
Genome-Wide Association Analyses Reveal Candidate Genes Controlling Harvest Index and Related Agronomic Traits in Brassica napus L.


Abstract
:1. Introduction
2. Materials and Methods
2.1. Material Planting and Field Management
2.2. Phenotype Collection
2.3. DNA Isolation, Sequencing, and Quality Controlling
2.4. Population Structure Analysis
2.5. Genome-Wide Associated Loci
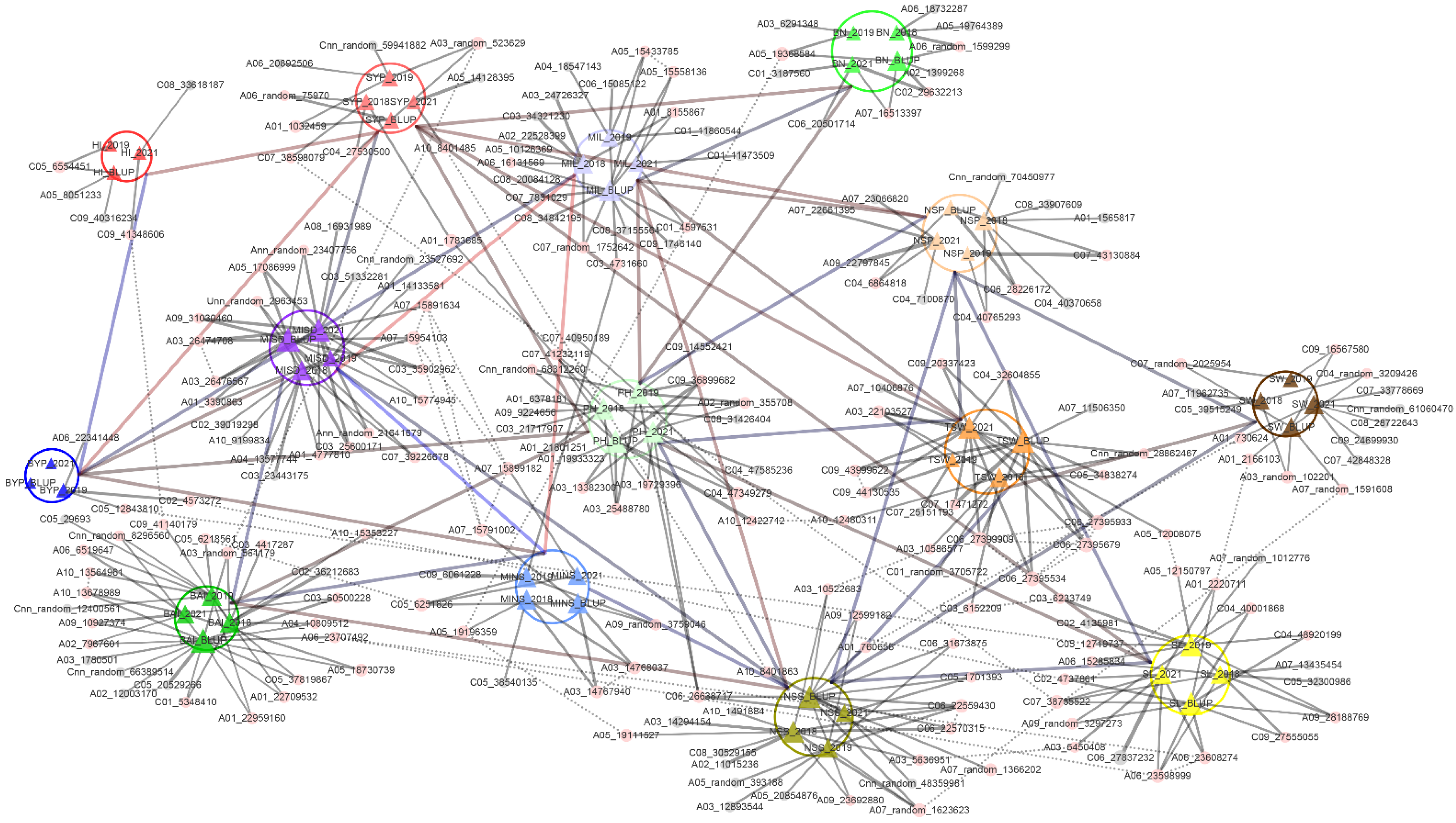
2.6. Network Construction
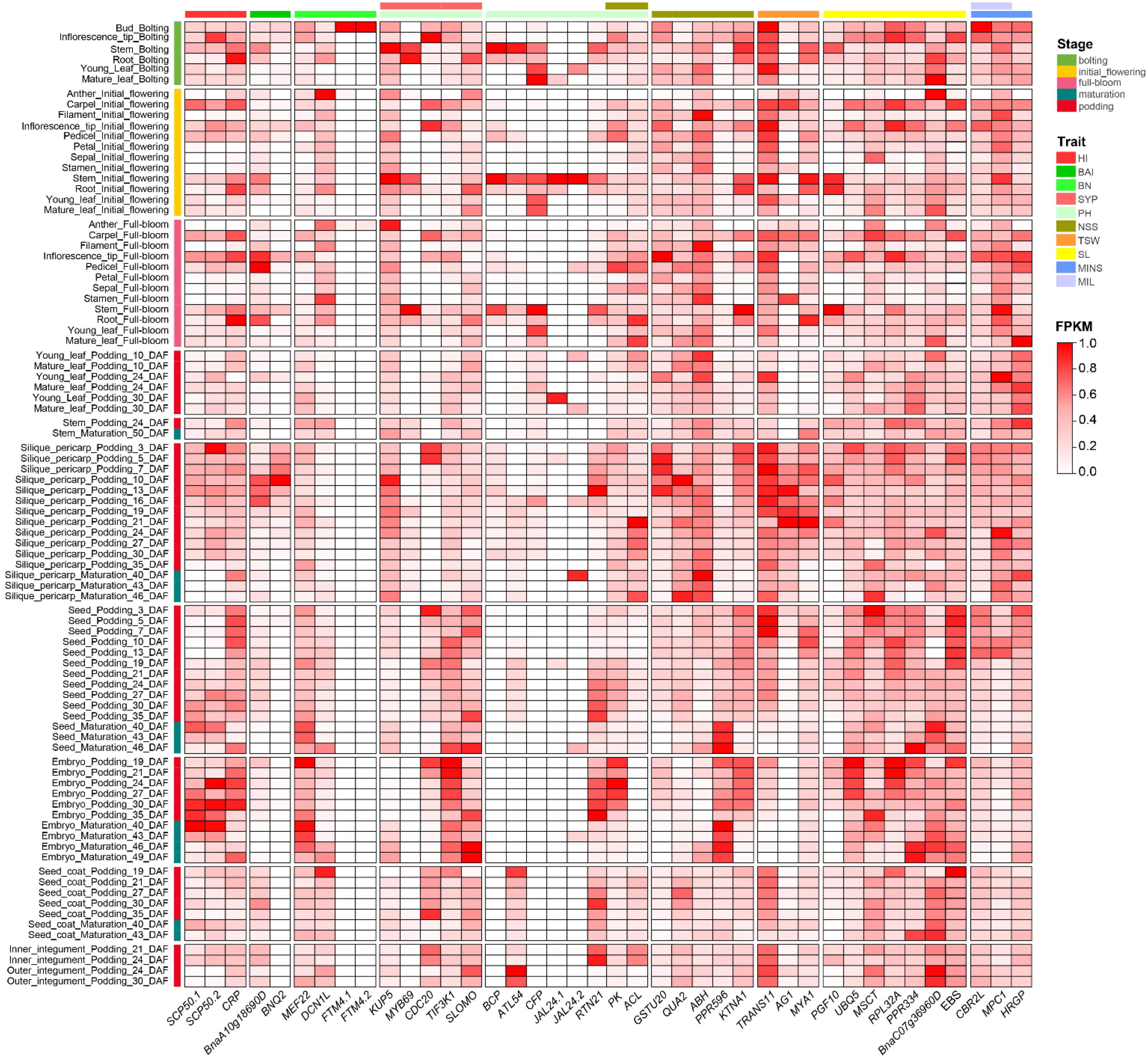
2.7. Candidate Gene Identification
2.8. Data Analyses and Visualization
3. Results
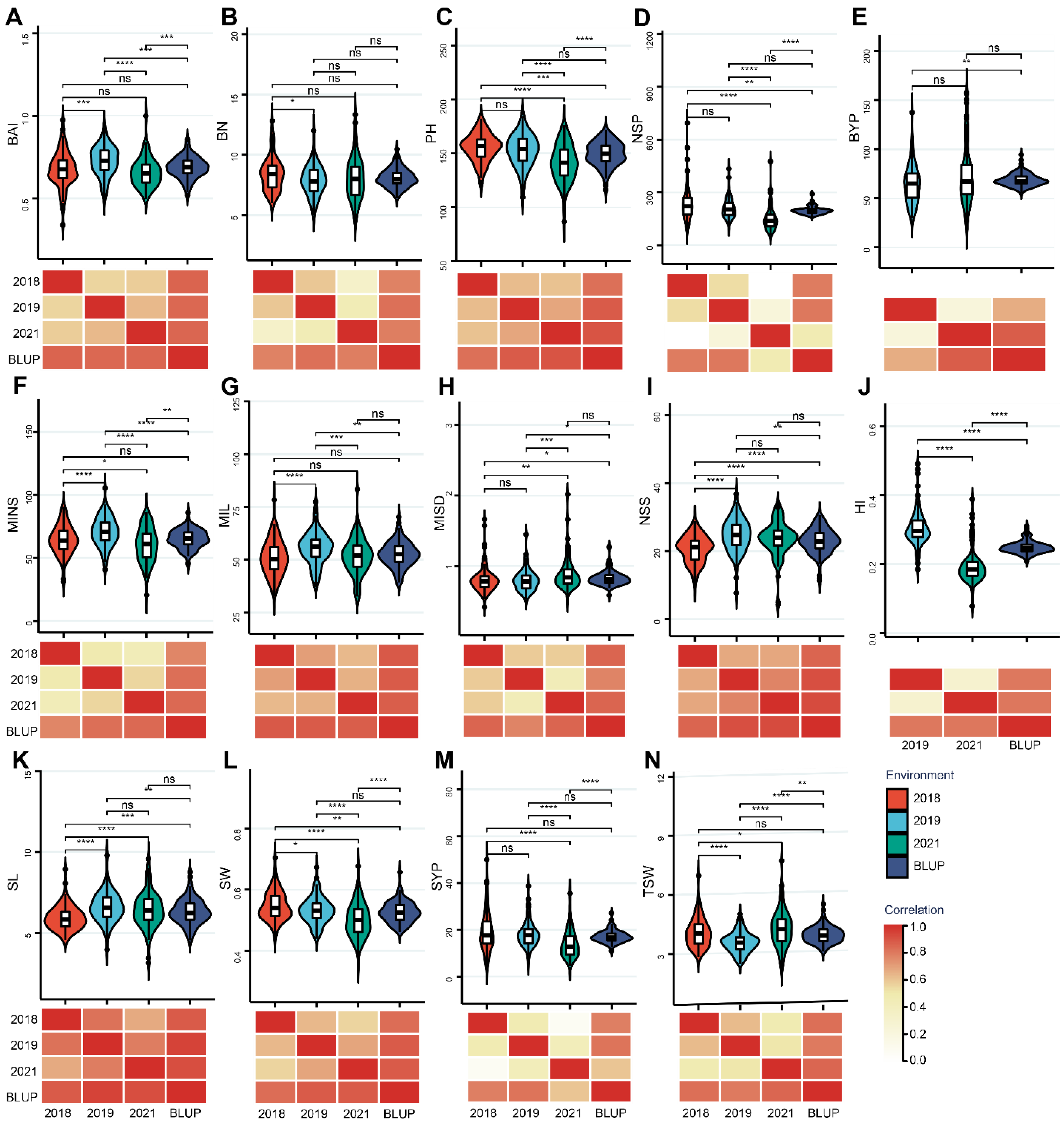
3.1. Phenotypic Variation for Different Agronomy Traits
3.2. Population Structure and Genome-Wide Association Analysis
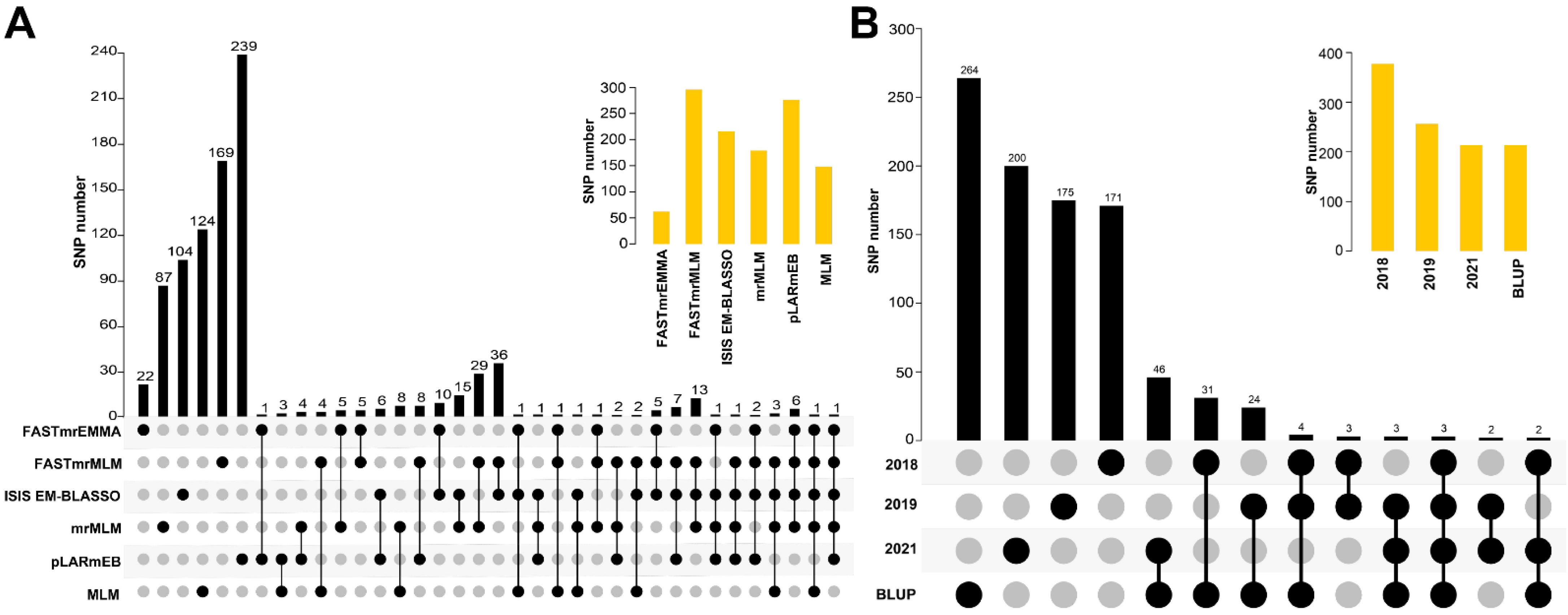
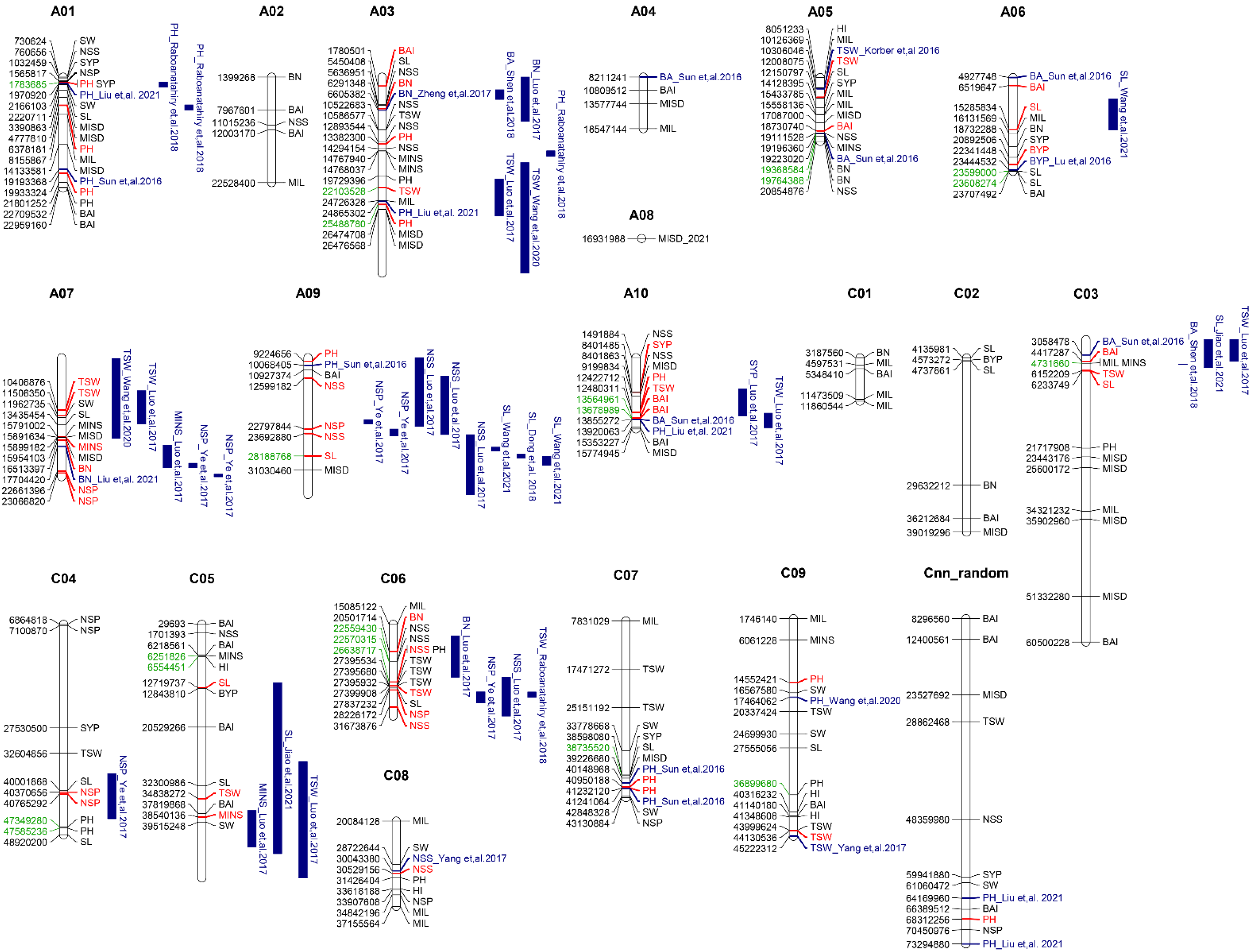
3.3. Comparison of the Stable SNPs
3.4. Network of Significant SNPs Associated with Phenotypes
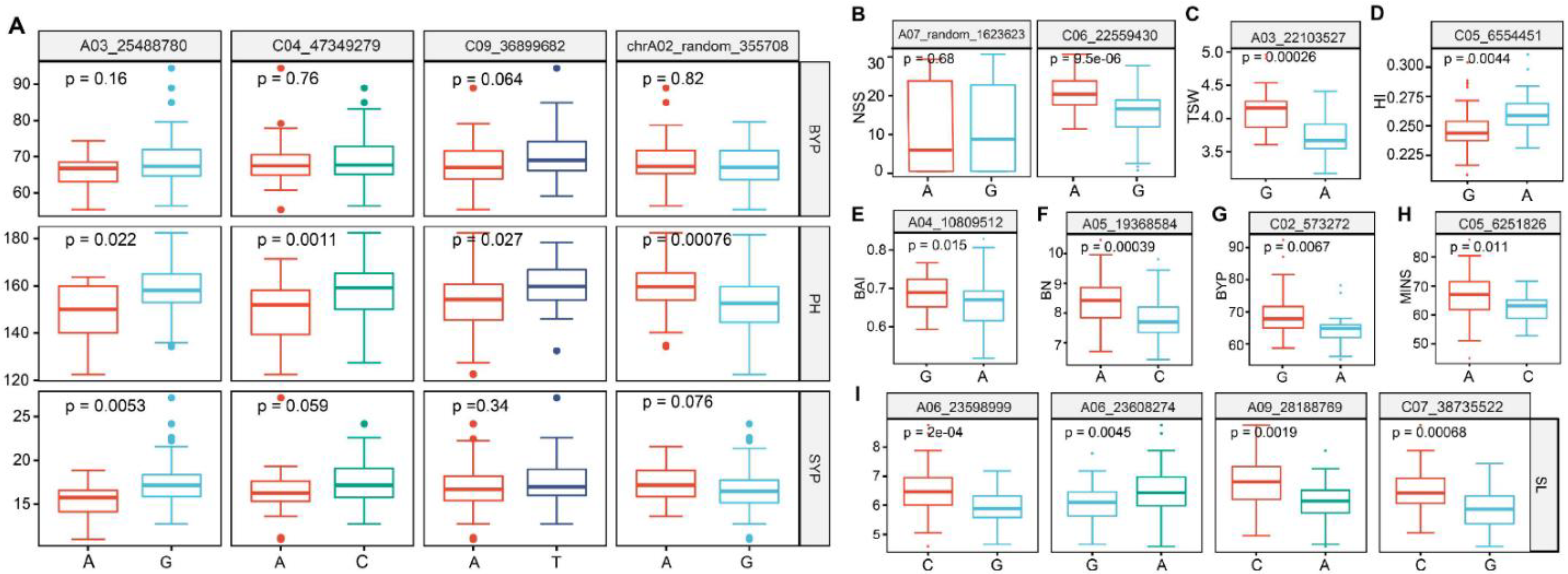
3.5. Identification of Candidate Genes for Stable Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BA | Branch angle |
| BAI | Branch angle index |
| BBA | Basal branch angle |
| BLUP | Best linear unbiased prediction |
| BN | Numbers of primary valid branch |
| BYP | Biomass yield per plant |
| CV | Coefficient of variation |
| GWAS | Genome-wide association study |
| HI | Harvest index |
| LD | Linkage disequilibrium |
| MBA | Middle branch angle |
| MIL | Main inflorescence length |
| MINS | Number of effective siliques on main inflorescence |
| MISD | Silique density of the main inflorescence |
| MLM | Mix linear model |
| NSP | Siliques per plant |
| NSS | Number of seeds per silique |
| PCA | Principal component analysis |
| PH | Plant height |
| QQ plot | Quantile-quantile plot |
| QTL | Quantitative trait locus/loci |
| SL | Silique length |
| SNP | Single nucleotide polymorphism |
| SW | Silique width |
| SYP | Seed yield per plant |
| TBA | Top branch angle |
| TSW | Thousand-seed weight |
References
- Richards, R.A. Selectable traits to increase crop photosynthesis and yield of grain crops. J. Exp. Bot. 2000, 51, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Raboanatahiry, N.; Wang, X.; Zhao, W.; Chen, L.; Guo, L.; Li, B.; Hou, D.; Pu, S.; Zhang, L.; et al. Genetic dissection of harvest index and related traits through genome-wide quantitative trait locus mapping in Brassica napus L. Breed Sci. 2019, 69, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, R.K.M. Harvest index: A review of its use in plant breeding and crop physiology. Ann. Appl. Biol. 1995, 126, 197–216. [Google Scholar] [CrossRef]
- Xie, G.; Han, D.; Wang, X.; Xue, S. Harvest index and residue factor of non-cereal crops in China. J. China Agric. Univ. 2011, 16, 9–17. [Google Scholar]
- Xie, G.; Han, D.; Wang, X.; Lv, R. Harvest index and residue factor of cereal crops in China. J. China Agric. Univ. 2011, 16, 1–8. [Google Scholar]
- Evenson, R.E.; Gollin, D. Assessing the impact of the green revolution, 1960 to 2000. Science 2003, 300, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Shen, G.-Z.; Liang, Y.; Fu, M.-L.; He, B.; Tie, L.-M.; Zhang, Y.; Peng, L.; Li, J.-N. Analysis of Yield Components with High Harvest Index in Brassica napus under Environments Fitting Different Yield Levels. Acta Agron. Sin. 2017, 43, 82–96. [Google Scholar] [CrossRef]
- Pal, L.; Sandhu, S.K.; Bhatia, D.; Sethi, S. Genome-wide association study for candidate genes controlling seed yield and its components in rapeseed (Brassica napus subsp. napus). Physiol. Mol. Biol. Plants 2021, 27, 1933–1951. [Google Scholar] [CrossRef]
- Hu, D.; Jing, J.; Snowdon, R.J.; Mason, A.S.; Shen, J.; Meng, J.; Zou, J. Exploring the gene pool of Brassica napus by genomics-based approaches. Plant Biotechnol. J. 2021, 19, 1693–1712. [Google Scholar] [CrossRef]
- Sun, C.; Wang, B.; Wang, X.; Hu, K.; Li, K.; Li, Z.; Li, S.; Yan, L.; Guan, C.; Zhang, J.; et al. Genome-Wide Association Study Dissecting the Genetic Architecture Underlying the Branch Angle Trait in Rapeseed (Brassica napus L.). Sci. Rep. 2016, 6, 33673. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Ma, C.; Yue, Y.; Hu, K.; Li, Y.; Duan, Z.; Wu, M.; Tu, J.; Shen, J.; Yi, B.; et al. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genom. 2015, 16, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Xiao, Z.; Jian, H.; Peng, L.; Qu, C.; Fu, M.; He, B.; Tie, L.; Liang, Y.; Xu, X.; et al. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in Brassica napus. Sci. Rep. 2016, 6, 36452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Wang, M.; Long, Y.; Huang, Y.; Shi, L.; Zhang, C.; Liu, X.; Fitt, B.D.L.; Xiang, J.; Mason, A.S.; et al. Incorporating pleiotropic quantitative trait loci in dissection of complex traits: Seed yield in rapeseed as an example. Theor. Appl. Genet. 2017, 130, 1569–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Wang, H.; Song, N.; Yu, Z.; Imran, K.; Xie, W.; Qiu, S.; Zhou, F.; Wen, J.; Dai, C.; et al. The Bnapus50K array: A quick and versatile genotyping tool for Brassica napus genomic breeding and research. G3 2021, 11, jkab241. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Han, D.; Mason, A.S.; Zhou, C.; Zheng, W.; Li, Y.; Wu, C.; Fu, D.; Huang, Y. Earliness traits in rapeseed (Brassica napus): SNP loci and candidate genes identified by genome-wide association analysis. DNA Res. 2018, 25, 229–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanzada, H.; Wassan, G.M.; He, H.; Mason, A.S.; Keerio, A.A.; Khanzada, S.; Faheem, M.; Solangi, A.M.; Zhou, Q.; Fu, D.; et al. Differentially evolved drought stress indices determine the genetic variation of Brassica napus at seedling traits by genome-wide association mapping. J. Adv. Res. 2020, 24, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Li, T.; Luo, C.; Huang, H.; Ruan, Y.; Li, X.; Niu, Y.; Fan, Y.; Sun, W.; Zhang, K.; et al. BrassicaEDB: A Gene Expression Database for Brassica Crops. Int. J. Mol. Sci. 2020, 21, 5831. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wang, J. Partial correlation coefficient. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 1634–1635. [Google Scholar]
- Knapp, S.J. Confidence intervals for heritability for two-factor mating design single environment linear models. Theor. Appl. Genet. 1986, 72, 587–591. [Google Scholar] [CrossRef]
- Raboanatahiry, N.; Chao, H.; Dalin, H.; Pu, S.; Yan, W.; Yu, L.; Wang, B.; Li, M. QTL Alignment for Seed Yield and Yield Related Traits in Brassica napus. Front. Plant Sci. 2018, 9, 1127. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, J.; Zhang, B.; Yang, X.; Hammond, J.P.; Ding, G.; Wang, S.; Cai, H.; Wang, C.; Xu, F.; et al. Genome-wide association study dissects the genetic control of plant height and branch number in response to low-phosphorus stress in Brassica napus. Ann. Bot. 2021, 128, 919–930. [Google Scholar] [CrossRef]
- Liu, W.; Hou, P.; Liu, G.; Yang, Y.; Guo, X.; Ming, B.; Xie, R.; Wang, K.; Liu, Y.; Li, S. Contribution of total dry matter and harvest index to maize grain yield—A multisource data analysis. Food Energy Secur. 2020, 9, e256. [Google Scholar] [CrossRef]
- Biabani, A.; Foroughi, A.; Karizaki, A.R.; Rassam, G.A.; Hashemi, M.; Afshar, R.K. Physiological traits, yield, and yield components relationship in winter and spring canola. J. Sci. Food Agric. 2021, 101, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-Genome Resequencing of a Worldwide Collection of Rapeseed Accessions Reveals the Genetic Basis of Ecotype Divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Tan, C.; Li, Y.; He, Y.; Wei, S.; Cui, Y.; Chen, Y.; Wei, D.; Fu, Y.; He, Y.; et al. Genome-Wide Association Study Reveals Both Overlapping and Independent Genetic Loci to Control Seed Weight and Silique Length in Brassica napus. Front. Plant Sci. 2018, 9, 921. [Google Scholar] [CrossRef]
- Zhang, Q. Strategies for developing Green Super Rice. Proc. Natl. Acad. Sci. USA 2007, 104, 16402–16409. [Google Scholar] [CrossRef] [Green Version]
- Körber, N.; Bus, A.; Li, J.; Parkin, I.A.P.; Wittkop, B.; Snowdon, R.J.; Stich, B. Agronomic and Seed Quality Traits Dissected by Genome-Wide Association Mapping in Brassica napus. Front. Plant Sci. 2016, 7, 386. [Google Scholar] [CrossRef]
- Sun, C.; Wang, B.; Yan, L.; Hu, K.; Liu, S.; Zhou, Y.; Guan, C.; Zhang, Z.; Li, J.; Zhang, J.; et al. Genome-Wide Association Study Provides Insight into the Genetic Control of Plant Height in Rapeseed (Brassica napus L.). Front. Plant Sci. 2016, 7, 1102. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shen, Y.; Li, S.; Ge, X.; Li, Z. High Density Linkage Map Construction and QTL Detection for Three Silique-Related Traits in Orychophragmus violaceus Derived Brassica napus Population. Front. Plant Sci. 2017, 8, 1512. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Yang, Y.; Chen, B.; Shi, J.; Luo, M.; Zhan, J.; Wang, X.; Liu, G.; Wang, H. An integrated analysis of QTL mapping and RNA sequencing provides further insights and promising candidates for pod number variation in rapeseed (Brassica napus L.). BMC Genom. 2017, 18, 71. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Xiang, Y.; Xu, E.; Ge, X.; Li, Z. Major Co-localized QTL for Plant Height, Branch Initiation Height, Stem Diameter, and Flowering Time in an Alien Introgression Derived Brassica napus DH Population. Front. Plant Sci. 2018, 9, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Xu, E.; Xiang, Y.; Li, Z.; Yang, Y.; Ge, X. Novel and major QTL for branch angle detected by using DH population from an exotic introgression in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2017, 131, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, M.; Xiong, M.; Wang, P.; Liu, Y.; Xin, Q.; Wan, L.; Yang, G.; Hong, D. Genetic dissection of thousand-seed weight and fine mapping of cqSW.A03-2 via linkage and association analysis in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2020, 133, 1321–1335. [Google Scholar] [CrossRef]
- Basu, U.; Parida, S.K. Restructuring plant types for developing tailor-made crops. Plant Biotechnol. J. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Tsutsui, H.; Shiina, K.; Sprunck, S.; Takeuchi, H.; Yui, R.; Kasahara, R.D.; Hamamura, Y.; Mizukami, A.; Susaki, D.; et al. Defensin-like polypeptide LUREs are pollen tube attractants secreted from synergid cells. Nature 2009, 458, 357–361. [Google Scholar] [CrossRef]
- Marshall, E.; Costa, L.M.; Gutierrez-Marcos, J. Cysteine-rich peptides (CRPs) mediate diverse aspects of cell-cell communication in plant reproduction and development. J. Exp. Bot. 2011, 62, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Aalen, R.B. Maturing peptides open for communication. J. Exp. Bot. 2013, 64, 5231–5235. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, D.; Stacey, N.; Breuninger, H.; Jikumaru, Y.; Muller, D.; Sicard, A.; Leyser, O.; Yamaguchi, S.; Lenhard, M. Slow Motion is required for within-plant auxin homeostasis and normal timing of lateral organ initiation at the shoot meristem in Arabidopsis. Plant Cell 2010, 22, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Panteris, E.; Kouskouveli, A.; Pappas, D.; Adamakis, I.-D. Cytokinesis in fra2 Arabidopsis thaliana p60-katanin Mutant: Defects in Cell Plate/Daughter Wall Formation. Int. J. Mol. Sci. 2021, 22, 1405. [Google Scholar] [CrossRef]
- Stiefel, V.; Ruiz-Avila, L.; Raz, R.; Pilar Valles, M.; Gomez, J.; Pages, M.; Martinez-Izquierdo, J.A.; Ludevid, M.D.; Langdale, J.A.; Nelson, T.; et al. Expression of a maize cell wall hydroxyproline-rich glycoprotein gene in early leaf and root vascular differentiation. Plant Cell 1990, 2, 785–793. [Google Scholar]
- Wang, Q.; Sajja, U.; Rosloski, S.; Humphrey, T.; Kim, M.C.; Bomblies, K.; Weigel, D.; Grbic, V. HUA2 Caused Natural Variation in Shoot Morphology of A. thaliana. Curr. Biol. 2007, 17, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.; Fornara, F.; Vincent, C.; Andres, F.; Nordstrom, K.; Gobel, U.; Knoll, D.; Schoof, H.; Coupland, G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell 2012, 24, 444–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mara, C.D.; Huang, T.; Irish, V.F. The Arabidopsis floral homeotic proteins APETALA3 and PISTILLATA negatively regulate the BANQUO genes implicated in light signaling. Plant Cell 2010, 22, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, K.; Lee, I.; Kim, E.; Park, S.K.; Soh, M.-S.; Lee, S. Paclobutrazol-resistance Gene Family Regulates Floral Organ Growth with Unequal Genetic Redundancy in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Qian, S.; Scheid, R.N.; Lu, L.; Chen, X.; Liu, R.; Du, X.; Lv, X.; Boersma, M.D.; Scalf, M.; et al. EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat. Genet. 2018, 50, 1247–1253. [Google Scholar] [CrossRef]
- Lionetti, V.; Fabri, E.; De Caroli, M.; Hansen, A.R.; Willats, W.G.; Piro, G.; Bellincampi, D. Three Pectin Methylesterase Inhibitors Protect Cell Wall Integrity for Arabidopsis Immunity to Botrytis. Plant Physiol 2017, 173, 1844–1863. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kuroyama, H.; Takeuchi, Y.; Tsumuraya, Y. Characterization of pectin ethyltransferase from soybean hypocotyls. Planta 2000, 210, 782–791. [Google Scholar] [CrossRef]
- Mouille, G.; Ralet, M.C.; Cavelier, C.; Eland, C.; Effroy, D.; Hematy, K.; McCartney, L.; Truong, H.N.; Gaudon, V.; Thibault, J.F.; et al. Homogalacturonan synthesis in Arabidopsis thaliana requires a Golgi-localized protein with a putative methyltransferase domain. Plant J. 2007, 50, 605–614. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Id | SNP | Trait | Trait & SNP | Gene Name | Protein Name |
|---|---|---|---|---|---|
| BnaA01g03380D | A01_1783685 | PH; SYP | PH-SYP.A01_1783685 | KUP5 | K+ uptake permease 5 |
| BnaA01g03490D | A01_1783685 | PH; SYP | PH-SYP.A01_1783685 | MYB69 | MYB domain protein 69 |
| BnaA01g03590D | A01_1783685 | PH; SYP | PH-SYP.A01_1783685 | CDC20 | Cell division cycle 20 |
| BnaA01g03600D | A01_1783685 | PH; SYP | PH-SYP.A01_1783685 | TIF3K1 | Eukaryotic translation initiation factor |
| BnaA01g03630D | A01_1783685 | PH; SYP | PH-SYP.A01_1783685 | SLOMO | Slow motion |
| BnaA02g35610D | A02_random_355708 | PH | PH.A02_random_355708 | BCP | Blue copper protein |
| BnaA02g35620D | A02_random_355708 | PH | PH.A02_random_355708 | ATL54 | RING-H2 finger protein ATL54 |
| BnaA03g43740D | A03_22103527 | TSW | TSW.A03_22103527 | TRANS11 | Translocase 11 |
| BnaA03g43820D | A03_22103527 | TSW | TSW.A03_22103527 | AG1 | Floral homeotic protein AGAMOUS |
| BnaA03g43840D | A03_22103527 | TSW | TSW.A03_22103527 | MYA1 | MYOSIN 1 |
| BnaA03g49330D | A03_25488780 | PH | PH.A03_25488780 | CFP | Cotton fiber protein |
| BnaA05g26410D | A05_19368584; A05_19764389 | BN | BN.A05_19368584; BN.A05_19764389 | DCN1L | Defective in cullin neddylation protein |
| BnaA05g26840D | A05_19368584; A05_19764389 | BN | BN.A05_19368584; BN.A05_19764389 | FTM4.1 | Leucine-rich repeat protein |
| BnaA05g26860D | A05_19368584; A05_19764389 | BN | BN.A05_19368584; BN.A05_19764389 | FTM4.2 | Leucine-rich repeat protein |
| BnaA06g35950D | A06_23598999; A06_23608274 | SL | SL.A06_23598999; SL.A06_23608274 | MSCT | Man1-Src1p-carboxy-terminal domain protein |
| BnaA06g35970D | A06_23598999; A06_23608274 | SL | SL.A06_23598999; SL.A06_23608274 | RPL32A | 60S ribosomal protein L32-1 |
| BnaA07g38220D | A07_random_1623623 | NSS | NSS.A07_random_1623623 | ABH | alpha/beta-Hydrolases superfamily protein |
| BnaA07g38350D | A07_random_1623623 | NSS | NSS.A07_random_1623623 | PPR596 | Pentatricopeptide repeat 596 |
| BnaA07g38370D | A07_random_1623623 | NSS | NSS.A07_random_1623623 | KTNA1 | Katanin p60 ATPase-containing subunit A1 |
| BnaA09g39690D | A09_28188769 | SL | SL.A09_28188769 | PGF10 | Polygalaturonase clade F 10 |
| BnaA09g39760D | A09_28188769 | SL | SL.A09_28188769 | UBQ5 | Ubiquitin 5 |
| BnaA10g18690D | A10_13564961; A10_13678989 | BAI | BAI.A10_13564961; BAI.A10_13678989 | BnaA10g18690D | Hypothetical protein |
| BnaA10g18940D | A10_13564961; A10_13678989 | BAI | BAI.A10_13564961; BAI.A10_13678989 | BNQ2 | Banquo 2 |
| BnaC03g09950D | C03_4731660 | MIL; MINS | MIL-MINS.C03_4731660 | CBR2L | NADH-cytochrome b5 reductase-like protein |
| BnaC03g09960D | C03_4731660 | MIL; MINS | MIL-MINS.C03_4731660 | MPC1 | Mitochondrial pyruvate carrier 1 |
| BnaC04g49260D | C04_47349279; C04_47585236 | PH | PH.C04_47349279; PH.C04_47585236 | JAL24.1 | Jacalin-related lectin 24 |
| BnaC04g49270D | C04_47349279; C04_47585236 | PH | PH.C04_47349279; PH.C04_47585236 | JAL24.2 | Jacalin-related lectin 24 |
| BnaC05g10890D | C05_6251826 | MINS | MINS.C05_6251826 | HRGP | Hydroxyproline-rich glycoprotein family protein |
| BnaC05g11300D | C05_6554451 | HI | HI.C05_6554451 | SCP50.1 | Serine carboxypeptidase-like 50 |
| BnaC05g11310D | C05_6554451 | HI | HI.C05_6554451 | SCP50.2 | Serine carboxypeptidase-like 50 |
| BnaC05g11350D | C05_6554451 | HI | HI.C05_6554451 | CRP | Cysteine-rich peptide family protein |
| BnaC06g20430D | C06_22559430; C06_22570315 | NSS | NSS.C06_22559430; NSS.C06_22570315 | GSTU20 | Glutathione S-transferase U20 |
| BnaC06g20510D | C06_22559430; C06_22570315 | NSS | NSS.C06_22559430; NSS.C06_22570315 | QUA2 | Pectin methyltransferase QUA2 |
| BnaC06g25100D | C06_26638717 | PH; NSS | PH-NSS.C06_26638717 | PK | Protein kinase superfamily protein |
| BnaC06g25110D | C06_26638717 | PH; NSS | PH-NSS.C06_26638717 | ACL | Actin cross-linking protein |
| BnaC07g36830D | C07_38735522 | SL | SL.C07_38735522 | PPR334 | Pentatricopeptide repeat 334 |
| BnaC07g36960D | C07_38735522 | SL | SL.C07_38735522 | BnaC07g36960D | Hypothetical protein |
| BnaC07g37120D | C07_38735522 | SL | SL.C07_38735522 | EBS | Early Bolting in Short Days |
| BnaC09g33520D | C09_36899682 | PH | PH.C09_36899682 | RTN21 | Reticulon protein 21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, M.; Song, J.; Guo, N.; Zhang, M.; Zhu, Y.; Huang, Z.; Xu, A. Genome-Wide Association Analyses Reveal Candidate Genes Controlling Harvest Index and Related Agronomic Traits in Brassica napus L. Agronomy 2022, 12, 814. https://doi.org/10.3390/agronomy12040814
Qin M, Song J, Guo N, Zhang M, Zhu Y, Huang Z, Xu A. Genome-Wide Association Analyses Reveal Candidate Genes Controlling Harvest Index and Related Agronomic Traits in Brassica napus L. Agronomy. 2022; 12(4):814. https://doi.org/10.3390/agronomy12040814
Chicago/Turabian StyleQin, Mengfan, Jia Song, Na Guo, Miao Zhang, Yunlin Zhu, Zhen Huang, and Aixia Xu. 2022. "Genome-Wide Association Analyses Reveal Candidate Genes Controlling Harvest Index and Related Agronomic Traits in Brassica napus L." Agronomy 12, no. 4: 814. https://doi.org/10.3390/agronomy12040814
APA StyleQin, M., Song, J., Guo, N., Zhang, M., Zhu, Y., Huang, Z., & Xu, A. (2022). Genome-Wide Association Analyses Reveal Candidate Genes Controlling Harvest Index and Related Agronomic Traits in Brassica napus L. Agronomy, 12(4), 814. https://doi.org/10.3390/agronomy12040814