
Genome-Wide Association Study (GWAS) Reveals an SNP Associated with Waxy Trait and Development of a Functional Marker for Predicting Waxy Maize (Zea mays L. var. ceratina)
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping
2.2. SNP Array Genotyping and Data Filtering
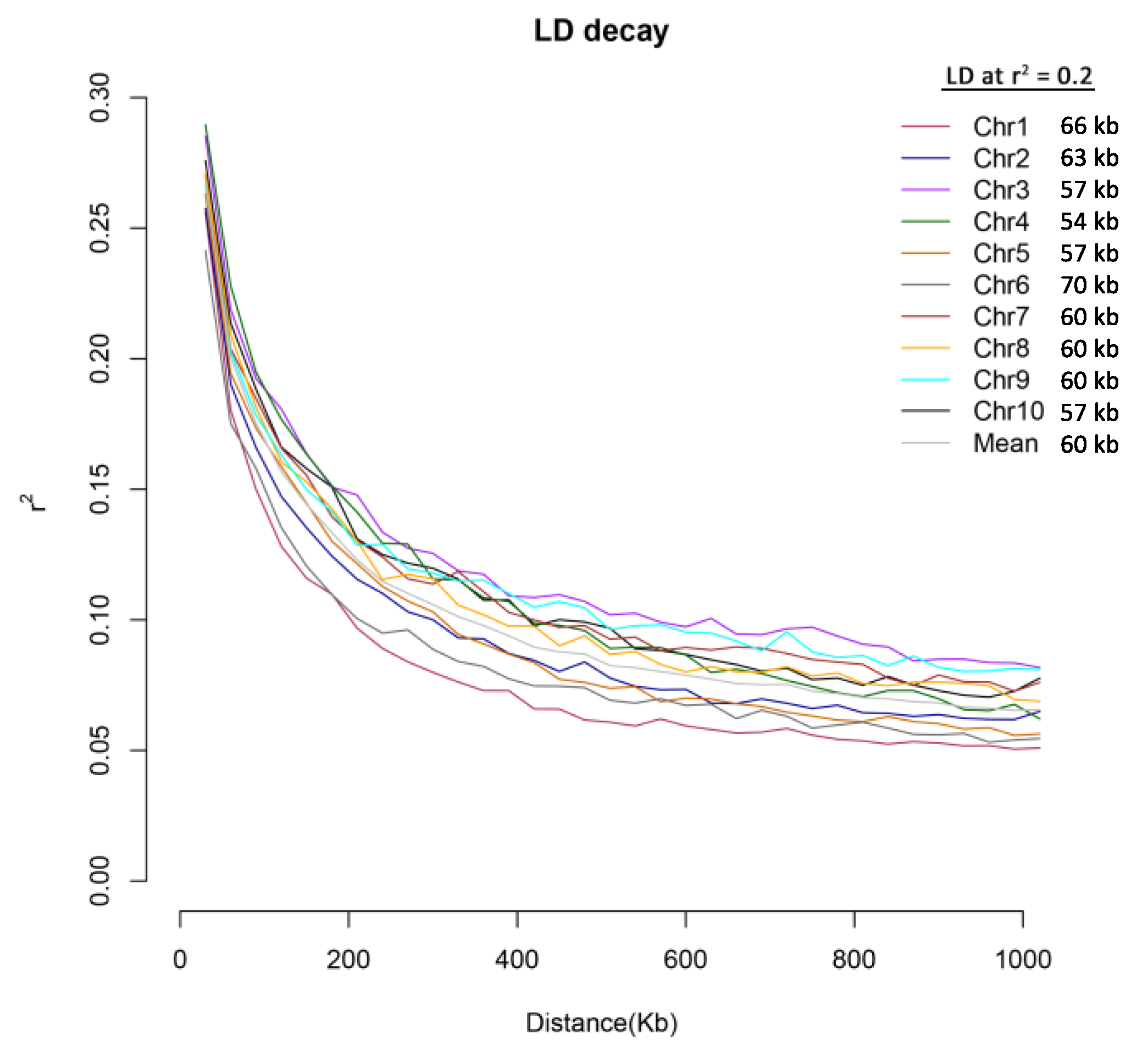
2.3. Analysis of Population Structure and Linkage Disequilibrium (LD) Decay
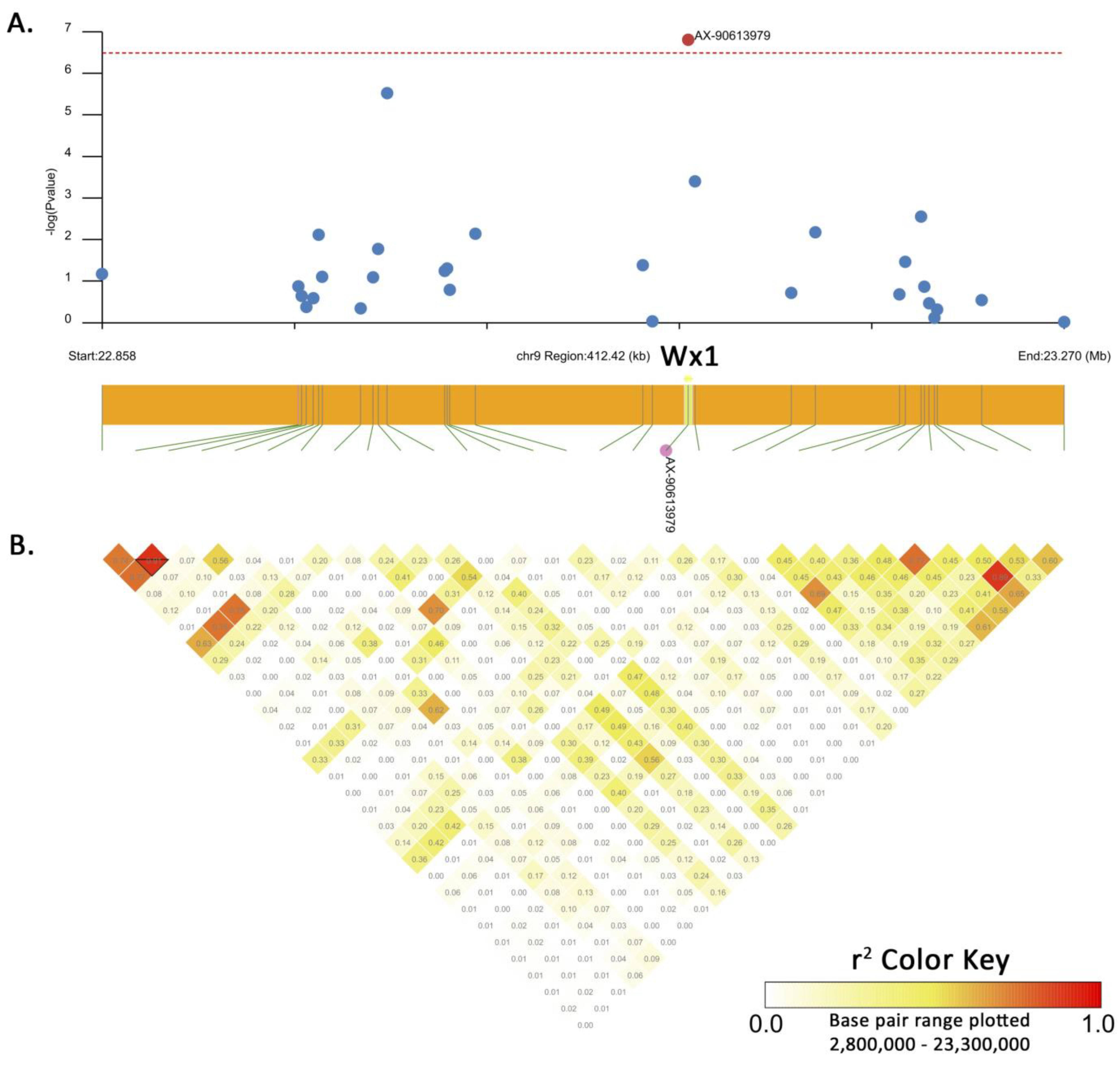
2.4. Genome-Wide Association Study (GWAS) and Candidate Gene Analysis
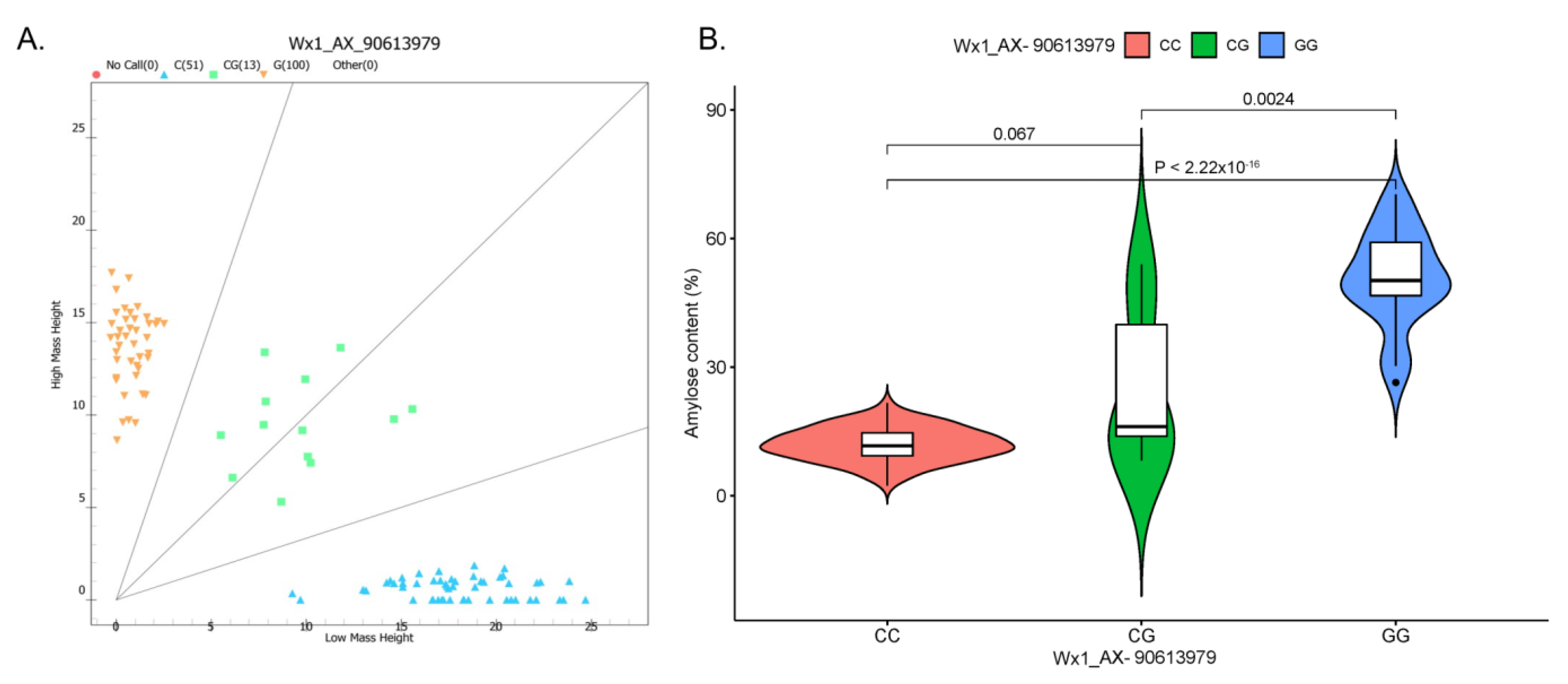
2.5. Development and Validation of SNP Marker
3. Results
3.1. Phenotyping, Genotyping, and Population Study in the Panel of 213 Maize Lines
3.2. Genome-Wide Association Study (GWAS) for Waxy Trait
3.3. Development of an SNP Marker Associated with Waxy Trait and Validation among Different Maize Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandhu, K.S.; Singh, N.; Malhi, N.S. Some properties of corn grains and their flours I: Physicochemical, functional and chapati-making properties of flours. Food Chem. 2007, 101, 938–946. [Google Scholar] [CrossRef]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, I.J. Starch biosynthesis in developing seeds. Seed Sci. Res. 2011, 21, 5–32. [Google Scholar] [CrossRef]
- Shannon, J.C.; Garwood, D.L.; Boyer, C.D. Genetics and physiology of starch development. In Starch; Elsevier: Amsterdam, The Netherlands, 2009; pp. 23–82. ISBN 9780127462752. [Google Scholar]
- Tian, M.; Tan, G.; Liu, Y.; Rong, T.; Huang, Y. Origin and evolution of Chinese waxy maize: Evidence from the Globulin-1 gene. Genet. Resour. Crop Evol. 2009, 56, 247–255. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, H.; Yang, H.; Wu, J.; Shi, B.; Cai, R.; Xu, Y.; Wu, A.; Luo, L. Genetic diversity and molecular evolution of Chinese waxy maize germplasm. PLoS ONE 2013, 8, e66606. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Yao, J.; Zhu, J.; Hu, W.; Cai, D.; Li, Y.; Shu, Q.; Fan, L. Identification of glutinous maize landraces and inbred lines with altered transcription of waxy gene. Mol. Breed. 2012, 30, 1707–1714. [Google Scholar] [CrossRef]
- Collins, G.N. A New Type of Indian Corn from China; Govt. Print. Off.: Washington, DC, USA, 1909.
- Fan, L.; Quan, L.; Leng, X.; Guo, X.; Hu, W.; Ruan, S.; Ma, H.; Zeng, M. Molecular evidence for post-domestication selection in the Waxy gene of Chinese waxy maize. Mol. Breed. 2008, 22, 329–338. [Google Scholar] [CrossRef]
- Jeon, J.-S.; Ryoo, N.; Hahn, T.-R.; Walia, H.; Nakamura, Y. Starch biosynthesis in cereal endosperm. Plant Physiol. Biochem. 2010, 48, 383–392. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.; Huang, R.; Wu, Y.; Wang, W. The genetic architecture of amylose biosynthesis in maize kernel. Plant Biotechnol. J. 2018, 16, 688–695. [Google Scholar] [CrossRef]
- James, M.G.; Robertson, D.S.; Myers, A.M. Characterization of the maize gene sugary1, a determinant of starch composition in kernels. Plant Cell 1995, 7, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Sprague, G.F.; Brimhall, B.; Hixon, R.M. Some effects of the waxy gene in corn on properties of the endosperm starch. Agron. J. 1943, 35, 817–822. [Google Scholar] [CrossRef]
- Demerec, M. A case of pollen dimorphism in maize. Am. J. Bot. 1924, 11, 461–464. [Google Scholar] [CrossRef]
- Nelson, O.E.; Rines, H.W. The enzymatic deficiency in the waxy mutant of maize. Biochem. Biophys. Res. Commun. 1962, 9, 297–300. [Google Scholar] [CrossRef]
- Wessler, S.R.; Varagona, M.J. Molecular basis of mutations at the waxy locus of maize: Correlation with the fine structure genetic map. Proc. Natl. Acad. Sci. USA 1985, 82, 4177–4181. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, Z.; Xue, Y.; Meng, S.; Huang, Y.; Li, W.; Huang, J.; Tang, J. Identification of Quantitative Trait Loci and Candidate Genes for Maize Starch Granule Size through Association Mapping. Sci. Rep. 2018, 8, 14236. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xue, Y.; Guo, Z.; Li, W.; Tang, J. Genome-Wide Association Study Identifies Candidate Genes for Starch Content Regulation in Maize Kernels. Front. Plant Sci. 2016, 7, 1046. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.L.; Carbas, B.; Gaspar, D.; Paulo, M.; Brites, C.; Mendes-Moreira, P.; Brites, C.M.; Malosetti, M.; van Eeuwijk, F.; Vaz Patto, M.C. Genome-wide association study for kernel composition and flour pasting behavior in wholemeal maize flour. BMC Plant Biol. 2019, 19, 123. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Wu, X.; Alexander, L.W. Genome-wide association studies for inflorescence type and remontancy in Hydrangea macrophylla. Hortic. Res. 2020, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Eizenga, G.C.; Jackson, A.K.; Edwards, J.D. Prototype for developing SNP markers from GWAS and biparental QTL for rice panicle and grain traits. Agric. Environ. Lett. 2021, 6, e20047. [Google Scholar] [CrossRef]
- Simla, S.; Lertrat, K.; Suriharn, B. Carbohydrate Characters of Six Vegetable Waxy Corn varieties as Affected by Harvest Time and Storage Duration. Asian J. Plant Sci. 2010, 9, 463–470. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Bandillo, N.B.; Lorenz, A.J.; Graef, G.L.; Jarquin, D.; Hyten, D.L.; Nelson, R.L.; Specht, J.E. Genome-wide Association Mapping of Qualitatively Inherited Traits in a Germplasm Collection. Plant Genome 2017, 10. [Google Scholar] [CrossRef]
- Ruanjaichon, V.; Khammona, K.; Thunnom, B.; Suriharn, K.; Kerdsri, C.; Aesomnuk, W.; Yongsuwan, A.; Chaomueang, N.; Thammapichai, P.; Arikit, S.; et al. Identification of Gene Associated with Sweetness in Corn (Zea mays L.) by Genome-Wide Association Study (GWAS) and Development of a Functional SNP Marker for Predicting Sweet Corn. Plants 2021, 10, 1239. [Google Scholar] [CrossRef]
- Nelson, O.; Pan, D. Starch synthesis in maize endosperms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 475–496. [Google Scholar] [CrossRef]
- Creech, R.G. Genetic control of carbohydrate synthesis in maize endosperm. Genetics 1965, 52, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Klösgen, R.B.; Gierl, A.; Schwarz-Sommer, Z.; Saedler, H. Molecular analysis of the waxy locus of Zea mays. Molec. Gen. Genet. 1986, 203, 237–244. [Google Scholar] [CrossRef]
- Lertrat, K.; Thongnarin, N. Novel approach to eating quality improvement in local waxy corn: Improvement of sweet taste in local waxy corn variety with mixed kernels from super sweet corn. Acta Hortic. 2008, 769, 145–150. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTL | Chr. | Flanking Region (Mb) | Significant SNP | Position (v.5) | −log10(P) | MAF | Candidate Gene |
|---|---|---|---|---|---|---|---|
| qWx9 | 9 | 25.06–25.18 | AX-90613979 | 25,128,753 | 6.80 | 0.333 | Zm00001eb378140 (Wx1 or GBSSI) |
| Marker | Allele | Unextended Primer (UEP) Sequence (5′-3′) a | UEP Mass (Da) | Call 1 | Mass 1 (Da) | Call 2 | Mass 2 (Da) |
|---|---|---|---|---|---|---|---|
| Wx-MassArray (Wx_AX-90613979) | C/G | atctAAGGACGACTTGAATCTCTC | 7311.8 | C | 7559 | G | 7599 |
| Marker | R | R2 | Adjusted R2 | Overall Model Test | |||
|---|---|---|---|---|---|---|---|
| F | df1 | df2 | p | ||||
| Wx-MassArray (Wx1_AX-90613979) | 0.904 | 0.817 | 0.814 | 304 | 2 | 136 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruanjaichon, V.; Yin, K.K.; Thunnom, B.; Khammona, K.; Suriharn, K.; Simla, S.; Kerdsri, C.; Aesomnuk, W.; Yongsuwan, A.; Chaomueang, N.; et al. Genome-Wide Association Study (GWAS) Reveals an SNP Associated with Waxy Trait and Development of a Functional Marker for Predicting Waxy Maize (Zea mays L. var. ceratina). Agronomy 2022, 12, 2289. https://doi.org/10.3390/agronomy12102289
Ruanjaichon V, Yin KK, Thunnom B, Khammona K, Suriharn K, Simla S, Kerdsri C, Aesomnuk W, Yongsuwan A, Chaomueang N, et al. Genome-Wide Association Study (GWAS) Reveals an SNP Associated with Waxy Trait and Development of a Functional Marker for Predicting Waxy Maize (Zea mays L. var. ceratina). Agronomy. 2022; 12(10):2289. https://doi.org/10.3390/agronomy12102289
Chicago/Turabian StyleRuanjaichon, Vinitchan, Khin Kyawt Yin, Burin Thunnom, Kanogporn Khammona, Khundej Suriharn, Sakunkan Simla, Chalong Kerdsri, Wanchana Aesomnuk, Arweewut Yongsuwan, Naraporn Chaomueang, and et al. 2022. "Genome-Wide Association Study (GWAS) Reveals an SNP Associated with Waxy Trait and Development of a Functional Marker for Predicting Waxy Maize (Zea mays L. var. ceratina)" Agronomy 12, no. 10: 2289. https://doi.org/10.3390/agronomy12102289
APA StyleRuanjaichon, V., Yin, K. K., Thunnom, B., Khammona, K., Suriharn, K., Simla, S., Kerdsri, C., Aesomnuk, W., Yongsuwan, A., Chaomueang, N., Oo, N. N., Unartngam, J., Arikit, S., Wanchana, S., & Toojinda, T. (2022). Genome-Wide Association Study (GWAS) Reveals an SNP Associated with Waxy Trait and Development of a Functional Marker for Predicting Waxy Maize (Zea mays L. var. ceratina). Agronomy, 12(10), 2289. https://doi.org/10.3390/agronomy12102289