Development of A Nested-MultiLocus Sequence Typing Approach for A Highly Sensitive and Specific Identification of Xylella fastidiosa Subspecies Directly from Plant Samples

 , , and
, , and
Abstract
:1. Introduction
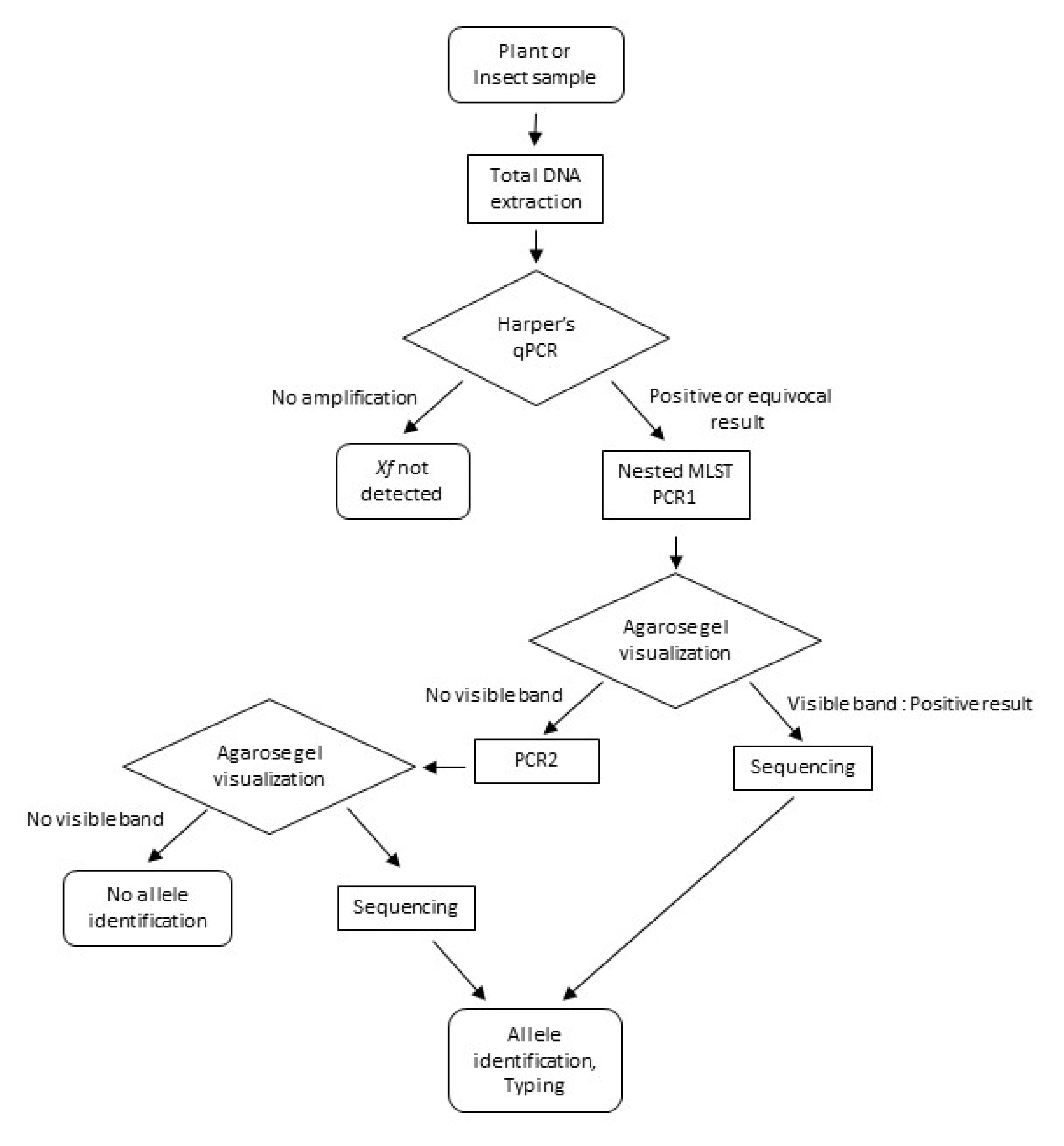
2. Materials and Methods
2.1. Strains and Media
2.2. DNA Extraction
2.3. Nested-MLST Primers and Reactions
2.4. Statistical Analysis
2.5. Sequence Acquisition, Alignment and Analyses
3. Results
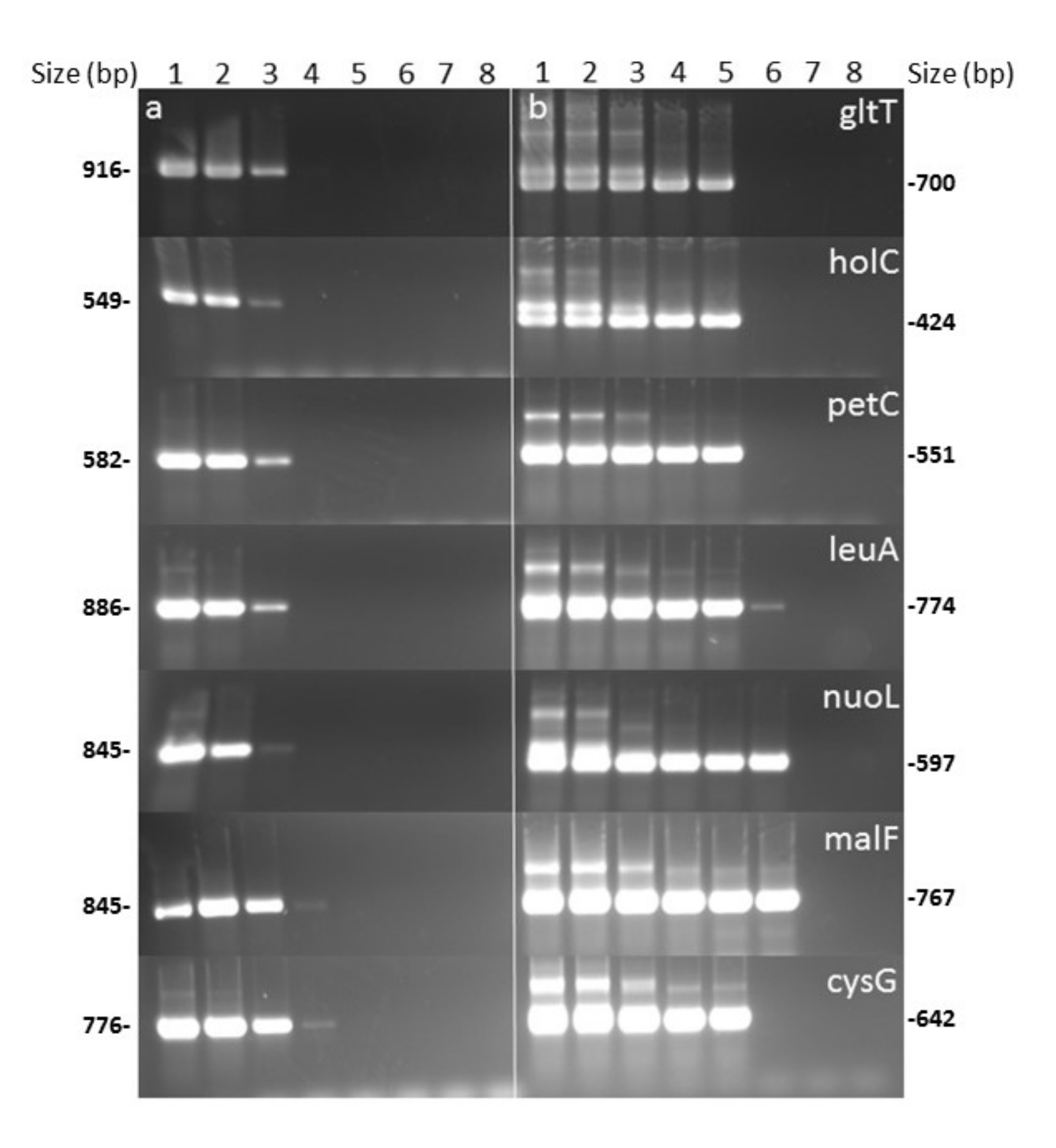
3.1. Nested-MLST Proved to be Specific
3.2. Nested-MLST Limit of Detection is Comparable to That of qPCR
3.3. Analysis of Naturally Infected Samples
3.4. Nested-MLST Improved Successful HKG Typing by Increasing Sensitivity Level
3.5. Nested-MLST Allowed Identification of New Alleles Among French Samples
3.6. Recombinants or Mixed Infections Were Identified by Nested-MLST
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| BLAST | Basic Local Alignment Search Tool |
| Cq | quantification cycle |
| HKG | housekeeping gene |
| INRA | French National Institute for Agricultural Research |
| IRHS | Research Institute of Horticulture and Seeds |
| LoD | Limit of Detection |
| MLST | Multilocus Sequence Typing |
| NCBI | National Center for Biotechnology Information |
| ST | Sequence Type |
| Xf | Xylella fastidiosa |
| WGS | Whole Genome Shotgun |
References
- Marcelletti, S.; Scortichini, M. Genome-Wide Comparison and Taxonomic Relatedness of Multiple Xylella Fastidiosa Strains Reveal the Occurrence of Three Subspecies and a New Xylella Species. Arch. Microbiol. 2016, 198, 803–812. [Google Scholar] [CrossRef]
- Denancé, N.; Briand, M.; Gaborieau, R.; Gaillard, S.; Jacques, M.-A. Identification of Genetic Relationships and Subspecies Signatures in Xylella Fastidiosa. BMC Genom. 2019, 20, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA (European Food Safety Authority). Scientific report on the update of the Xylella spp. host plant database. EFSA J. 2018, 16, 5408. [Google Scholar]
- Janse, J.D.; Obradovic, A. XYLELLA FASTIDIOSA: ITS BIOLOGY, DIAGNOSIS, CONTROL AND RISKS. J. Plant Pathol. 2010, 14, S35–S48. [Google Scholar]
- Almeida, R.P.P.; Nascimento, F.E.; Chau, J.; Prado, S.S.; Tsai, C.-W.; Lopes, S.A.; Lopes, J.R.S. Genetic Structure and Biology of Xylella Fastidiosa Strains Causing Disease in Citrus and Coffee in Brazil. AEM 2008, 74, 3690–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta-filho, H.D.; Francisco, C.S.; Lopes, J.R.S.; de Oliveira, A.F.; Da Silva, L.F.d.O. First Report of Olive Leaf Scorch in Brazil, Associated with Xylella Fastidiosa subsp. pauca. Phytopathol. Mediterr. 2016, 55, 130–135. [Google Scholar] [CrossRef]
- Haelterman, R.M.; Tolocka, P.A.; Roca, M.E.; Guzmán, F.A.; Fernández, F.D.; Otero, M.L. FIRST PRESUMPTIVE DIAGNOSIS OF XYLELLA FASTIDIOSA CAUSING OLIVE SCORCH IN ARGENTINA. J. Plant Pathol. 2015, 1. [Google Scholar] [CrossRef]
- Saponari, M.; Boscia, D.; Nigro, F.; Martelli, G.P. IDENTIFICATION OF DNA SEQUENCES RELATED TO XYLELLA FASTIDIOSA IN OLEANDER, ALMOND AND OLIVE TREES EXHIBITING LEAF SCORCH SYMPTOMS IN APULIA (SOUTHERN ITALY). J. Plant Pathol. 2013, 95. [Google Scholar] [CrossRef]
- Wells, J.M.; Raju, B.C.; Hung, H.-Y.; Weisburg, W.G.; Mandelco-Paul, L.; Brenner, D.J. Xylella Fastidiosa Gen. Nov., Sp. Nov: Gram-Negative, Xylem-Limited, Fastidious Plant Bacteria Related to Xanthomonas Spp. Int. J. Syst. Bacteriol. 1987, 37, 136–143. [Google Scholar] [CrossRef]
- PM 7/24 (4) Xylella Fastidiosa. EPPO Bull. 2019, 49, 175–227. [CrossRef] [Green Version]
- Ouyang, P.; Arif, M.; Fletcher, J.; Melcher, U.; Ochoa Corona, F.M. Enhanced Reliability and Accuracy for Field Deployable Bioforensic Detection and Discrimination of Xylella Fastidiosa Subsp. Pauca, Causal Agent of Citrus Variegated Chlorosis Using Razor Ex Technology and TaqMan Quantitative PCR. PLoS ONE 2013, 8, e81647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and Real-Time PCR Methods for the Rapid Detection of Xylella Fastidiosa for Quarantine and Field Applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Bonants, P.; Griekspoor, Y.; Houwers, I.; Krijger, M.; van der Zouwen, P.; van der Lee, T.A.J.; van der Wolf, J. Development and Evaluation of a Triplex TaqMan Assay and Next-Generation Sequence Analysis for Improved Detection of Xylella in Plant Material. Plant Dis. 2019, 103, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Waliullah, S.; Hudson, O.; Oliver, J.E.; Brannen, P.M.; Ji, P.; Ali, M.E. Comparative Analysis of Different Molecular and Serological Methods for Detection of Xylella Fastidiosa in Blueberry. PLoS ONE 2019, 14, e0221903. [Google Scholar] [CrossRef] [Green Version]
- Yaseen, T.; Drago, S.; Valentini, F.; Elbeaino, T.; Stampone, G.; Digiaro, M.; D’onghia, A.M. On-Site Detection of Xylella Fastidiosa in Host Plants and in “Spy Insects” Using the Real-Time Loop-Mediated Isothermal Amplification Method. Phytopathol. Mediterr. 2015, 54, 17–25. [Google Scholar] [CrossRef]
- Burbank, L.P.; Ortega, B.C. Novel Amplification Targets for Rapid Detection and Differentiation of Xylella Fastidiosa Subspecies Fastidiosa and Multiplex in Plant and Insect Tissues. J. Microbiol. Methods 2018, 155, 8–18. [Google Scholar] [CrossRef]
- Dupas, E.; Legendre, B.; Olivier, V.; Poliakoff, F.; Manceau, C.; Cunty, A. Comparison of Real-Time PCR and Droplet Digital PCR for the Detection of Xylella Fastidiosa in Plants. J. Microbiol. Methods 2019, 162, 86–95. [Google Scholar] [CrossRef]
- Ciapina, L.P.; Carareto Alves, L.M.; Lemos, E.G.M. A Nested-PCR Assay for Detection of Xylella Fastidiosa in Citrus Plants and Sharpshooter Leafhoppers. J. Appl. Microbiol. 2004, 96, 546–551. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Shao, J.; Singh, R.; Davis, R.E.; Zhao, T.; Huang, Q. A TaqMan-Based Real Time PCR Assay for Specific Detection and Quantification of Xylella Fastidiosa Strains Causing Bacterial Leaf Scorch in Oleander. J. Microbiol. Methods 2013, 92, 108–112. [Google Scholar] [CrossRef]
- Dupas, E.; Briand, M.; Jacques, M.-A.; Cesbron, S. Novel Tetraplex Quantitative PCR Assays for Simultaneous Detection and Identification of Xylella Fastidiosa Subspecies in Plant Tissues. Front. Plant Sci. 2019, 10, 1732. [Google Scholar] [CrossRef] [Green Version]
- Scally, M.; Schuenzel, E.L.; Stouthamer, R.; Nunney, L. Multilocus Sequence Type System for the Plant Pathogen Xylella Fastidiosa and Relative Contributions of Recombination and Point Mutation to Clonal Diversity. AEM 2005, 71, 8491–8499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Morano, L.; Bromley, R.; Spring-Pearson, S.; Stouthamer, R.; Nunney, L. Multilocus Sequence Typing of Xylella Fastidiosa Causing Pierce’s Disease and Oleander Leaf Scorch in the United States. Phytopathology 2010, 100, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denancé, N.; Legendre, B.; Briand, M.; Olivier, V.; de Boisseson, C.; Poliakoff, F.; Jacques, M.-A. Several Subspecies and Sequence Types Are Associated with the Emergence of Xylella Fastidiosa in Natural Settings in France. Plant Pathol. 2017, 66, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Cruaud, A.; Gonzalez, A.-A.; Godefroid, M.; Nidelet, S.; Streito, J.-C.; Thuillier, J.-M.; Rossi, J.-P.; Santoni, S.; Rasplus, J.-Y. Using Insects to Detect, Monitor and Predict the Distribution of Xylella Fastidiosa: A Case Study in Corsica. Sci. Rep. 2018, 8, 15628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Menezes, A.; Woods, K.; Chanthongthip, A.; Dittrich, S.; Opoku-Boateng, A.; Kimuli, M.; Chalker, V. An Extended Multilocus Sequence Typing (MLST) Scheme for Rapid Direct Typing of Leptospira from Clinical Samples. PLoS Negl. Trop Dis. 2016, 10, e0004996. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, C.; Himschoot, M.; Bruisten, S.M. Multilocus Sequence Typing of Trichomonas Vaginalis Clinical Samples from Amsterdam, the Netherlands. BMJ Open 2016, 6, e013997. [Google Scholar] [CrossRef] [Green Version]
- Mougel, C.; Cournoyer, B.; Nesme, X. Novel Tellurite-Amended Media and Specific Chromosomal and Ti Plasmid Probes for Direct Analysis of Soil Populations of Agrobacterium Biovars 1 and 2. Appl. Environ. Microbiol. 2001, 67, 65–74. [Google Scholar] [CrossRef] [Green Version]
- King, E.O.; Ward, M.K.; Raney, D.E. Two simple media for the demonstration of pyocyanin and fluorescin. J. Lab. Clin. Med. 1954, 44, 301–307. [Google Scholar]
- Doležel, J.; Bartos, J.; Voglmayr, H.; Greilhuber, J. Letter to the Editor. Cytometry 2003, 51, 127–128. [Google Scholar] [CrossRef]
- Drevinek, P.; Vosahlikova, S.; Dedeckova, K.; Cinek, O.; Mahenthiralingam, E. Direct Culture-Independent Strain Typing of Burkholderia Cepacia Complex in Sputum Samples from Patients with Cystic Fibrosis. J. Clin. Microbiol. 2010, 48, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Diggle, M.A.; Bell, C.M.; Clarke, S.C. Nucleotide Sequence-Based Typing of Meningococci Directly from Clinical Samples. J. Med. Microbiol. 2003, 52, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.; Huggett, J. QPCR Primer Design Revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Jacques, M.-A.; Denancé, N.; Legendre, B.; Morel, E.; Briand, M.; Mississipi, S.; Durand, K.; Olivier, V.; Portier, P.; Poliakoff, F.; et al. New Coffee Plant-Infecting Xylella Fastidiosa Variants Derived via Homologous Recombination. Appl. Environ. Microbiol. 2016, 82, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Nunney, L.; Hopkins, D.L.; Morano, L.D.; Russell, S.E.; Stouthamer, R. Intersubspecific Recombination in Xylella Fastidiosa Strains Native to the United States: Infection of Novel Hosts Associated with an Unsuccessful Invasion. Appl. Environ. Microbiol. 2014, 80, 1159–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saponari, M.; D’Attoma, G.; Abou Kubaa, R.; Loconsole, G.; Altamura, G.; Zicca, S.; Rizzo, D.; Boscia, D. A New Variant of Xylella Fastidiosa Subspecies Multiplex Detected in Different Host Plants in the Recently Emerged Outbreak in the Region of Tuscany, Italy. Eur. J. Plant Pathol. 2019, 154, 1195–1200. [Google Scholar] [CrossRef] [Green Version]
- Nunney, L.; Schuenzel, E.L.; Scally, M.; Bromley, R.E.; Stouthamer, R. Large-Scale Intersubspecific Recombination in the Plant-Pathogenic Bacterium Xylella Fastidiosa Is Associated with the Host Shift to Mulberry. Appl. Environ. Microbiol. 2014, 80, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Potnis, N.; Kandel, P.P.; Merfa, M.V.; Retchless, A.C.; Parker, J.K.; Stenger, D.C.; Almeida, R.P.P.; Bergsma-Vlami, M.; Westenberg, M.; Cobine, P.A.; et al. Patterns of Inter- and Intrasubspecific Homologous Recombination Inform Eco-Evolutionary Dynamics of Xylella Fastidiosa. ISME J. 2019, 13, 2319–2333. [Google Scholar] [CrossRef]
- Kandel, P.P.; Almeida, R.P.P.; Cobine, P.A.; De La Fuente, L. Natural Competence Rates Are Variable among Xylella Fastidiosa Strains and Homologous Recombination Occurs in Vitro Between Subspecies Fastidiosa and Multiplex. MPMI 2017, 30, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Kandel, P.P.; Lopez, S.M.; Almeida, R.P.P.; De La Fuente, L. Natural Competence of Xylella Fastidiosa Occurs at a High Frequency Inside Microfluidic Chambers Mimicking the Bacterium’s Natural Habitats. Appl. Environ. Microbiol. 2016, 82, 5269–5277. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| CFBP Code | Bacterial Species | Host Plant | Origin |
|---|---|---|---|
| 6448 | Agrobacterium rubi | Rubus ursinus var. loganobaccus | USA (1942) |
| 2413 | Agrobacterium tumefaciens | Malus sp. | NA (1935) |
| 5523 | Agrobacterium vitis | Vitis vinifera | Australia (1977) |
| 2404 | Clavibacter insidiosus | Medicago sativa | USA (1955) |
| 4999 | Clavibacter michiganensis | Lycopersicon esculentum | Hungary (1957) |
| 3418 | Curtobacterium flaccumfaciens pv. flaccumfaciens | Phaseolus vulgaris | Hungary (1957) |
| 1200 | Dickeya dianthicola | Dianthus caryophyllus | United Kingdom (1956) |
| 5561 | Ensifer meliloti | Medicago sativa | VA, USA (1984) |
| 1232 | Erwinia amylovora | Pyrus communis | United Kingdom (1959) |
| 3845 | Pantoea agglomerans | Knee laceration | Zimbabwe (1956) |
| 3167 | Pantoea stewartii pv. stewartii | Zea mays var. rugosa | USA (1970) |
| 3205 | Pseudomonas amygdali | Prunus amygdalus | Greece (1967) |
| 8305 | Pseudomonas cerasi | Prunus cerasus | Poland (2007) |
| 7019 | Pseudomonas congelans | na 1 | Germany (1994) |
| 1573 | Pseudomonas syringae pv. persicae | Prunus persica | France (1974) |
| 1392 | Pseudomonas syringae pv. syringae | Syringa vulgaris | United Kingdom (1950) |
| 7436 | Rhizobium nepotum | Prunus ceresifera myrobolan | Hungary (1989) |
| 13100 | Stenotrophomas maltophilia | Phaseolus vulgaris | Cameroon (2009) |
| 3371 | Xanthomonas euvesicatoria pv. citrumelonis | Citrus sp. | USA (1989) |
| 2528 | Xanthomonas arboricola pv. juglandis | Juglans regia | New Zealand (1956) |
| 2535 | Xanthomonas arboricola pv. pruni | Prunus salicina | New Zealand (1953) |
| 4924 | Xanthomonas axonopodis pv. axonopodis | Axonopus scoparius | Colombia (1949) |
| 5241 | Xanthomonas campestris pv. campestris | Brassica oleracea var. gemmifera | United Kingdom (1957) |
| 2901 | Xanthomonas citri pv. aurantifolii | Citrus limon | Argentina (1988) |
| 2525 | Xanthomonas citri pv. citri | Citrus limon | New Zealand (1956) |
| 7660 | Xanthomonas citri pv. viticola | Vitis vinifera | India (1969) |
| 2625 | Xanthomonas gardneri | Medicago sativa | Reunion Island (1986) |
| 4925 | Xanthomonas hortorum pv. hederae | Hedera helix | USA (1944) |
| 2533 | Xanthomonas hortorum pv. pelargonii | Pelargonium peltatum | New Zealand (1974) |
| 1156 | Xanthomonas hyacinthi | Hyacinthus orientalis | Netherlands (1958) |
| 2532 | Xanthomonas oryzae pv. oryzae | Oryza sativa | India (1965) |
| 2054 | Xanthomonas translucens | Hordeum vulgare | USA (1933) |
| 2543 | Xanthomonas vasicola pv. holcicola | Sorghum vulgare | New Zealand (1969) |
| 7970 | Xylella fastidiosa subsp. fastidiosa | Vitis vinifera | USA (1987) |
| 8416 | Xylella fastidiosa subsp. multiplex | Polygala myrtifolia | France (2015) |
| 8084 | Xylella fastidiosa subsp. morus | Morus alba | USA (na1) |
| 8070 | Xylella fastidiosa subsp. multiplex | Prunus spp. | USA (2004) |
| 8402 | Xylella fastidiosa subsp. pauca | Olea europea | Italy (2014) |
| 1192 | Xylophilus ampelinus | Vitis vinifera | Greece (1966) |
| Locus | PCR Round | 5′-Forward Primer-3′ | 5′-Reverse Primer-3′ | Position on Xf M12 Genome (CP000941.1) | Annealing Temperature (°C) | Size (pb) of Reaction Product |
|---|---|---|---|---|---|---|
| cysG | 1 | CCAAACATAGAAGCACGCCG | GCGAGTGTTTTCAGCGTTCC | 2111116–2111891 | 64 | 776 |
| 2 | GCCGAAGCAGTGCTGGAAG 1 | GCCATTTTCGATCAGTGCAAAAG 1 | 2111203–2111844 | 56 | 642 | |
| gltT | 1 | GGTGCCATCCAATCCGTTTT | TCAGGATGTCCCAATTCCAACG | 1731589–1732504 | 60 | 916 |
| 2 | TCATGATCCAAATCACTCGCTT 1 | TTACTGGACGCTGCCTCG | 1731783–1732482 | 56 | 700 | |
| holC | 1 | CCGATGGTGAAGAACAGTAGACA | GCTCGAGAAACTSGATTAATGG | 133166–133714 | 62 | 549 |
| 2 | GGTCACATGTCGTGTTTGTTC | CACGCGCCGACTTCTATTT | 133269–133692 | 59 | 424 | |
| leuA | 1 | CGAAGGTGCAAACAAAGTGA | CGCACTGGCTTCGATAATGTCT | 1271664–1272549 | 58 | 886 |
| 2 | GGTGCACGCCAAATCGAATG 1 | ACTGGTCCCTGTACCTTCGT | 1271752–1272525 | 60 | 774 | |
| malF | 1 | AACGTCGTCACCCCAAGAA | ATGAGGCGGGCTTCTTTGG | 1680264–1681108 | 56 | 845 |
| 2 | AGCAGAAGCACGTCCCAGAT | CTGGTCCTGCGGTGTTGG | 1680308–1681074 | 60 | 767 | |
| nuoL | 1 | TTGGTACGTTGGCTTTGGTG | GACAAAACCAGATTGCGTGC | 325347–326191 | 60 | 845 |
| 2 | GCGACTTACGGTTACTGGGC | ACCACCGATCCACAACGCAT 1 | 325454–326050 | 54 | 597 | |
| petC | 1 | TCAATGCACGTCCTCCCAAT | GGCTGCCATTCGTTGAAGTA | 2020498–2021079 | 60 | 582 |
| 2 | ACGTCCTCCCAATAAGCCT | CGTTATTCACGTATCGCTGC | 2020505–2021055 | 56 | 551 |
| Percentage of Successful Amplifications Obtained for Each Locus in Conventional and Nested MLST-PCR | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample Type | Country | Year | Number of Samples | qPCR Harper Number of Samples | cysG | gltT | holC | leuA | malF | nuoL | petC | Average per Year | |||||||||
| Cq < 35 | Cq ≥ 35 | conv | nest | conv | nest | conv | nest | conv | nest | conv | nest | conv | nest | conv | nest | conv | nest | ||||
| Plant | France | 2017 | 106 | 22 | 70 | 1.1 | 28.3 | 2.2 | 26.1 | 4.3 | 55.4 | 4.3 | 34.8 | 1.1 | 35.9 | 0 | 26.1 | 1.1 | 46.7 | 2 | 36.2 |
| Plant | France | 2018 | 162 | 8 | 36 | 0 | 11.4 | 0 | 9.1 | 0 | 27.3 | 0 | 27.3 | 0 | 15.9 | 0 | 27.3 | 0 | 25 | 0 | 20.5 |
| Plant | Spain | 2018 | 40 | 38 | 2 | 55 | 90 * | 10 | 77.5 * | 15 | 80 * | 12.5 | 75 * | 30 | 75 * | 40 | 85 * | 15 | 85 * | 25.4 | 81.1 |
| Plant | Spain | 2019 | 30 | 30 | 0 | 30 | 90 * | 13.3 | 90 * | 16.7 | 93.3 * | 16.7 | 90 * | 20 | 90 * | 66.7 | 90 * | 20 | 90 * | 26.2 | 90.5 |
| Insect | Spain | 2018 | 26 | 18 | 8 | 65.4 | 80.8 | 7.7 | 73.1 * | 19.2 | 69.2 * | 11.5 | 57.7 * | 7.7 | 53.8 * | 26.9 | 57.7 * | 15.4 | 73.1 * | 22 | 66.5 |
| Country | Sample Names | cysG | gltT | holC | leuA | malF | nuoL | petC | Sequence Type (ST) |
|---|---|---|---|---|---|---|---|---|---|
| France | Spartium junceum 2 | 7 | 3 | 3 | 3 | 3 | 3 | 3 | ST7 |
| France | Polygala myrtifolia 3, 4 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| France | Genista corsica 1 | 7 | 3 | 3 | 3 | 3 | 3 | 3 | ST7 |
| France | Polygala myrtifolia 5, 6 | 7 | 3 | 3 | 3 | 3 | 3 | 3 | ST7 |
| Spain | Cistus albidus 2 | 31 | 15 | 10 | 7 | 17 | 16 | 6 | ST80 |
| Spain | Ficus carica 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ST1 |
| Spain | Helichrysum italicum 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Juglans regia 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ST1 |
| Spain | Lavandula angustifolia 1 | 32 | 3 | 3 | 3 | 3 | 3 | 3 | ST81 |
| Spain | Olea europaea 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Phagnalon saxatile 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Polygala myrtifolia 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Prunus armeniaca 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Prunus domestica 1 | 32 | 3 | 3 | 3 | 3 | 3 | 3 | ST81 |
| Spain | Prunus domestica 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Prunus dulcis 4–8,10,11,15,18–26,30–47 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Prunus dulcis 9 | 31 | 15 | 10 | 7 | 17 | 16 | 6 | ST80 |
| Spain | Prunus dulcis 1,2 | 32 | 3 | 3 | 3 | 3 | 3 | 3 | ST81 |
| Spain | Prunus dulcis 3 | 7 | 3 | 3 | 3 | 3 | 3 | 3 | ST7 |
| Spain | Rhamnus alaternus 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Rosmarinus officinalis 4 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Rosmarinus officinalis 1,3 | 31 | 15 | 10 | 7 | 17 | 16 | 6 | ST80 |
| Spain | Prunus domestica 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Philaenus spumarius 6,7,8,10,11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ST1 |
| Spain | Philaenus spumarius 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
| Spain | Philaenus spumarius 22 | 32 | 3 | 3 | 3 | 3 | 3 | 3 | ST81 |
| Spain | Neophilaenus campestris 1,2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | ST6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesbron, S.; Dupas, E.; Beaurepère, Q.; Briand, M.; Montes-Borrego, M.; Velasco-Amo, M.d.P.; Landa, B.B.; Jacques, M.-A. Development of A Nested-MultiLocus Sequence Typing Approach for A Highly Sensitive and Specific Identification of Xylella fastidiosa Subspecies Directly from Plant Samples. Agronomy 2020, 10, 1099. https://doi.org/10.3390/agronomy10081099
Cesbron S, Dupas E, Beaurepère Q, Briand M, Montes-Borrego M, Velasco-Amo MdP, Landa BB, Jacques M-A. Development of A Nested-MultiLocus Sequence Typing Approach for A Highly Sensitive and Specific Identification of Xylella fastidiosa Subspecies Directly from Plant Samples. Agronomy. 2020; 10(8):1099. https://doi.org/10.3390/agronomy10081099
Chicago/Turabian StyleCesbron, Sophie, Enora Dupas, Quentin Beaurepère, Martial Briand, Miguel Montes-Borrego, Maria del Pilar Velasco-Amo, Blanca B. Landa, and Marie-Agnès Jacques. 2020. "Development of A Nested-MultiLocus Sequence Typing Approach for A Highly Sensitive and Specific Identification of Xylella fastidiosa Subspecies Directly from Plant Samples" Agronomy 10, no. 8: 1099. https://doi.org/10.3390/agronomy10081099
APA StyleCesbron, S., Dupas, E., Beaurepère, Q., Briand, M., Montes-Borrego, M., Velasco-Amo, M. d. P., Landa, B. B., & Jacques, M. -A. (2020). Development of A Nested-MultiLocus Sequence Typing Approach for A Highly Sensitive and Specific Identification of Xylella fastidiosa Subspecies Directly from Plant Samples. Agronomy, 10(8), 1099. https://doi.org/10.3390/agronomy10081099