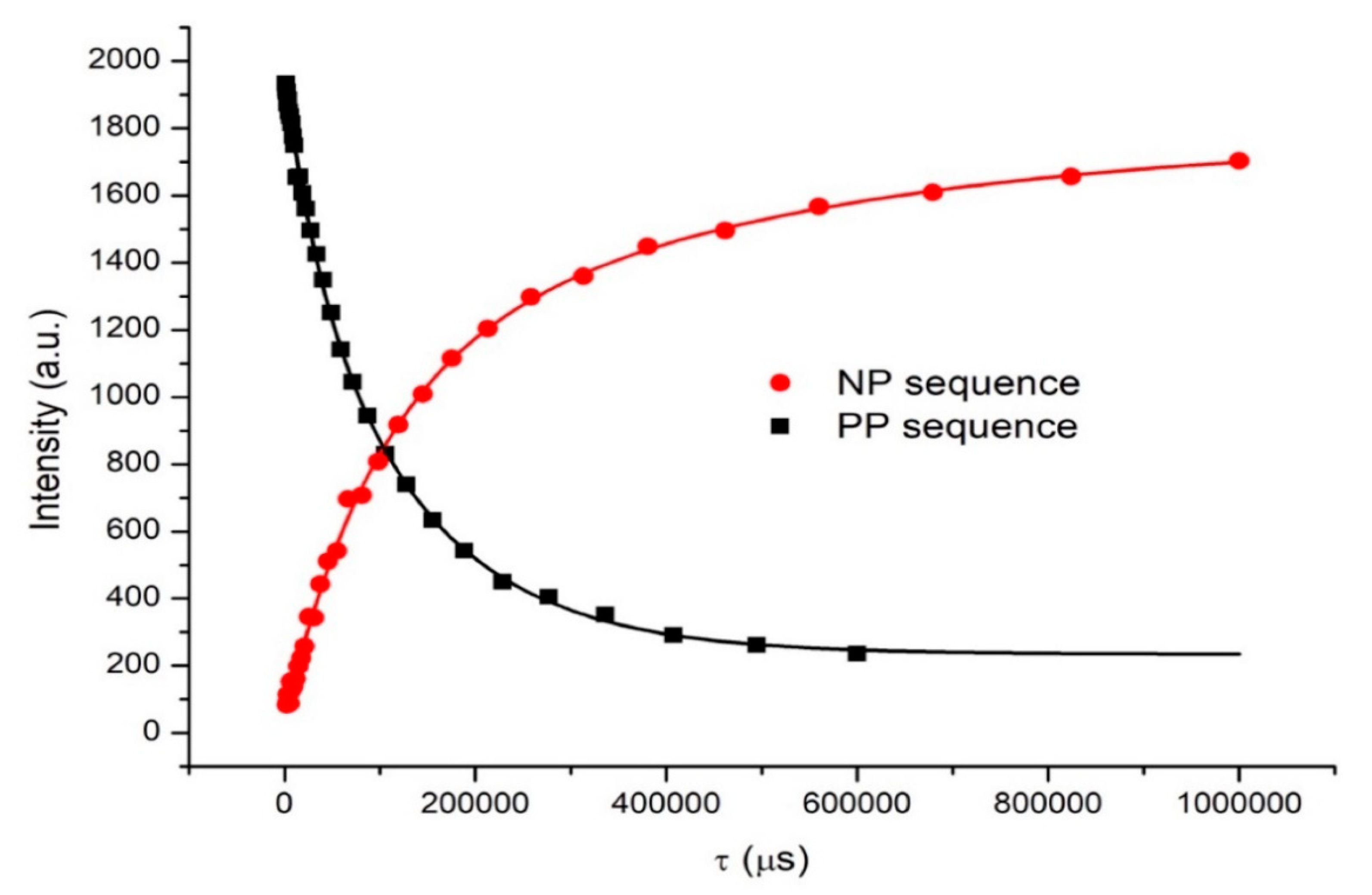
3.2. How to Obtain the T1 Value from the FFC NMR Experiment
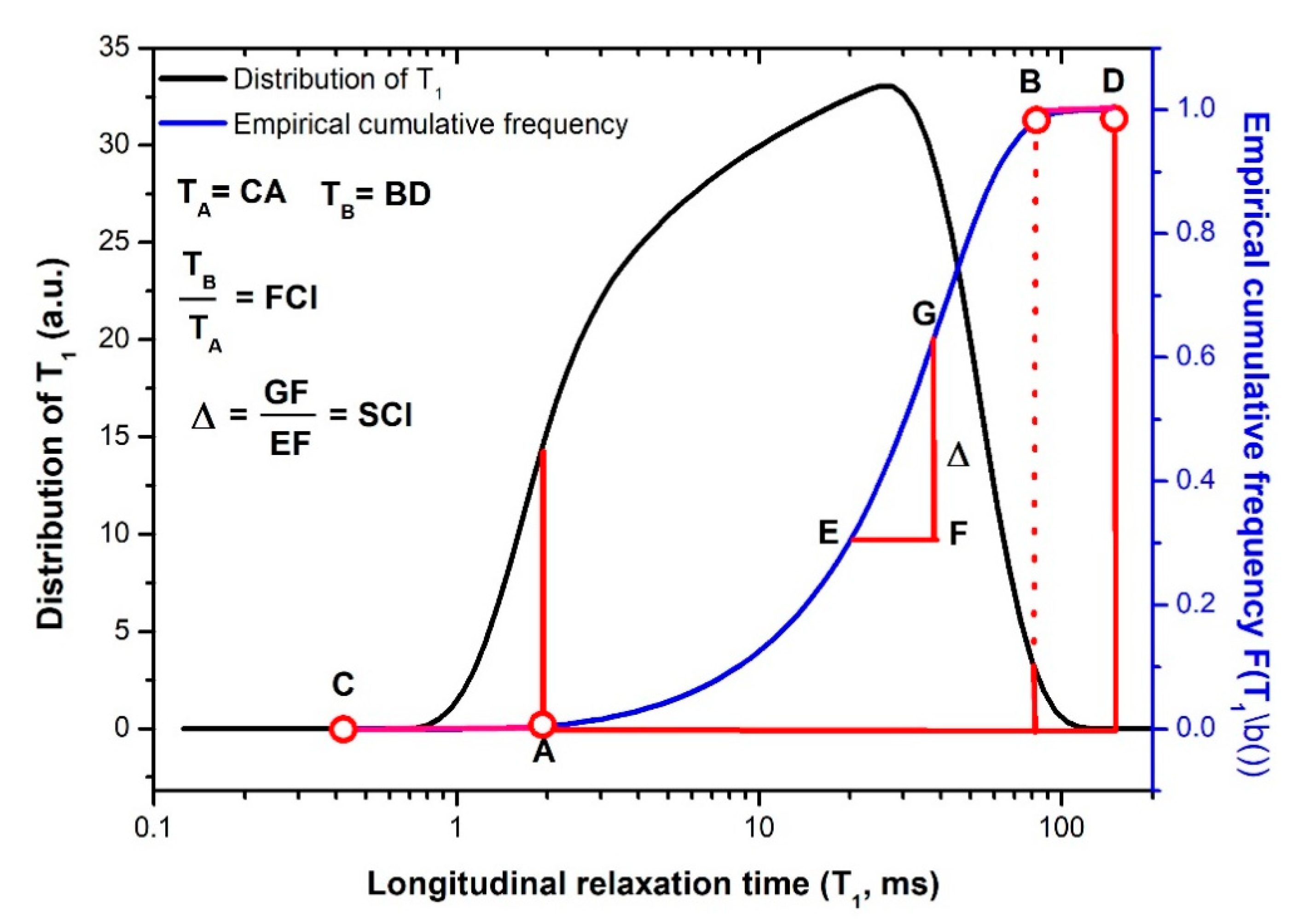
4. From Time Domain to Time Domain
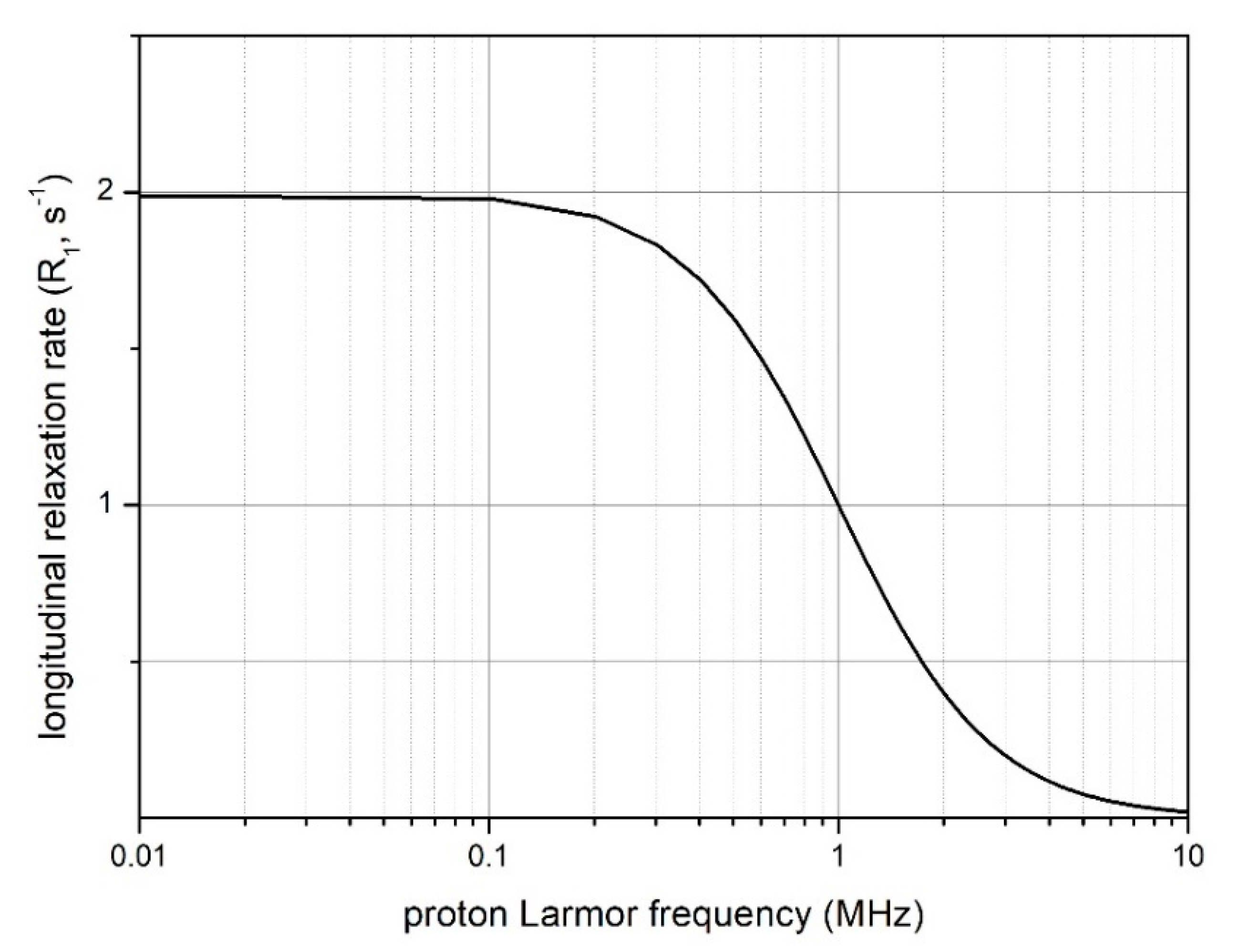
5. The Nuclear Magnetic Resonance Dispersion (NMRD) Profile and Its Modeling
6. How to Choose the Right Model to Correctly Interpret NMRD Profiles in Soil Science
6.1. The Application of the BPP Model: The Quadrupolar-Less Nuclei Quartz Sand
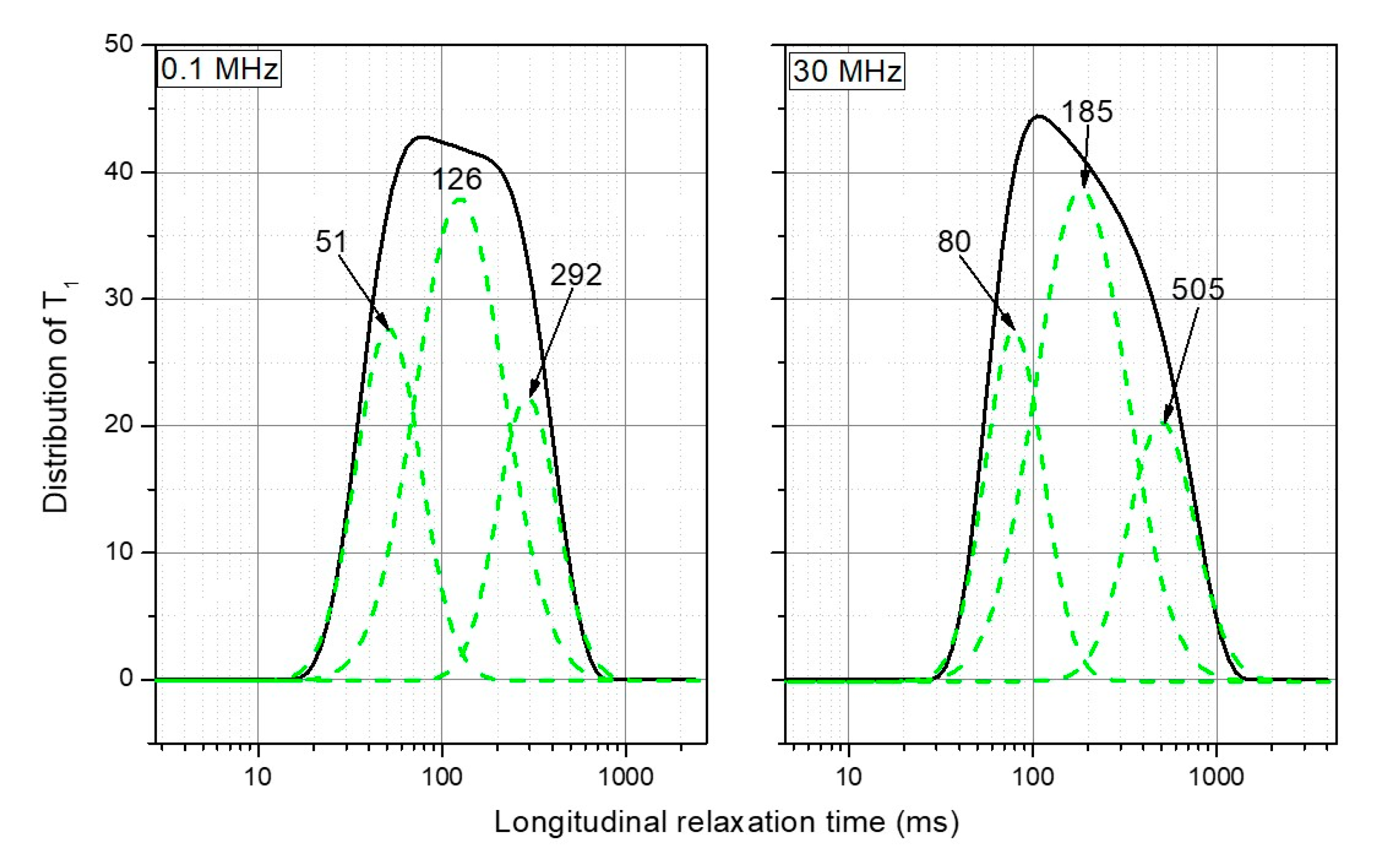
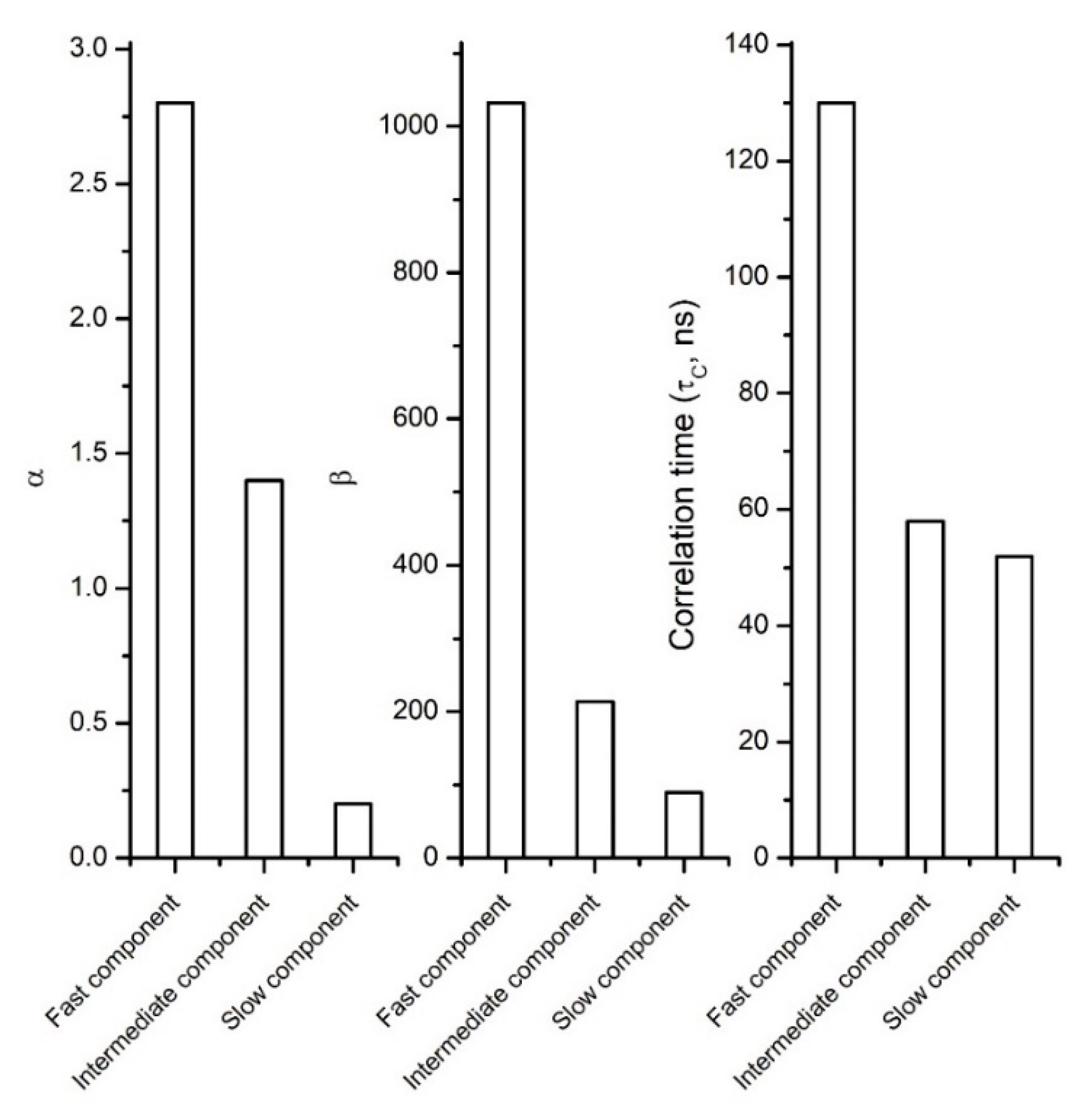
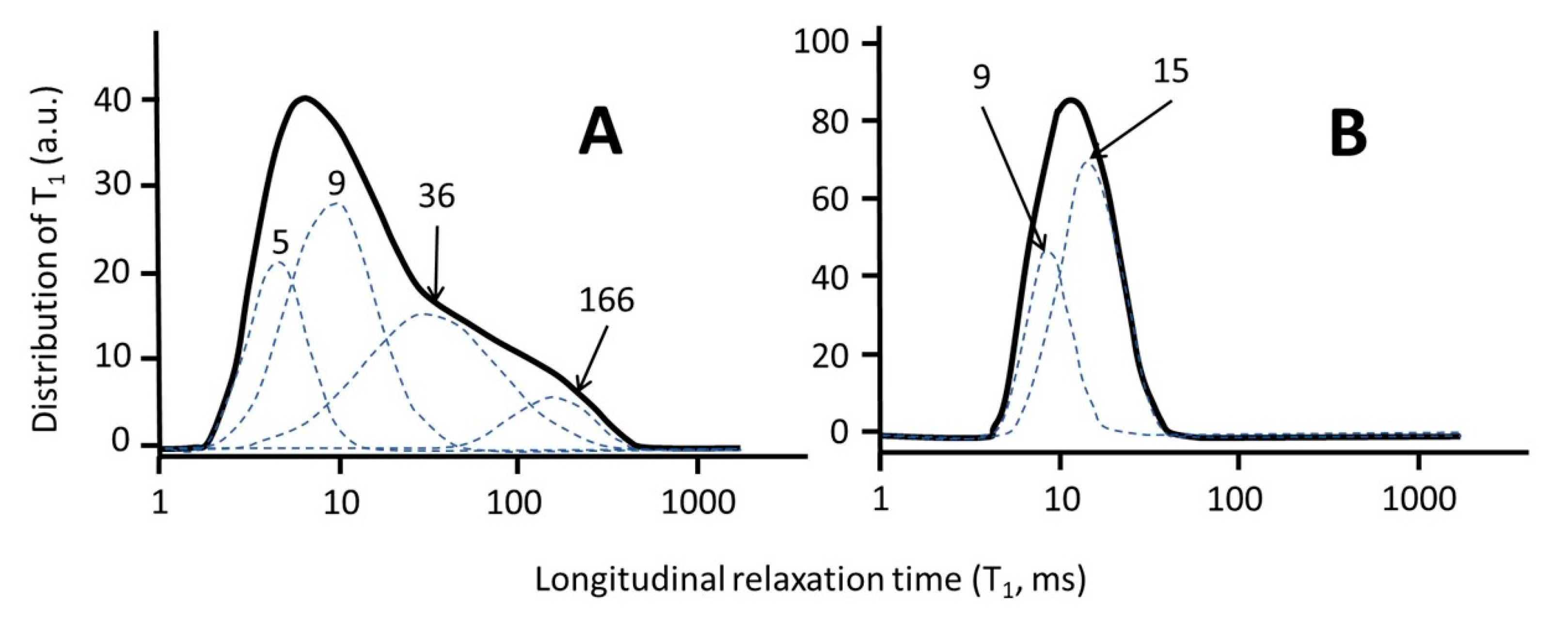
6.2. When the Free Model Analysis is the Best Choice. Two Case Studies: Soil and Biochar
6.3. The Combination between the BPP Model and the Free Model Analysis: The Saltmasrh Sediment Case Study
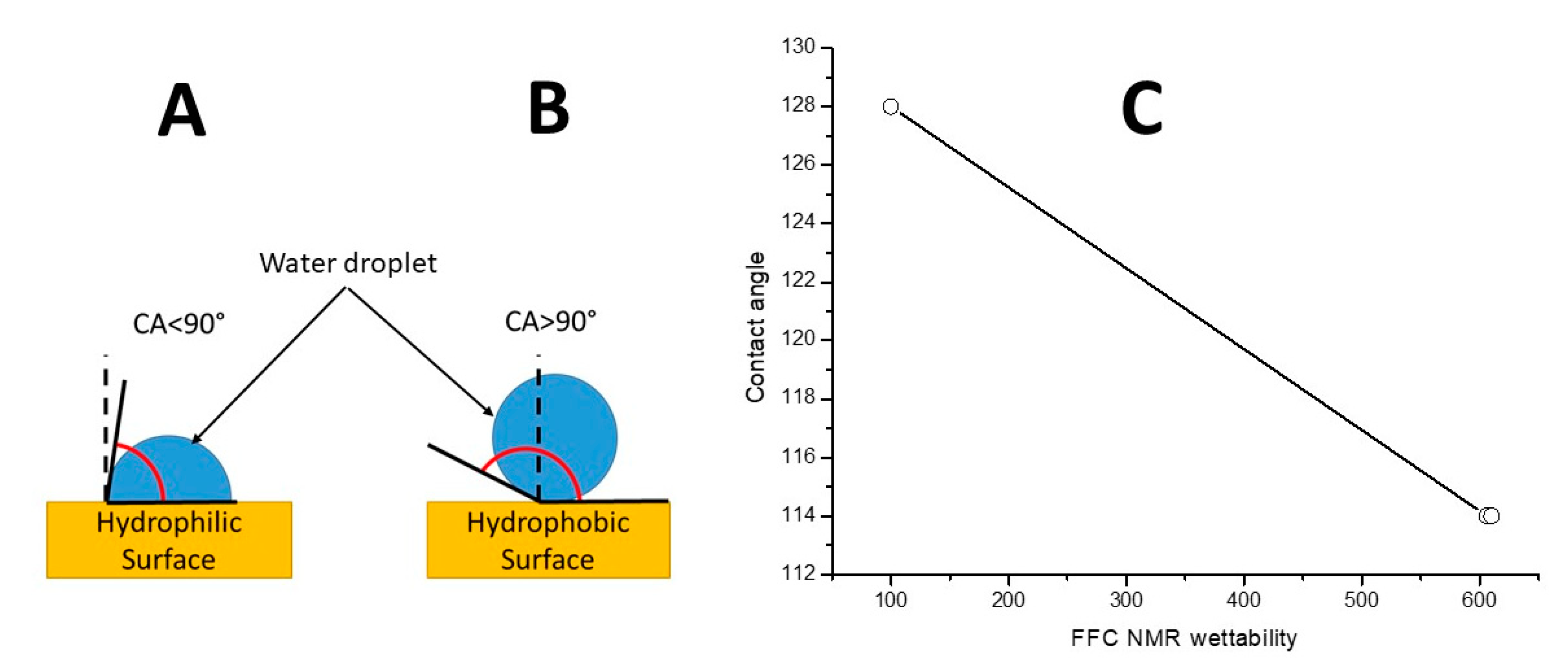
6.4. NMRD Evaluation by the Wettability Model: Application to Biochar
6.5. The FFC NMR Modeling via Molecular Dynamics Simulations
7. Understanding Soils by Fast Field-Cycling NMR Relaxometry
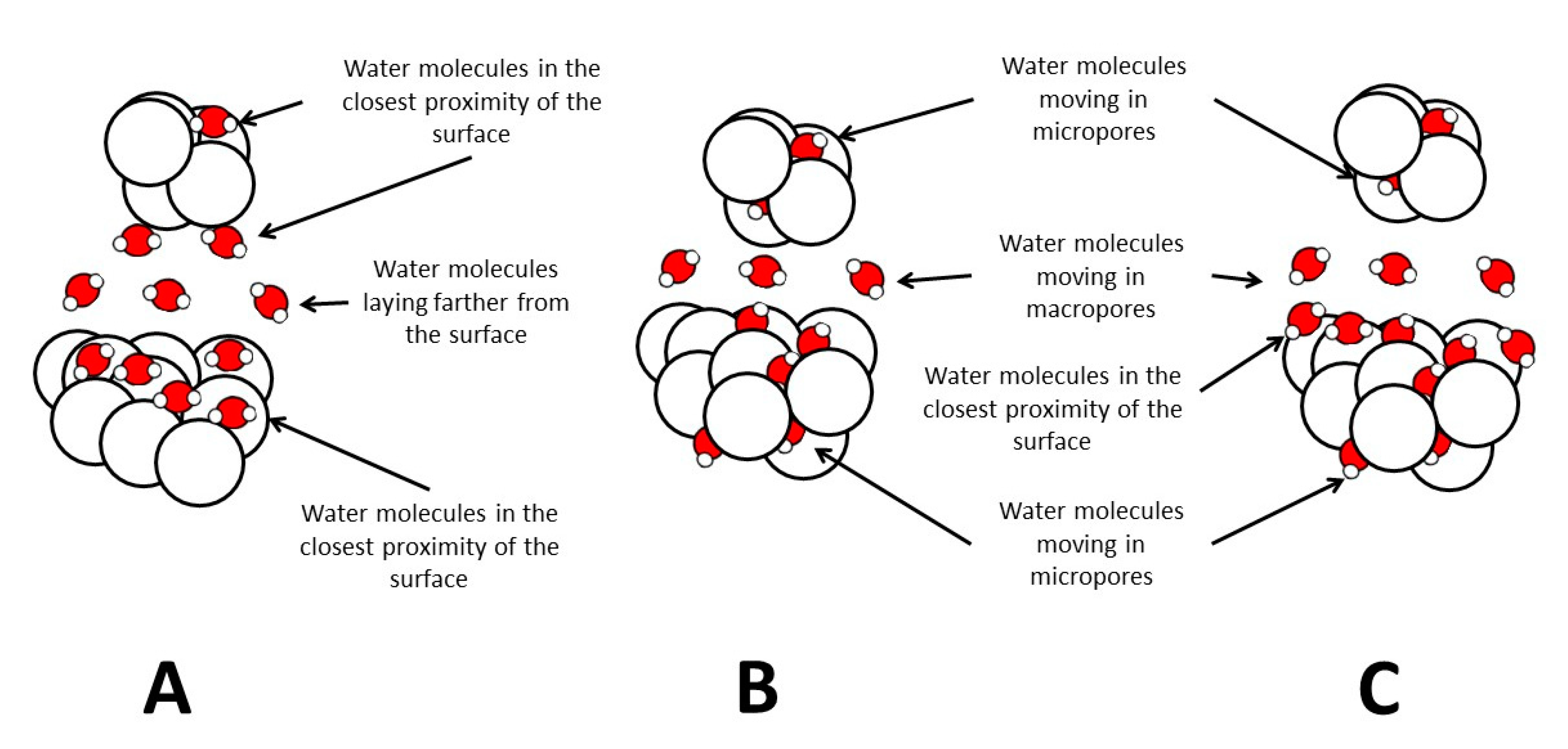
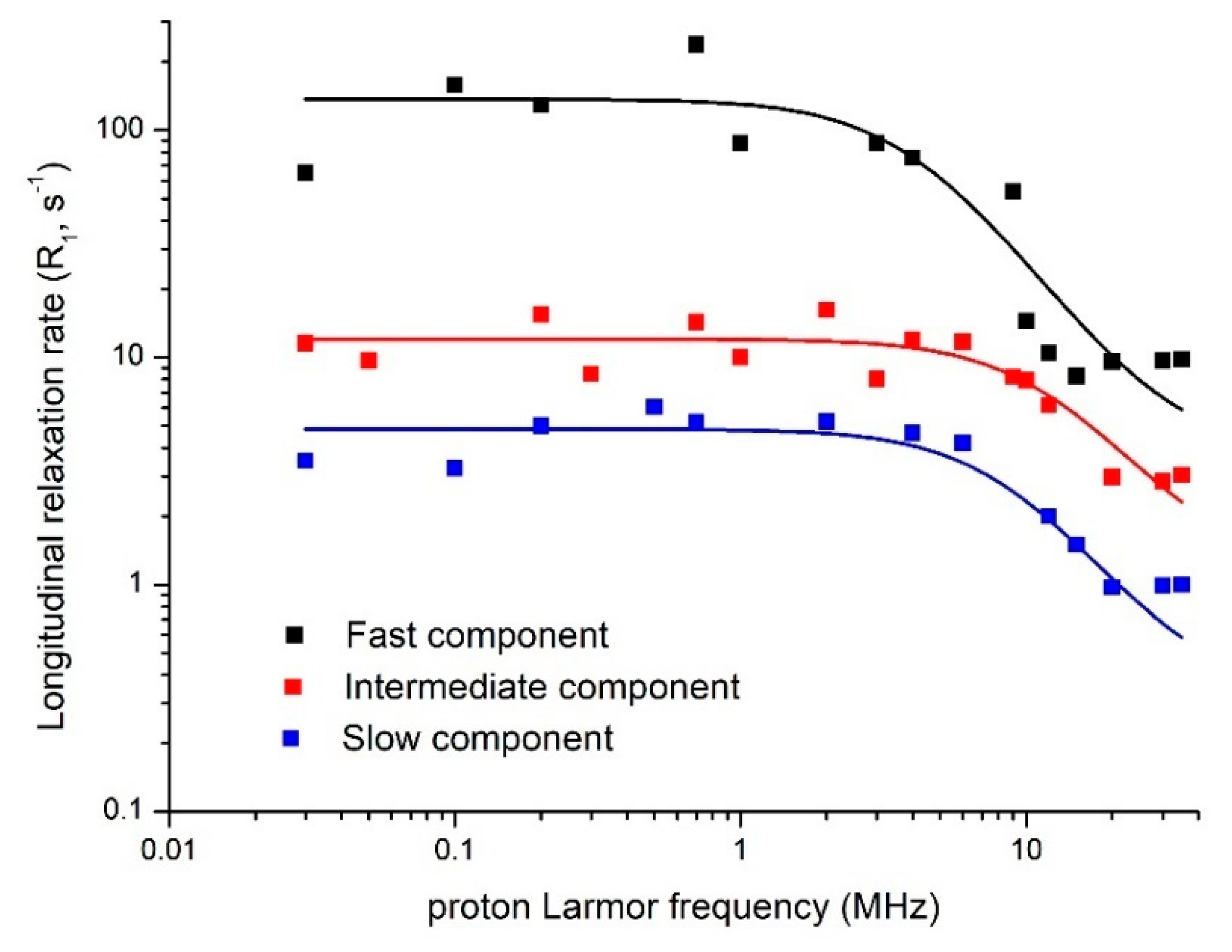
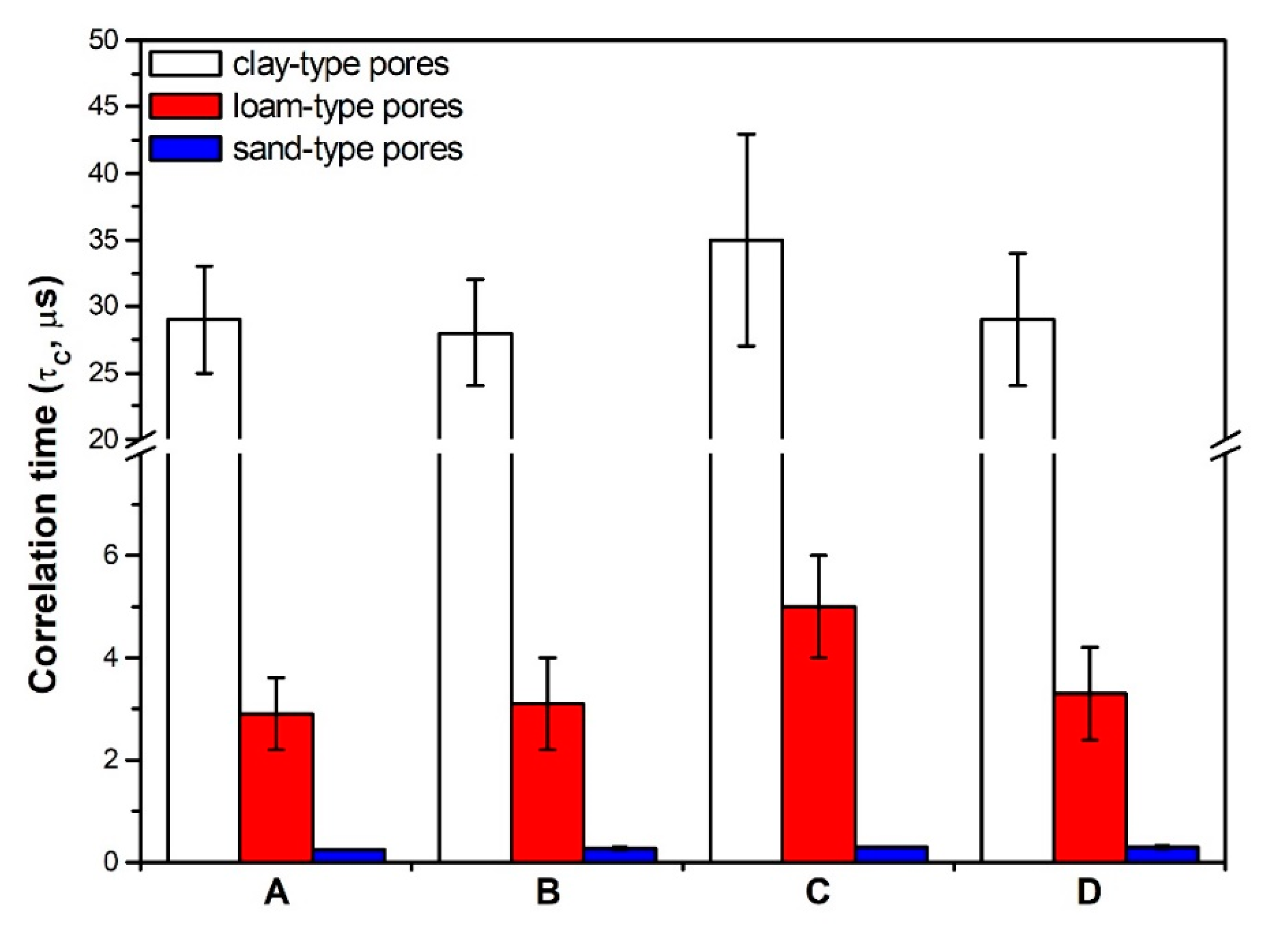
7.1. The Role of Soil Pores in Water Dynamics
7.2. FFC NMR to Quantify Soil Erosion
7.3. The Behaviour of Dissolved Organic Matter (DOM)
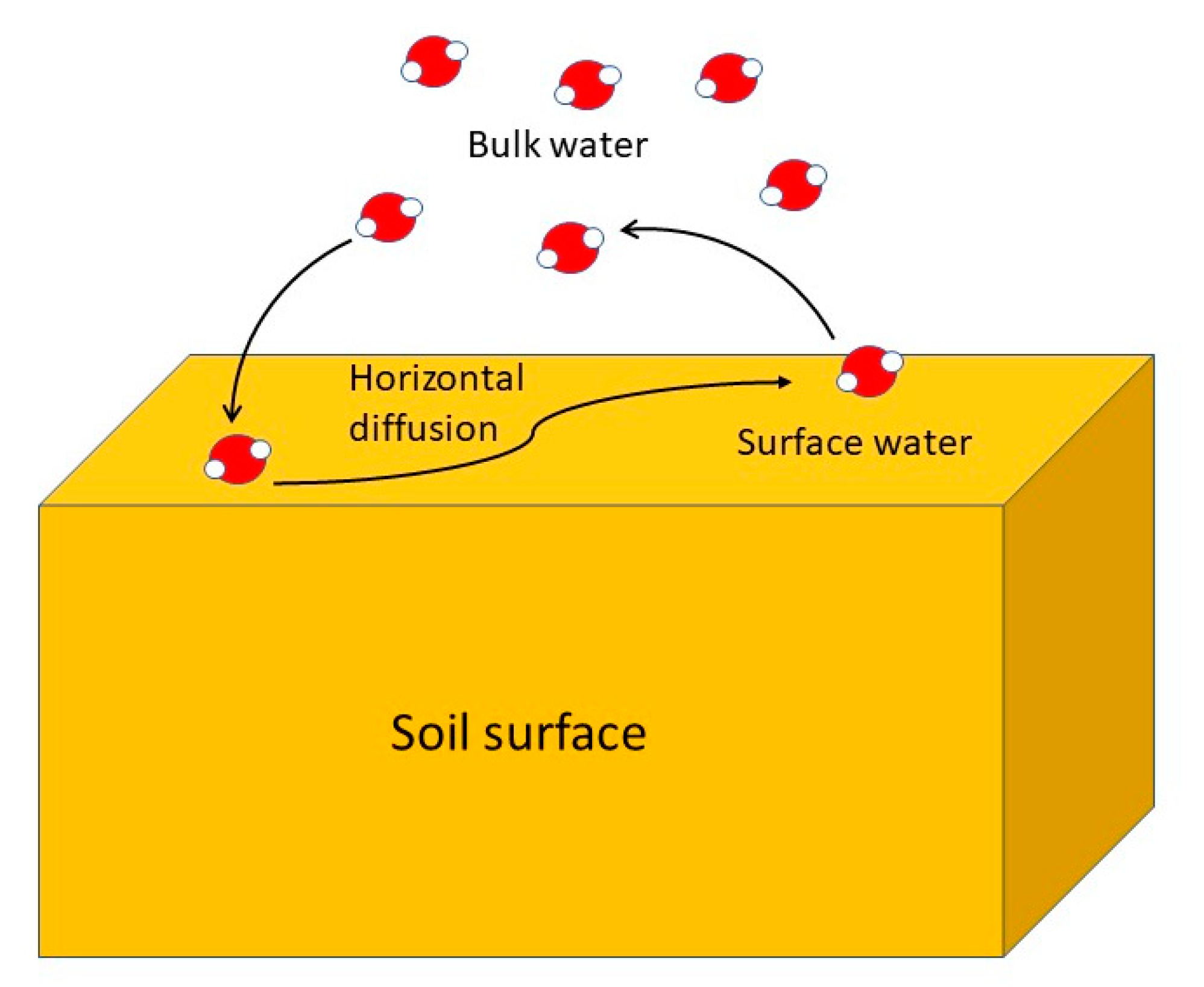
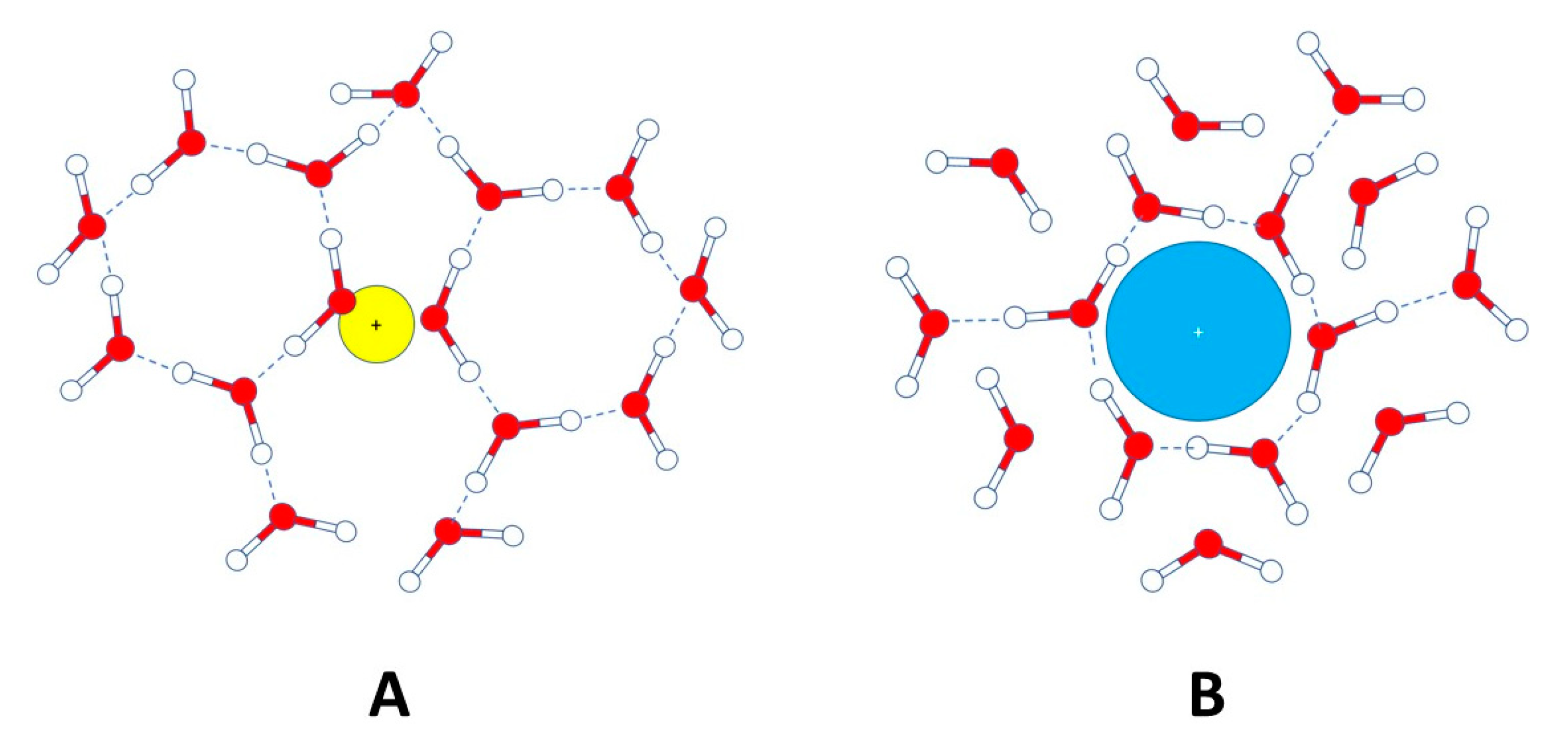
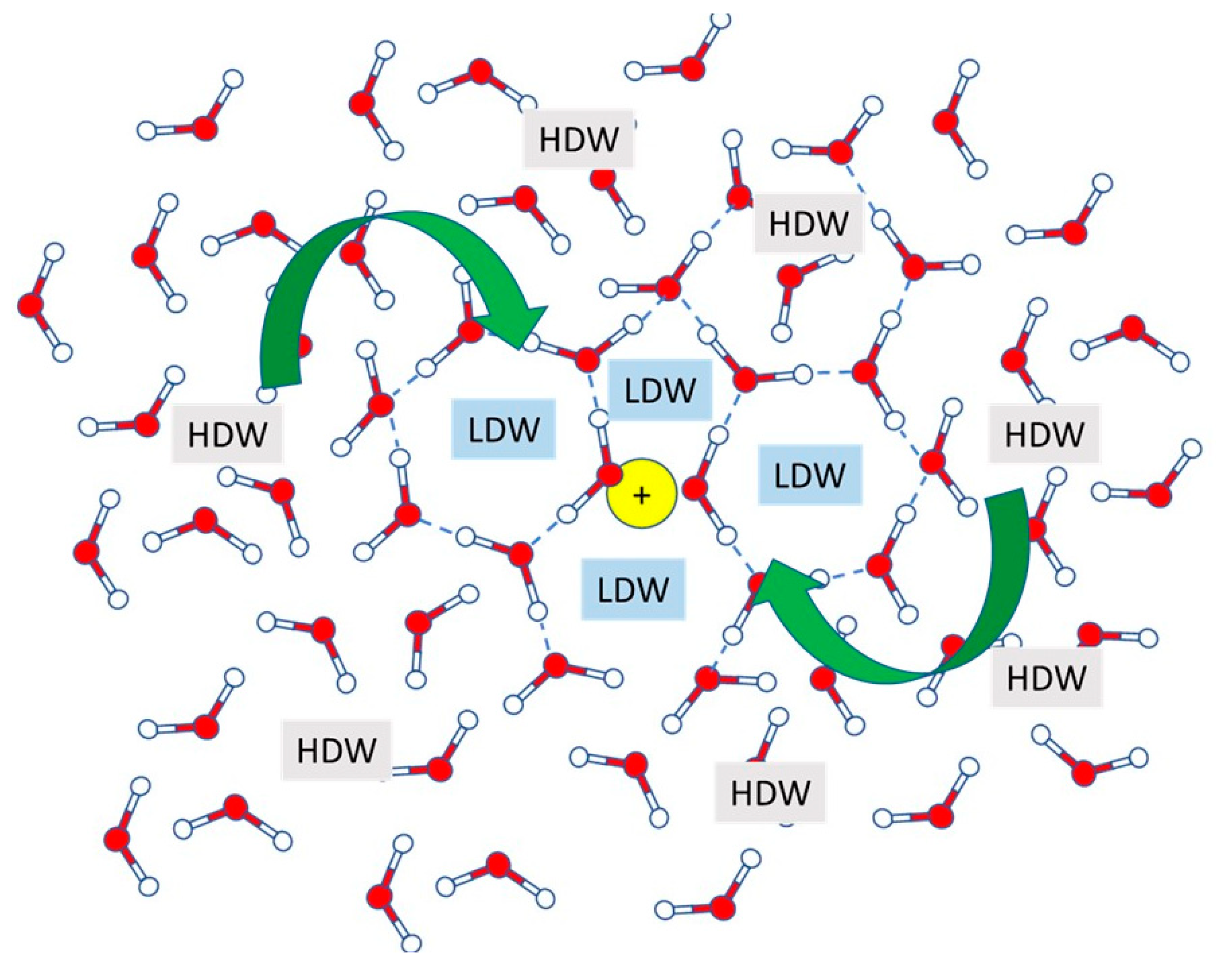
7.4. Water Behavior in the Presence of Inorganic Ions: The Dynamics of Soil Solution
7.5. A FFC NMR-Based Model for Nutrient Dynamics in Soils
7.6. The Mechanism of Soil Ionic Exchange Capacity: The FFC NMR Relaxometry Point of View
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Simpson, A.J.; Simpson, M.J.; Soong, R. Nuclear magnetic resonance spectroscopy and its key role in environmental research. Environ. Sci. Technol. 2012, 46, 11488–11496. [Google Scholar] [CrossRef] [PubMed]
- Farooq, H.; Courtier-Murias, D.; Soong, R.; Bermel, W.; Kingery, W.; Simpson, A. HR-MAS NMR spectroscopy: A practical guide for natural samples. Curr. Org. Chem. 2013, 17, 3013–3031. [Google Scholar] [CrossRef]
- Conte, P.; Spaccini, R.; Piccolo, A. State of the art of CPMAS13C-NMR spectroscopy applied to natural organic matter. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 215–223. [Google Scholar] [CrossRef]
- Jameson, C.J. Gas-Phase NMR Spectroscopy. Chem. Rev. 1991, 91, 1375–1395. [Google Scholar] [CrossRef]
- Elyashberg, M. Trends in Analytical Chemistry Identification and structure elucidation by NMR spectroscopy. Trends Anal. Chem. 2015, 69, 88–97. [Google Scholar] [CrossRef]
- Simpson, A.J. Environmental NMR. Magn. Reson. Chem. 2015, 53, 633–634. [Google Scholar] [CrossRef]
- Lurie, D.J.; Aime, S.; Baroni, S.; Booth, N.A.; Broche, L.M.; Choi, C.; Davies, G.R.; Ismail, S.; Dara, Ó.; Pine, K.J. Comptes Rendus Physique Fast field-cycling magnetic resonance imaging Imagerie de resonance magnétique en champ cyclé. Comptes Rendus Phys. 2010, 11, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Bastawrous, M.; Jenne, A.; Tabatabaei Anaraki, M.; Simpson, A.J. In-vivo NMR spectroscopy: A powerful and complimentary tool for understanding environmental toxicity. Metabolites 2018, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Dais, P.; Spyros, A. Nuclear magnetic resonance. In Chemical Analysis of Food: Techniques and Applications; Picó, Y., Ed.; Academic Press—Elsevier: Waltham, MA, USA, 2012; pp. 91–115. ISBN 9780123848628. [Google Scholar]
- Bakhmutov, V.I. Practical NMR Relaxation for Chemists, 1st ed.; John Wiley & Sons, Inc.: Chichester, UK, 2004; ISBN 0470094451. [Google Scholar]
- Berns, A.E.; Conte, P. Effect of rf field inhomogeneity and sample restriction on spectral resolution of CP/MAS- 13C NMR spectra of natural organic matter. Open Magn. Reson. J. 2010, 3, 75–83. [Google Scholar] [CrossRef]
- Berns, A.E.; Conte, P. Effect of ramp size and sample spinning speed on CPMAS13C NMR spectra of soil organic matter. Org. Geochem. 2011, 42. [Google Scholar] [CrossRef]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Uniform-penalty inversion of multiexponential decay data. J. Magn. Reson. 1998, 132, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Uniform-penalty inversion of multiexponential decay data: II. Data spacing, T2 data, systematic data errors, and diagnostics. J. Magn. Reson. 2000, 147, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Examples of marginal resolution of NMR relaxation peaks using UPEN and diagnostics. Magn. Reson. Imaging 2001, 19, 473–475. [Google Scholar] [CrossRef]
- Bortolotti, V.; Brown, R.J.S.; Fantazzini, P.; Landi, G.; Zama, F. Uniform Penalty inversion of two-dimensional NMR relaxation data. Inverse Probl. 2017, 33, 015003. [Google Scholar] [CrossRef] [Green Version]
- Bortolotti, V.; Brown, R.J.S.; Fantazzini, P.; Landi, G.; Zama, F. I2DUPEN: Improved 2DUPEN algorithm for inversion of two-dimensional NMR data. Microporous Mesoporous Mater. 2018, 269, 195–198. [Google Scholar] [CrossRef]
- Conte, P. Environmental Applications of Fast Field-cycling NMR Relaxometry. In Field-cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Kimmich, R., Ed.; The Royal Society of Chemistry: Croydon, UK, 2019; pp. 229–254. ISBN 9781788011549. [Google Scholar]
- Kimmich, R. Field cycling in NMR relaxation spectroscopy: Applications in biological, chemical and polymer physics. Bull. Magn. Reson 1979, 1, 195–218. [Google Scholar]
- Kimmich, R.; Anoardo, E. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Schaumann, G.E.; Jaeger, F.; Bayer, J. V Relaxometry in soil science. In Proceedings of the Geophysical Research Abstracts, Wien, Austria, 19–24 April 2009; Volume 11, p. EGU2009-3208-1. [Google Scholar]
- Buchmann, C.; Meyer, M.; Schaumann, G.E. Characterization of wet aggregate stability of soils by H-NMR relaxometry. Magn. Reson. Chem. 2015, 53, 694–703. [Google Scholar] [CrossRef]
- Schaumann, G.E.; Diehl, D.; Bertmer, M.; Jaeger, A.; Conte, P.; Alonzo, G.; Bachmann, J. Combined proton NMR wideline and NMR relaxometry to study SOM-water interactions of cation-treated soils. J. Hydrol. Hydromechanics 2013, 61, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Anoardo, E.; Galli, G.; Ferrante, G. Fast-Field-Cycling NMR: Applications and Instrumentation. Appl. Magn. Reson. 2001, 20, 365–404. [Google Scholar] [CrossRef]
- Conte, P.; Bubici, S.; Palazzolo, E.; Alonzo, G. Solid-state 1H-NMR relaxation properties of the fruit of a wild relative of eggplant at different proton Larmor frequencies. Spectrosc. Lett. 2009, 42, 235–239. [Google Scholar] [CrossRef]
- Conte, P.; Maccotta, A.; De Pasquale, C.; Alonzo, G. Supramolecular organization of triglycerides in extra-virgin olive oils as assessed by NMR relaxometry. Fresenius Environ. Bull. 2010, 19, 2077–2082. [Google Scholar]
- Baroni, S.; Consonni, R.; Ferrante, G.; Aime, S. Relaxometric studies for food characterization: The case of balsamic and traditional balsamic vinegars. J. Agric. Food Chem. 2009, 57, 3028–3032. [Google Scholar] [CrossRef] [PubMed]
- Conte, P.; Mineo, V.; Bubici, S.; De Pasquale, C.; Aboud, F.; MacCotta, A.; Planeta, D.; Alonzo, G. Dynamics of pistachio oils by proton nuclear magnetic resonance relaxation dispersion. Anal. Bioanal. Chem. 2011, 400, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Bodart, P.R.; Rachocki, A.; Tritt-Goc, J.; Michalke, B.; Schmitt-Kopplin, P.; Karbowiak, T.; Gougeon, R.D. Quantification of manganous ions in wine by NMR relaxometry. Talanta 2020, 209, 120561. [Google Scholar] [CrossRef]
- Conte, P.; Cuccurullo, G.; Metallo, A.; Micalizzi, A.; Cinquanta, L.; Corona, O. Comparing different processing methods in apple slice drying. Part 2 solid-state Fast Field Cycling 1H-NMR relaxation properties, shrinkage and changes in volatile compounds. Biosyst. Eng. 2019, 188, 345–354. [Google Scholar] [CrossRef]
- Cimò, G.; Conte, P. Conformational redistribution of honey components following different storage conditions. Int. J. Spectrosc. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lo Scalzo, R.; Fibiani, M.; Francese, G.; D’Alessandro, A.; Rotino, G.L.; Conte, P.; Mennella, G. Cooking influence on physico-chemical fruit characteristics of eggplant (Solanum melongena L.). Food Chem. 2016, 194. [Google Scholar] [CrossRef] [Green Version]
- Ladd-Parada, M.; Povey, M.J.; Vieira, J.; Ries, M.E. Fast field cycling NMR relaxometry studies of molten and cooled cocoa butter. Mol. Phys. 2018, 117, 1020–1027. [Google Scholar] [CrossRef]
- Uguz, S.S.; Ozvural, E.B.; Beira, M.J.; Oztop, M.H.; Sebastião, P.J. Use of NMR Relaxometry to identify frankfurters of different meat sources. Mol. Phys. 2018, 117, 1015–1019. [Google Scholar] [CrossRef]
- Płowaś-Korus, I.; Masewicz, Ł.; Szwengiel, A.; Rachocki, A.; Baranowska, H.M.; Medycki, W. A novel method of recognizing liquefied honey. Food Chem. 2018, 245, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Tavares, M.I.B.; da Silva, E.O.; Silva, P.S.R.C.; Sebastião, P.J. The use of fast field cycling to evaluate the time domain relaxation of starches from tropical fruit seeds. Mol. Phys. 2019, 117, 1028–1033. [Google Scholar] [CrossRef]
- Ladd Parada, M.; Povey, M.J.; Vieira, J.; Rappolt, M.; Ries, M.E. Early stages of fat crystallisation evaluated by low-field NMR and small-angle X-ray scattering. Magn. Reson. Chem. 2019, 57, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Meo, P.; Mundo, F.; Terranova, S.; Conte, P.; Chillura Martino, D. water dynamics at the solid-liquid interface to unveil the textural features of synthetic nanosponges. J. Phys. Chem. B 2020, 124, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Ó Hógáin, D.; Davies, G.R.; Baroni, S.; Aime, S.; Lurie, D.J. The use of contrast agents with fast field-cycling magnetic resonance imaging. Phys. Med. Biol. 2011, 56, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Gossuin, Y.; Serhan, Z.; Sandiford, L.; Henrard, D.; Marquardsen, T.; de Rosales, R.T.M.; Sakellariou, D.; Ferrage, F. sample shuttling relaxometry of contrast agents: nmrd profiles above 1 T with a single device. Appl. Magn. Reson. 2016, 47, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.M.; Wilds, J.M.; Carniato, F.; Botta, M.; Woods, M. On water and its effect on the performance of T1-Shortening Contrast Agents. JourIsrael J. Chem. 2017, 57, 880–888. [Google Scholar] [CrossRef]
- Baroni, S.; Ruggiero, M.R.; Aime, S.; Geninatti Crich, S. Exploring the tumour extracellular matrix by in vivo Fast Field Cycling relaxometry after the administration of a Gadolinium-based MRI contrast agent. Magn. Reson. Chem. 2019, 57, 845–851. [Google Scholar] [CrossRef]
- Conte, P.; Alonzo, G. Environmental NMR: Fast-field-cycling relaxometry. eMagRes 2013, 2, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Conte, P.; Schmidt, H.-P. Soil-Water Interactions Unveiled by Fast Field Cycling NMR Relaxometry; Wiley Online Library: Hoboken, NJ, USA, 2017; Volume 6. [Google Scholar]
- Steele, R.M.; Korb, J.P.; Ferrante, G.; Bubici, S. New applications and perspectives of fast field cycling NMR relaxometry. Magn. Reson. Chem. 2016, 54, 502–509. [Google Scholar] [CrossRef]
- Stelar Book of Applications. Available online: https://www.stelar.it/pdf/Stelar_BOOKLET-web.pdf (accessed on 13 May 2020).
- Bleam, W.F. Soil science applications of nuclear magnetic resonance spectroscopy. Adv. Agron. 1991. [Google Scholar] [CrossRef]
- Akitt, J.W.; McDonald, W.S. Arrangements of ligands giving low electric field gradients. J. Magn. Reson. 1984, 58, 401–412. [Google Scholar] [CrossRef]
- Conte, P.; Nestle, N. Water dynamics in different biochar fractions. Magn. Reson. Chem. 2015, 53, 726–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, J. Understanding NMR Spectroscopy, 1st ed.; John Wiley & Sons, Inc.: Chichester, UK, 2010; ISBN 1119964938. [Google Scholar]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Zampetoulas, V.; Lurie, D.J.; Broche, L.M. Correction of environmental magnetic fields for the acquisition of Nuclear magnetic relaxation dispersion profiles below Earth’s field. J. Magn. Reson. 2017, 282, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provencher, S.W. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput. Phys. Commun. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Provencher, S.W. Contin: A general purpose constrainded regularization program for inverting noisy linear algebraic and integral equations. Comput. Phys. Commun. 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Luchinat, C.; Parigi, G. Nuclear relaxometry helps designing systems for solution DNP on proteins. Appl. Magn. Reson. 2008, 34, 379–392. [Google Scholar] [CrossRef]
- Maccotta, A.; De Pasquale, C.; Caruso, A.; Cosentino, C.; Alonzo, G.; Conte, P. Reconstruction of the environmental evolution of a Sicilian saltmarsh (Italy). Environ. Sci. Pollut. Res. 2013, 20, 4847–4858. [Google Scholar] [CrossRef]
- Conte, P.; Ferro, V. Standardizing the use of fast-field cycling NMR relaxometry for measuring hydrological connectivity inside the soil. Magn. Reson. Chem. 2020, 58, 41–50. [Google Scholar] [CrossRef]
- Halle, B.; Jóhannesson, H.; Venu, K. Model-Free Analysis of Stretched Relaxation Dispersions. J. Magn. Reson. 1998, 135, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Korb, J.P.; Whaley-Hodges, M.; Bryant, R.G. Translational diffusion of liquids at surfaces of microporous materials: Theoretical analysis of field-cycling magnetic relaxation measurements. Phys. Rev. E—Stat. Physics, Plasmas, Fluids, Relat. Interdiscip. Top. 1997, 56, 1934–1945. [Google Scholar] [CrossRef]
- Bubici, S.; Korb, J.-P.; Kučerik, J.; Conte, P. Evaluation of the surface affinity of water in three biochars using fast field cycling NMR relaxometry. Magn. Reson. Chem. 2016, 54, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, J.P. Multiscale nuclear magnetic relaxation dispersion of complex liquids in bulk and confinement. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 104, 12–55. [Google Scholar] [CrossRef] [PubMed]
- Laudicina, V.A.; de Pasquale, C.; Conte, P.; Badalucco, L.; Alonzo, G.; Palazzolo, E. Effects of afforestation with four unmixed plant species on the soil-water interactions in a semiarid Mediterranean region (Sicily, Italy). J. Soils Sediments 2012, 12, 1222–1230. [Google Scholar] [CrossRef]
- Conte, P.; Marsala, V.; De Pasquale, C.; Bubici, S.; Valagussa, M.; Pozzi, A.; Alonzo, G. Nature of water-biochar interface interactions. GCB Bioenergy 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Conte, P.; Schmidt, H.-P.; Cimò, G. Research and Application of Biochar in Europe. In Agricultural and Environmental Applications of Biochar: Advances and Barriers; Guo, M., He, Z., Uchimiya, S.M., Eds.; Soil Science Society of America, Inc.: Madison, WI, USA, 2015; pp. 409–422. ISBN 9780891189640. [Google Scholar]
- Schmidt, H.; Pandit, B.; Martinsen, V.; Cornelissen, G.; Conte, P.; Kammann, C. Fourfold increase in pumpkin yield in response to low-dosage root zone application of urine-enhanced biochar to a fertile tropical soil. Agriculture 2015, 5, 723–741. [Google Scholar] [CrossRef] [Green Version]
- Kammann, C.I.; Schmidt, H.-P.; Messerschmidt, N.; Linsel, S.; Steffens, D.; Müller, C.; Koyro, H.-W.; Conte, P.; Stephen, J. Plant growth improvement mediated by nitrate capture in co-composted biochar. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Hagemann, N.; Joseph, S.; Schmidt, H.-P.; Kammann, C.I.; Harter, J.; Borch, T.; Young, R.B.; Varga, K.; Taherymoosavi, S.; Elliott, K.W.; et al. Organic coating on biochar explains its nutrient retention and stimulation of soil fertility. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, B.; Lin, Y.; Guan, Y. Aromatic and hydrophobic surfaces of wood-derived biochar enhance perchlorate adsorption via hydrogen bonding to oxygen-containing organic groups. Environ. Sci. Technol. 2014, 48, 279–288. [Google Scholar] [CrossRef]
- Joseph, S.; Kammann, C.I.; Shepherd, J.G.; Conte, P.; Schmidt, H.-P.; Hagemann, N.; Rich, A.M.; Marjo, C.E.; Allen, J.; Munroe, P.; et al. Microstructural and associated chemical changes during the composting of a high temperature biochar: Mechanisms for nitrate, phosphate and other nutrient retention and release. Sci. Total Environ. 2018, 618, 1210–1223. [Google Scholar] [CrossRef] [Green Version]
- Baiamonte, G.; De Pasquale, C.; Marsala, V.; Cimò, G.; Alonzo, G.; Crescimanno, G.; Conte, P. Structure alteration of a sandy-clay soil by biochar amendments. J. Soils Sediments 2015, 15, 816–824. [Google Scholar] [CrossRef]
- Juriga, M.; Šimanský, V. Effect of biochar on soil structure—review. Acta Fytotech. Zootech. 2018, 21, 11–19. [Google Scholar] [CrossRef]
- Blas, E.D.; Almendros, G.; Sanz, J. Geoderma Molecular characterization of lipid fractions from extremely water-repellent pine and eucalyptus forest soils. Geoderma 2013, 206, 75–84. [Google Scholar] [CrossRef]
- Diehl, D. Soil water repellency: Dynamics of heterogeneous surfaces. Colloids Surfaces A Physicochem. Eng. Asp. 2013, 432, 8–18. [Google Scholar] [CrossRef]
- Hajnos, M.; Calka, A.; Jozefaciuk, G. Wettability of mineral soils. Geoderma 2013, 206, 63–69. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, W.; Hao, X.; Zhou, D. Transport of biochar particles in saturated granular media: Effects of pyrolysis temperature and particle size. Environ. Sci. Technol. 2013, 47, 821–828. [Google Scholar] [CrossRef]
- Yi, S.; Witt, B.; Chiu, P.; Guo, M.; Imhoff, P. The Origin and Reversible Nature of Poultry Litter Biochar Hydrophobicity. J. Environ. Qual. 2015, 44, 963–971. [Google Scholar] [CrossRef]
- Yi, S.; Chang, N.Y.; Imhoff, P.T. Predicting water retention of biochar-amended soil from independent measurements of biochar and soil properties. Adv. Water Resour. 2020. Accepted. [Google Scholar] [CrossRef]
- Honegger, P.; Overbeck, V.; Strate, A.; Appelhagen, A.; Sappl, M.; Heid, E.; Schröder, C.; Ludwig, R.; Steinhauser, O. Understanding the nature of nuclear magnetic resonance relaxation by means of fast-field-cycling relaxometry and molecular dynamics simulations—the validity of relaxation models. J. Phys. Chem. Lett. 2020, 11, 2165–2170. [Google Scholar] [CrossRef]
- Mengistu, A.G.; Mavimbela, S.S.W.; van Rensburg, L.D. Characterisation of the soil pore system in relation to its hydraulic functions in two South African aeolian soil groups. South African J. Plant Soil 2019, 36, 107–116. [Google Scholar] [CrossRef]
- Pagliai, M.; Vignozzi, N.; Pellegrini, S. Soil structure and the effect of management practices. Soil Tillage Res. 2004, 79, 131–143. [Google Scholar] [CrossRef]
- Pohlmeier, A.; Haber-Pohlmeier, S.; Stapf, S. A Fast Field Cycling Nuclear Magnetic Resonance Relaxometry Study of Natural Soils. Vadose Zo. J. 2009, 8, 735–742. [Google Scholar] [CrossRef]
- Bayer, J.V.; Jaeger, F.; Schaumann, G.E. Proton nuclear magnetic resonance (NMR) relaxometry in soil science applications. Open Magn. Reson. J. 2010, 3, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Stingaciu, L.R.; Weihermller, L.; Haber-Pohlmeier, S.; Stapf, S.; Vereecken, H.; Pohlmeier, A. Determination of pore size distribution and hydraulic properties using nuclear magnetic resonance relaxometry: A comparative study of laboratory methods. Water Resour. Res. 2010, 46, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Conte, P.; Abbate, C.; Baglieri, A.; Nègre, M.; Pasquale, C.D.; Alonzo, G.; Gennari, M. Adsorption of dissolved organic matter on clay minerals as assessed by infra-red, CPMAS13C NMR spectroscopy and low field T<inf>1</inf>NMR relaxometry. Org. Geochem. 2011, 42. [Google Scholar] [CrossRef]
- Haber-Pohlmeier, S.; Stapf, S.; Van Dusschoten, D.; Pohlmeier, A. Relaxation in a Natural Soil: Comparison of Relaxometric Imaging, T 1-T 2 Correlation and Fast-Field Cycling NMR. Open Magn. Reson. J. 2010, 3, 57–62. [Google Scholar] [CrossRef]
- Haber-Pohlmeier, S.; Stapf, S.; Pohlmeier, A. NMR Fast Field Cycling Relaxometry of Unsaturated Soils. Appl. Magn. Reson. 2014, 45, 1099–1115. [Google Scholar] [CrossRef]
- Collins, A.L.; Walling, D.E.; Sichingabula, H.M.; Leeks, G.J.L. Using 137Cs measurements to quantify soil erosion and redistribution rates for areas under different land use in the Upper Kaleya River basin, southern Zambia. Geoderma 2001, 104, 299–323. [Google Scholar] [CrossRef]
- Conte, P.; Di Stefano, C.; Ferro, V.; Laudicina, V.A.; Palazzolo, E. Assessing hydrological connectivity inside a soil by fast-field-cycling nuclear magnetic resonance relaxometry and its link to sediment delivery processes. Environ. Earth Sci. 2017, 76. [Google Scholar] [CrossRef]
- Conte, P.; Ferro, V. Measuring hydrological connectivity inside a soil by low field nuclear magnetic resonance relaxometry. Hydrol. Process. 2018, 32. [Google Scholar] [CrossRef]
- Marchamalo, M.; Hooke, J.M.; Sandercock, P.J. Flow and Sediment Connectivity in Semi-arid Landscapes in SE Spain: Patterns and Controls. L. Degrad. Dev. 2016, 27, 1032–1044. [Google Scholar] [CrossRef]
- Bracken, L.J.; Wainwright, J.; Ali, G.A.; Tetzlaff, D.; Smith, M.W.; Reaney, S.M.; Roy, A.G. Concepts of hydrological connectivity: Research approaches, Pathways and future agendas. Earth-Science Rev. 2013, 119, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Reaney, S.M.; Bracken, L.J.; Kirkby, M.J. The importance of surface controls on overland flow connectivity in semi-arid environments: Results from a numerical experimental approach. Hydrol. Process. 2014, 28, 2116–2128. [Google Scholar] [CrossRef] [Green Version]
- López-Vicente, M.; Nadal-Romero, E.; Cammeraat, E.L.H. Hydrological connectivity does change over 70 years of abandonment and afforestation in the spanish pyrenees. L. Degrad. Dev. 2017, 28, 1298–1310. [Google Scholar] [CrossRef] [Green Version]
- Bracken, L.J.; Croke, J. The concept of hydrological connectivity and its contribution to understanding runoff-dominated geomorphic systems. Hydrol. Process. 2007, 21, 1749–1763. [Google Scholar] [CrossRef]
- Wainwright, J.; Parsons, A.J.; Cooper, J.R.; Gao, P.; Gillies, J.A.; Mao, L.; Orford, J.D.; Knight, P.G. The concept of transport capacity in geomorphology. Rev. Geophys. 2015, 53, 1155–1202. [Google Scholar] [CrossRef]
- Olson, K.R. The effects of erosion on soil pore distributions and root ramification in fine-textured Illinois soils. Soil Sci. 1988, 145, 365–373. [Google Scholar] [CrossRef]
- Zsolnay, Á. Dissolved organic matter: Artefacts, definitions, and functions. Geoderma 2003, 113, 187–209. [Google Scholar] [CrossRef]
- Kopinke, F.D.; Georgi, A.; Mackenzie, K. Sorption of pyrene to dissolved humic substances and related model polymers. 1. Structure—Property correlation. Environ. Sci. Technol. Sci. Technol. 2001, 35, 2536–2542. [Google Scholar] [CrossRef]
- Philippe, A.; Schaumann, G.E. Interactions of dissolved organic matter with natural and engineered inorganic colloids: A review. Environ. Sci. Technol. 2014, 48, 8946–8962. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Guggenberger, G.; Zech, W. Organically bound nutrients in dissolved organic matter fractions in seepage and pore water of weakly developed forest soils. Acta Hydrochim. Hydrobiol. 2001, 28, 411–419. [Google Scholar] [CrossRef]
- Raulund-Rasmussen, K.; Borggaard, O.K.; Hansen, H.C.B.; Olsson, M. Effect of natural organic soil solutes on weathering rates of soil minerals. Eur. J. Soil Sci. 1998, 49, 397–406. [Google Scholar] [CrossRef]
- Michalzik, B.; Kalbitz, K.; Park, J.H.; Solinger, S.; Matzner, E. Fluxes and concentrations of dissolved organic carbon and nitrogen—A synthesis for temperate forests. Biogeochemistry 2001, 52, 173–205. [Google Scholar] [CrossRef]
- Jansen, B.; Kalbitz, K.; McDowell, W.H. Dissolved Organic Matter: Linking Soils and Aquatic Systems. Vadose Zo. J. 2014, 13. [Google Scholar] [CrossRef] [Green Version]
- Gmach, M.R.; Cherubin, M.R.; Kaiser, K.; Cerri, C.E.P. Processes that influence dissolved organic matter in the soil: A review. Sci. Agric. 2020, 77, e20180164. [Google Scholar] [CrossRef]
- Tombàcz, E. Colloidal properties of humic acids and spontaneous changes of their state under varable solution conditions. Soil Sci. 1999, 164, 814–824. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular structure in soil humic substances: The new view. Environ. Sci. Technol. 2005, 39, 9009–9015. [Google Scholar] [CrossRef]
- Benedetti, M.F.; Van Riemsdijk, W.H.; Koopal, L.K. Humic substances considered as a heterogeneous Donnan gel phase. Environ. Sci. Technol. 1996, 30, 1805–1813. [Google Scholar] [CrossRef]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M. Simultaneous presence of diverse molecular patterns in humic substances in solution. J. Phys. Chem. B 2007, 111, 10577–10582. [Google Scholar] [CrossRef]
- Conte, P.; Kucerik, J. Water dynamics and its role in structural hysteresis of dissolved organic matter. Environ. Sci. Technol. 2016, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, P. Effects of ions on water structure: A low-field 1H T1 NMR relaxometry approach. Magn. Reson. Chem. 2015, 53, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, N.E.; Von Wandruszka, R. Dynamic light scattering measurements of particle size development in aqueous humic materials. Anal. Bioanal. Chem. 2001, 371, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; You, C.; Pan, G. Effect of temperature on the sorption and desorption of perfluorooctane sulfonate on humic acid. J. Environ. Sci. 2010, 22, 355–361. [Google Scholar] [CrossRef]
- Drastík, M.; Novák, F.; Kučerík, J. Origin of heat-induced structural changes in dissolved organic matter. Chemosphere 2013, 90, 789–795. [Google Scholar] [CrossRef]
- Conte, P.; Piccolo, A. Conformational arrangement of dissolved humic substances. Influence of solution composition on association of humic molecules. Environ. Sci. Technol. 1999, 33. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of ion hydration and its interpretation in terms of a common model. Pure Appl. Chem. 1987, 59, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Marcus, Y. Ionic Radii in Aqueous Solutions. Chem. Rev. Rev. 1988, 88, 1475–1498. [Google Scholar] [CrossRef]
- Plumridge, T.H.; Waigh, R.D. Water structure theory and some implications for drug design. J. Pharm. Pharmacol. 2002, 54, 1155–1179. [Google Scholar] [CrossRef]
- Bock, C.W.; Markham, G.D.; Katz, A.K.; Glusker, J.P. The arrangement of first- and second-shell water molecules around metal ions: Effects of charge and size. Theor. Chem. Acc. 2006, 115, 100–112. [Google Scholar] [CrossRef]
- Wiggins, P.M. Role of water in some biological processes. Microbiol. Rev. 1990, 54, 432–449. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, P.M. High and low density water in gels. Prog. Polym. Sci. 1995, 20, 1121–1163. [Google Scholar] [CrossRef]
- Wiggins, P.M. High and low density water and resting, active and transformed cells. Cell Biol. Int. 1996, 20, 429–435. [Google Scholar] [CrossRef]
- Wiggins, P.M. Water in complex environments such as living systems. Phys. A Stat. Mech. its Appl. 2002, 314, 485–491. [Google Scholar] [CrossRef]
- Wiggins, P. Life depends upon two kinds of water. PLoS ONE 2008, 3, e1406. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Choudhary, A.; Chandra, A. A first-principles molecular dynamics study of the solvation shell structure, vibrational spectra, polarity, and dynamics around a nitrate ion in aqueous solution. J. Phys. Chem. B 2017, 121, 9032–9044. [Google Scholar] [CrossRef] [PubMed]
- Farashishiko, A.; Slack, J.R.; Botta, M.; Woods, M. ParaCEST agents encapsulated in Reverse Nano-Assembled Capsules (RACs): How slow molecular tumbling can quench CEST contrast. Front. Chem. 2018, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Chandra, A. Born-oppenheimer molecular dynamics simulations of a bromate ion in water reveal its dual kosmotropic and chaotropic behavior. J. Phys. Chem. B 2018, 122, 2090–2101. [Google Scholar] [CrossRef]
- Holtzer, A. The use of flory–huggins theory in interpreting partitioning of solutes between organic liquids and water. Biopolymers 1992, 32, 711–715. [Google Scholar] [CrossRef]
- Holtzer, A. Does Flory–Huggins theory help in interpreting solute partitioning experiments? Biopolymers 1994, 34, 315–320. [Google Scholar] [CrossRef]
- Huang, X.; Chaparro, J.M.; Reardon, K.F.; Zhang, R.; Shen, Q.; Vivanco, J.M. Rhizosphere interactions: Root exudates, microbes, and microbial communities 1. Botany 2014, 275, 267–275. [Google Scholar] [CrossRef]
- Rudolph-Mohr, N.; Tötzke, C.; Kardjilov, N.; Oswald, S.E. Mapping water, oxygen, and pH dynamics in the rhizosphere of young maize roots. Zeitschrift fur Pflanzenernahrung und Bodenkd. 2017, 180, 336–346. [Google Scholar] [CrossRef]
- Hinsinger, P.; Plassard, C.; Tang, C.; Jaillard, B. Origins of root-mediated pH changes in the rhizosphere and their responses to environmental constraints: A review. Plant Soil 2003, 248, 43–59. [Google Scholar] [CrossRef]
- Nye, P.H. Changes of pH across the rhizosphere induced by roots. Plant Soil 1981, 61, 7–26. [Google Scholar] [CrossRef]
- Wang, X.; Tang, C. The role of rhizosphere pH in regulating the rhizosphere priming effect and implications for the availability of soil-derived nitrogen to plants. Ann. Bot. 2018, 121, 143–151. [Google Scholar] [CrossRef]
- Youssef, R.A.; Chino, M. Root-induced changes in the rhizosphere of plants. I. pH changes in relation to the bulk soil. Soil Sci. Plant Nutr. 1989, 35, 461–468. [Google Scholar] [CrossRef]
- Gallo, P.; Corradini, D.; Rovere, M. Ion hydration and structural properties of water in aqueous solutions at normal and supercooled conditions: A test of the structure making and breaking concept. Phys. Chem. Chem. Phys. 2011, 13, 19814–19822. [Google Scholar] [CrossRef]
- Henderson, M.A. The interaction of water with solid surfaces: Fundamental aspects. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Conte, P.; Piccolo, A.; Van Lagen, B.; Buurman, P.; Hemminga, M.A. Elemental quantitation of natural organic matter by CPMAS 13C NMR spectroscopy. Solid State Nucl. Magn. Reson. 2002, 21, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Ball, P. Life’s matrix: Water in the cell. Cell. Mol. Biol. 2001, 47, 717–720. [Google Scholar]
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible Effect of Ions on the Hydrogen-Bond Structure in Liquid Water. Science (80-. ) 2003, 301, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, K.R.; Tarr, C.E. Importance of classical diffusion in NMR studies of water in biological cells. Phys. Rev. A 1979, 19, 2446–2453. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0.1 | 129 | 34 |
| 30 | 445 | 96 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, P.; Lo Meo, P. Nuclear Magnetic Resonance with Fast Field-Cycling Setup: A Valid Tool for Soil Quality Investigation. Agronomy 2020, 10, 1040. https://doi.org/10.3390/agronomy10071040
Conte P, Lo Meo P. Nuclear Magnetic Resonance with Fast Field-Cycling Setup: A Valid Tool for Soil Quality Investigation. Agronomy. 2020; 10(7):1040. https://doi.org/10.3390/agronomy10071040
Chicago/Turabian StyleConte, Pellegrino, and Paolo Lo Meo. 2020. "Nuclear Magnetic Resonance with Fast Field-Cycling Setup: A Valid Tool for Soil Quality Investigation" Agronomy 10, no. 7: 1040. https://doi.org/10.3390/agronomy10071040
APA StyleConte, P., & Lo Meo, P. (2020). Nuclear Magnetic Resonance with Fast Field-Cycling Setup: A Valid Tool for Soil Quality Investigation. Agronomy, 10(7), 1040. https://doi.org/10.3390/agronomy10071040