
Integrating Genome-Wide Association Study with Transcriptomic Analysis to Predict Candidate Genes Controlling Storage Root Flesh Color in Sweet Potato


Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Plant Material and Storage Root Phenotyping
2.2. Read Alignment and Variation Calling
2.3. GWAS Analysis
2.4. RNA-Seq Analysis
2.5. qRT-PCR
2.6. Statistical Analysis
3. Results
3.1. SLAF-Sequencing of 300 Sweet Potato Accessions
3.2. Population Structure and Genomic Variation among the 300 Sweet Potato Accessions
3.3. GWAS for Identifying Significant SNPs Associated with SRFC
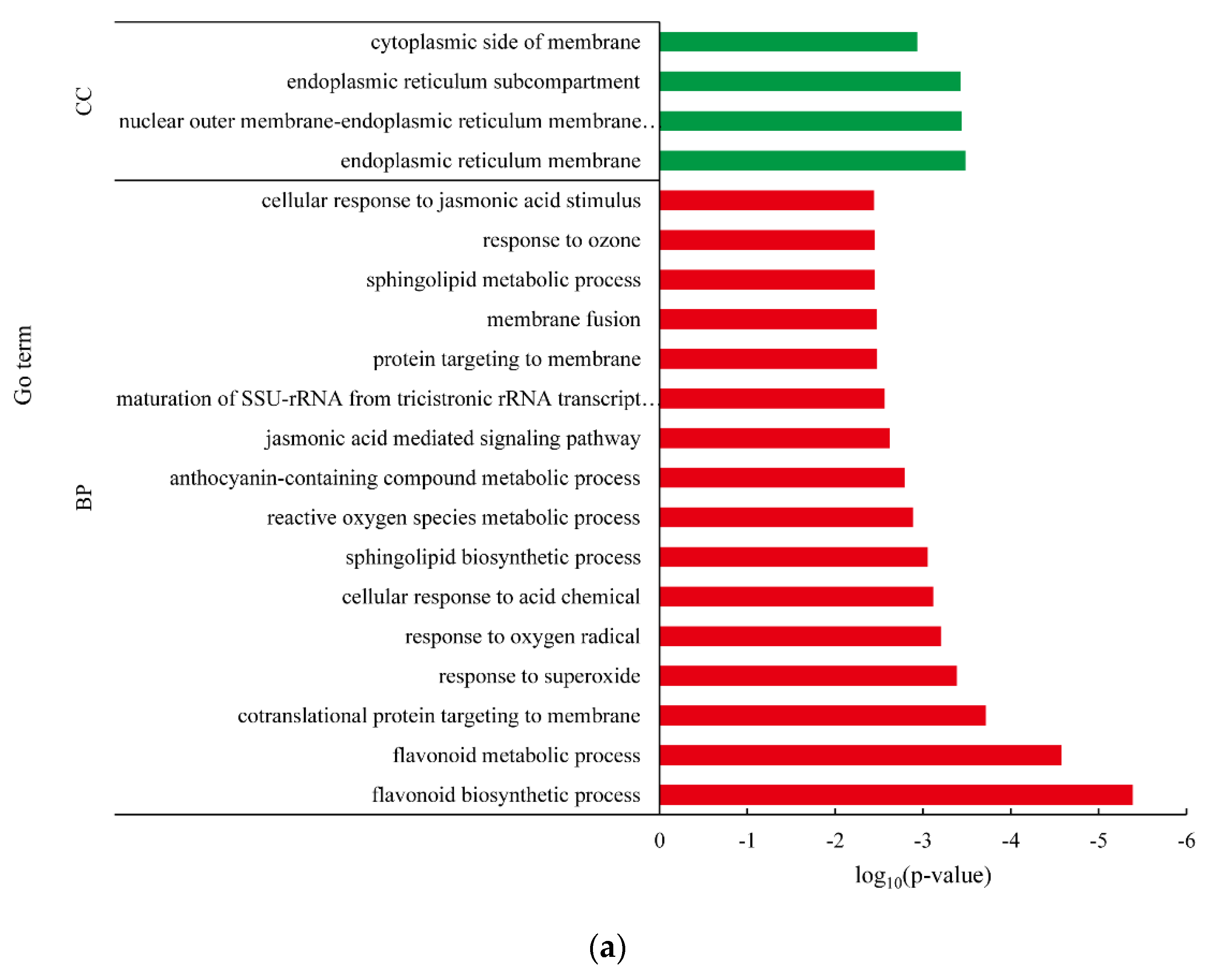
3.4. Differentially Expressed Gene between White-Fleshed and Purple-Fleshed Sweet Potato
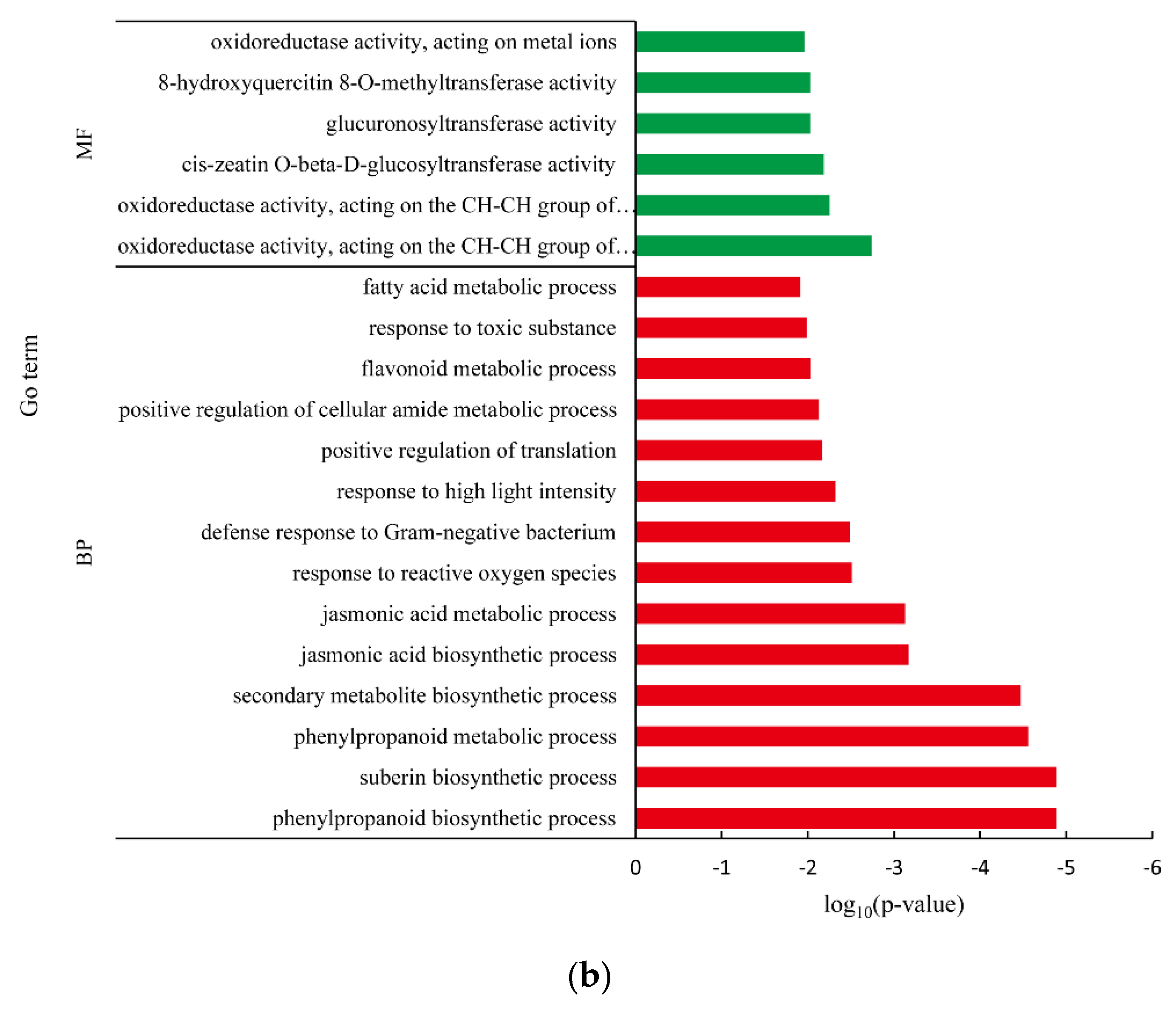
3.5. Differentially Expressed Gene between White Fleshed and Yellow-Fleshed Sweet Potato
3.6. Exploring Candidate Genes for SRFC by RNA-Seq
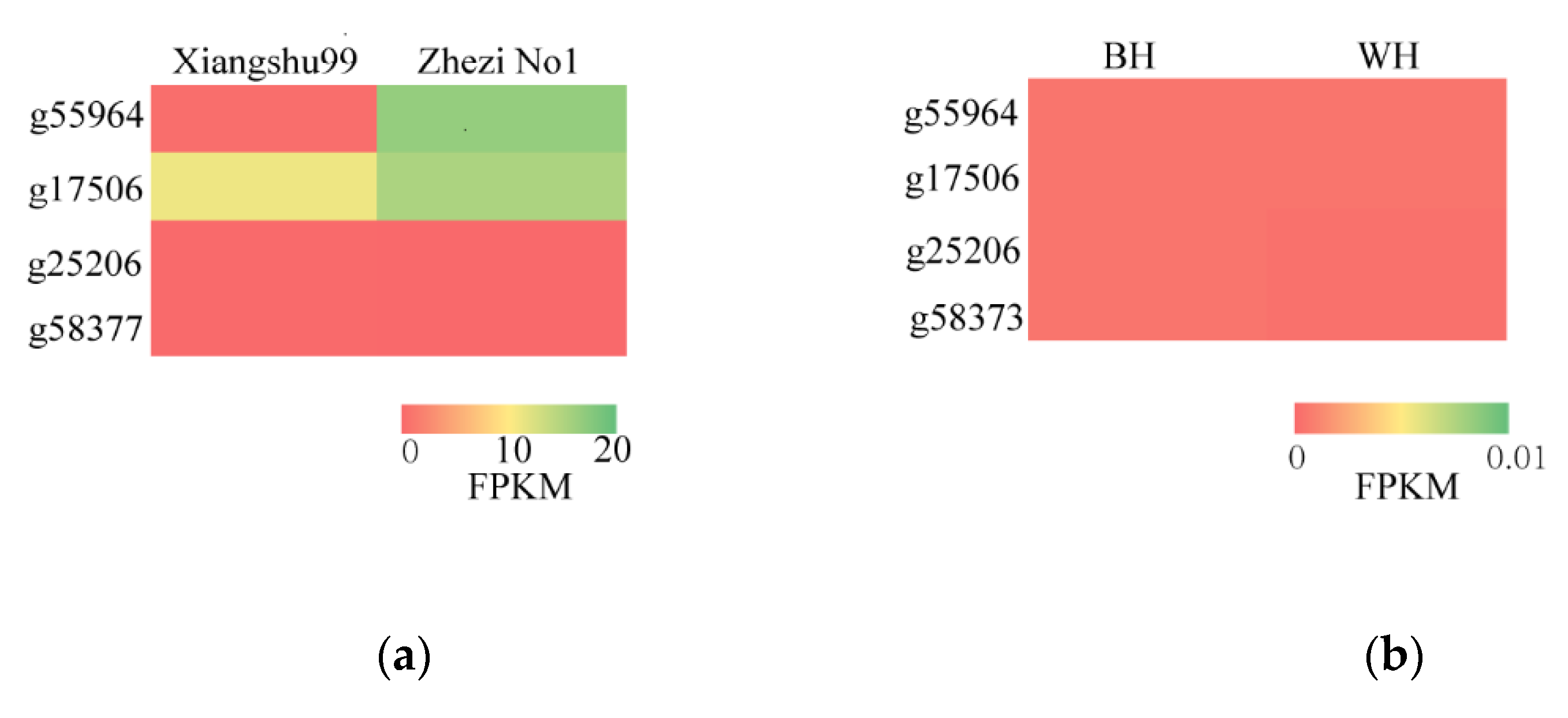
3.7. Candidate Genes Expression Analysis
4. Discussion
4.1. Genetic Variation among the 300 Accessions
4.2. GWAS Analysis of SRFC in Sweet Potato
4.3. Functional Analysis of DEGs in Different SRFC Varieties
4.4. Quantitative Trait Loci Analysis of Flesh Color in Sweet Potato
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Moeinzadeh, M.H.; Kuhl, H.; Helmuth, J.; Xiao, P.; Haas, S.; Liu, G.; Zheng, J.; Sun, Z.; Fan, W.; et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 2017, 3, 696–703. [Google Scholar] [CrossRef]
- Leighton, C.S.; Schönfeldt, H.; Kruger, R. Quantitative descriptive sensory analysis of five different cultivars of sweet potato to determine sensory and textural profiles. J. Sens. Stud. 2010, 25, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, K.; Rwiza, E.; Nyango, A.; Amour, R.; Ngendello, T.; Kapinga, R.; Rees, D.; Jolliffe, F. The use of sensory evaluation and consumer preference for the selection of sweetpotato cultivars in East Africa. J. Sci. Food Agric. 2004, 84, 791–799. [Google Scholar] [CrossRef]
- Leksrisompong, P.P.; Whitson, M.E.; Truong, V.D.; Drake, M.A. Sensory attributes and consumer acceptance of sweet potato cultivars with varying flesh colors. J. Sens. Stud. 2012, 27, 59–69. [Google Scholar] [CrossRef]
- Lado, J.; Vicente, E.; Moltini, A.I.; Alcaire, F.; Ares, G. Integrating consumer perception in the selection of purple skin sweet potato cultivars. J. Sci. Food Agric. 2021, 101, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, D.; Wang, Y.; Zhang, L.; Chen, X.; Yang, X.; Xiong, H.; Bhattarai, G.; Ravelombola, W.; Olaoye, D.; et al. Transcript profiling for regulation of sweet potato skin color in Sushu8 and its mutant Zhengshu20. Plant Physiol. Biochem. 2020, 148, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Teow, C.C.; Truong, V.D.; McFeeters, R.F.; Thompson, R.L.; Pecota, K.V.; Yencho, G.C. Antioxidant activities, phenolic and β-carotene contents of sweet potato genotypes with varying flesh colours. Food Chem. 2007, 103, 829–838. [Google Scholar] [CrossRef]
- Azeem, M.; Mu, T.H.; Zhang, M. Effects of high hydrostatic pressure and soaking solution on proximate composition, polyphenols, anthocyanins, β-carotene, and antioxidant activity of white, orange, and purple fleshed sweet potato flour. Food Sci. Technol. Int. 2020, 26, 388–402. [Google Scholar] [CrossRef]
- Castañeda-Ovando, A.; Pacheco-Hernandez, M.D.L.; Páez-Hernández, M.E.; Rodríguez, J.A.; Galán-Vidal, C.A. Chemical studies of anthocyanins: A review. Food Chem. 2009, 113, 859–871. [Google Scholar] [CrossRef]
- Zhao, J.; Dixon, R.A. The “ins” and “outs” of flavonoid transport. Trends Plant Sci. 2010, 15, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J. Flavonoid transport mechanisms: How to go, and with whom. Trends Plant Sci. 2015, 20, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.Z.; Hu, K.D.; Zhao, D.L.; Tang, J.; Huang, Z.Q.; Jin, P.; Li, Y.H.; Han, Z.; Hu, L.Y.; Yao, G.F.; et al. MYB44 competitively inhibits the formation of the MYB340-bHLH2-NAC56 complex to regulate anthocyanin biosynthesis in purple-fleshed sweet potato. BMC Plant Biol. 2020, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Kim, Y.H.; Kim, S.H.; Jeong, Y.J.; Kim, C.Y.; Lee, J.S.; Bae, J.Y.; Ahn, M.J.; Jeong, J.C.; Lee, H.S.; et al. Overexpression of the IbMYB1 gene in an orange-fleshed sweet potato cultivar produces a dual-pigmented transgenic sweet potato with improved antioxidant activity. Physiol. Plant. 2015, 153, 525–537. [Google Scholar] [CrossRef]
- Ning, Z.; Hu, K.; Zhou, Z.; Zhao, D.; Tang, J.; Wang, H.; Li, L.; Ding, C.; Chen, X.; Yao, G.; et al. IbERF71, with IbMYB340 and IbbHLH2, coregulates anthocyanin accumulation by binding to the IbANS1 promoter in purple-fleshed sweet potato (Ipomoea batatas L.). Plant Cell Rep. 2021, 40, 157–169. [Google Scholar] [CrossRef]
- Rosas-Saavedra, C.; Stange, C. Biosynthesis of Carotenoids in Plants: Enzymes and Color. Subcell Biochem. 2016, 79, 35–69. [Google Scholar]
- Wang, Y.H.; Li, T.; Zhang, R.R.; Khadr, A.; Tian, Y.S.; Xu, Z.S.; Xiong, A.S. Transcript profiling of genes involved in carotenoid biosynthesis among three carrot cultivars with various taproot colors. Protoplasma. 2020, 257, 949–963. [Google Scholar] [CrossRef]
- Jourdan, M.; Gagné, S.; Dubois-Laurent, C.; Maghraoui, M.; Huet, S.; Suel, A.; Hamama, L.; Briard, M.; Peltier, D.; Geoffriau, E. Carotenoid content and root color of cultivated carrot: A candidate-gene association study using an original broad unstructured population. PLoS ONE. 2015, 10, e0116674. [Google Scholar] [CrossRef] [Green Version]
- Drapal, M.; Rossel, G.; Heider, B.; Fraser, P.D. Metabolic diversity in sweet potato (Ipomoea batatas, Lam.) leaves and storage roots. Hortic. Res. 2019, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Park, S.C.; Ji, C.Y.; Kim, H.S.; Lee, H.S.; Kwak, S.S. Metabolic engineering of carotenoids in transgenic sweetpotato. Breed. Sci. 2017, 67, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Park, S.C.; Kim, S.H.; Park, S.; Lee, H.U.; Lee, J.S.; Park, W.S.; Ahn, M.J.; Kim, Y.H.; Jeong, J.C.; Lee, H.S.; et al. Enhanced accumulation of carotenoids in sweetpotato plants overexpressing IbOr-Ins gene in purple-fleshed sweetpotato cultivar. Plant Physiol. Biochem. 2015, 86, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.J.; Yang, Q. The future of genetic studies of complex human diseases. Epidemiology. 1998, 9, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Zhongqu, D.; Jun, Z. Research progress of genome-wide association study. J. Zhejiang Univ. 2015, 41, 385–393. [Google Scholar]
- Pang, Y.; Liu, C.; Wang, D.; St Amand, P.; Bernardo, A.; Li, W.; He, F.; Li, L.; Wang, L.; Yuan, X.; et al. High-Resolution Genome-Wide Association Study Identifies Genomic Regions and Candidate Genes for Important Agronomic Traits in Wheat. Mol. Plant 2020, 13, 1311–1327. [Google Scholar] [CrossRef]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Haijun, L.; Jianbing, Y. Crop genome-wide association study a harvest of biological relevance. Plant J. 2019, 97, 8–18. [Google Scholar]
- Su, W.; Wang, L.; Lei, J.; Chai, S.; Liu, Y.; Yang, Y.; Yang, X.; Jiao, C. Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PLoS ONE. 2017, 12, e0172066. [Google Scholar] [CrossRef]
- Su, W.J.; Zhao, N.; Lei, J.; Wang, L.J.; Chai, S.S.; Yang, X.S. SNP Sites Developed by Specific Length Amplification Fragment Sequencing (SLAF-seq) in Sweetpotato. Sci. Agric. Sin. 2016, 49, 27–34. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one penny imputed genome from next generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tan, Y.; Dong, F.; Zhang, Y.; Huang, Y.; Zhou, Y.; Zhao, Z.J.; Yin, Q.; Xie, X.; Gao, X.; et al. The Expression of IbMYB1 Is Essential to Maintain the Purple Color of Leaf and Storage Root in Sweet Potato [Ipomoea batatas (L.) Lam]. Front. Plant Sci. 2021, 12, 688707. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, K.; Tanaka, M.; Kurata, R.; Kai, Y. Comparative transcriptome analysis implied a ZEP paralog was a key gene involved in carotenoid accumulation in yellow-fleshed sweetpotato. Sci. Rep. 2020, 10, 20607. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg1, S.L. HISAT: A fast spliced aligner with low memory requirements Daehwan HHS Public Access. Nat. Methods. 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 217. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Liu, Q.; Wang, Y.; Zhai, H.; He, X.Q.; Liu, Q.C.; Wang, Y.P.; Zhai, H. Analysis of genetic diversity among sweetpotato landraces in China. Sci. Agric. Sin. 2005, 38, 250–257. [Google Scholar]
- Holton, T.A.; Cornish, E.C. Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell. 1995, 7, 1071–1083. [Google Scholar] [CrossRef]
- Ross, J.; Li, Y.; Lim, E.; Bowles, D.J. Higher plant glycosyltransferases. Genome Biol. 2001, 2, 3004.1–3004.6. [Google Scholar] [CrossRef]
- Ono, E.; Homma, Y.; Horikawa, M.; Kunikane-Doi, S.; Imai, H.; Takahashi, S.; Kawai, Y.; Ishiguro, M.; Fukui, Y.; Nakayama, T. Functional differentiation of the glycosyltransferases that contribute to the chemical diversity of bioactive flavonol glycosides in grapevines (Vitis vinifera). Plant Cell. 2010, 22, 2856–2871. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.; Yuan, X.X.; Murata, J.; De Luca, V. Molecular cloning and biochemical characterization of the UDP-glucose: Flavonoid 3-O-glucosyltransferase from Concord grape (Vitis labrusca). Phytochemistry 2012, 74, 90–99. [Google Scholar] [CrossRef]
- Doukhanina, E.V.; Chen, S.; Van Der Zalm, E.; Godzik, A.; Reed, J.; Dickman, M.B. Identification and functional characterization of the BAG protein family in Arabidopsis thaliana. J. Biol. Chem. 2006, 281, 18793–18801. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Yang, Y.; Zhou, Y.; Zhou, J.; Fan, B.; Yu, J.Q.; Chen, Z. Protein-protein interactions in the regulation of WRKY transcription factors. Mol. Plant 2013, 6, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zhou, Y.; Yang, Y.; Chi, Y.J.; Zhou, J.; Chen, J.Y.; Wang, F.; Fan, B.; Shi, K.; Zhou, Y.H.; et al. Structural and functional analysis of VQ motif-containing proteins in Arabidopsis as interacting proteins of WRKY transcription factors. Plant Physiol. 2012, 159, 810–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluttenhofer, C.; Yuan, L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Kou, M.; Li, C.; Zhang, Y.G. Comparative transcriptome analysis reveals candidate genes involved in anthocyanin biosynthesis in sweetpotato (Ipomoea batatas L.). Plant Physiol. Biochem. 2021, 158, 508–517. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Y.; Shi, T.; Kou, M.; Sun, J.; Xu, T.; Li, Q.; Wu, S.; Cao, Q. Genome-wide analysis of expression quantitative trait loci ( eQTLs ) reveals the regulatory architecture of gene expression variation in the storage roots of sweet potato. Hortic. Res. 2020, 7, 90. [Google Scholar] [CrossRef]
- Gemenet, D.C.; da Silva Pereira, G.; De Boeck, B.; Wood, J.C.; Mollinari, M.; Olukolu, B.A.; Diaz, F.; Mosquera, V.; Ssali, R.T.; David, M.; et al. Quantitative trait loci and differential gene expression analyses reveal the genetic basis for negatively associated β-carotene and starch content in hexaploid sweetpotato [Ipomoea batatas (L.) Lam.]. Theor. Appl. Genet. 2020, 133, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Ma, M.; Ahmad, M.Q.; Arisha, M.H.; Tang, W.; Li, C.; Zhang, Y.; Kou, M.; Wang, X.; Gao, R.; et al. High-Density Single Nucleotide Polymorphisms Genetic Map Construction and Quantitative Trait Locus Mapping of Color-Related Traits of Purple Sweet Potato [Ipomoea batatas (L.) Lam.]. Front. Plant Sci. 2022, 12, 797041. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphological Traits | Criteria for Recording |
|---|---|
| Storage root flesh color, SRFC | 1 = White, 2 = Light yellow, 3 = Yellow, 4 = Orange, 5 = Jacinth ikaika 6 = Light red, 7 = Red, 8 = Fuchsia, 9 = Purple, 10 = Dark purple, 11 = Black purple |
| Marker Name | Candidate Genes | Annotated | p-Value | R2 | LG | Allele | Position |
|---|---|---|---|---|---|---|---|
| LG14: 4,467,066 | g55964 | F3′H | 3.63 × 10−8 | 0.096366 | LG14 | A/G | 4,467,066 |
| LG5: 5,867,009 | g17506 | BAG2-like | 6.31 × 10−8 | 0.093125 | LG5 | A/G | 5,867,009 |
| LG6: 31,267,101 | g25206 | UGT-73D1-like | 6.41 × 10−8 | 0.093027 | LG6 | C/G | 31,267,101 |
| LG6: 31,267,107 | g25206 | UGT-73D1-like | 7.99 × 10−8 | 0.091735 | LG6 | T/C | 31,267,107 |
| LG14: 21,791,164 | g58377 | VQ25-isoform X2 | 9.43 × 10−8 | 0.090762 | LG14 | C/T | 21,791,164 |
| Gene ID | GO |
|---|---|
| g55964 | GO:0016705; GO:0016020; GO:0055114; GO:0016709; GO:0004497; GO:0009733; GO:0042221; GO:0019748; GO:0016491; GO:0050896; GO:0044550; GO:0008150; GO:0008152; GO:0009725; GO:0010033; GO:0009719; GO:0003674; GO:0005575; GO:0009058 |
| g17506 | none |
| g25206 | GO:0046527; GO:0005622; GO:0080044; GO:0043226; GO:0043231; GO:0043229; GO:0044424; GO:0035251; GO:0005623; GO:0016740; GO:0016758; GO:0016757; GO:0044464; GO:0080043; GO:0003674; GO:0043227; GO:0005575; GO:0008194 |
| g58377 | none |
| Pathway_ID | Pathway_Name | Gene_ID | KO_Entry | EC |
|---|---|---|---|---|
| ko00941 | Flavonoid biosynthesis | g55964 | K05280 | EC:1.14.14.82 |
| ko00944 | Flavone and flavonol biosynthesis | g55964 | K05280 | EC:1.14.14.82 |
| ko01100 | Metabolic pathways | g55964 | K05280 | EC:1.14.14.82 |
| ko01110 | Biosynthesis of secondary metabolites | g55967 | K05280 | EC:1.14.14.82 |
| ko01110 | Biosynthesis of secondary metabolites | g25206 | K13496 | EC:2.4.1. |
| Xiangshu99 | Zhezi No1 | BH | WH | |
|---|---|---|---|---|
| g55964 | 0.46 | 16.86 | 0.00 | 0.00 |
| g17506 | 11.17 | 15.35 | 0.00 | 0.00 |
| g25206 | 0.11 | 0.00 | 0.00 | 0.00 |
| g58377 | 0.12 | 0.00 | 0.00 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Pan, R.; Zhang, W.; Lei, J.; Wang, L.; Chai, S.; Jin, X.; Jiao, C.; Yang, X. Integrating Genome-Wide Association Study with Transcriptomic Analysis to Predict Candidate Genes Controlling Storage Root Flesh Color in Sweet Potato. Agronomy 2022, 12, 991. https://doi.org/10.3390/agronomy12050991
Liu Y, Pan R, Zhang W, Lei J, Wang L, Chai S, Jin X, Jiao C, Yang X. Integrating Genome-Wide Association Study with Transcriptomic Analysis to Predict Candidate Genes Controlling Storage Root Flesh Color in Sweet Potato. Agronomy. 2022; 12(5):991. https://doi.org/10.3390/agronomy12050991
Chicago/Turabian StyleLiu, Yi, Rui Pan, Wenying Zhang, Jian Lei, Lianjun Wang, Shasha Chai, Xiaojie Jin, Chunhai Jiao, and Xinsun Yang. 2022. "Integrating Genome-Wide Association Study with Transcriptomic Analysis to Predict Candidate Genes Controlling Storage Root Flesh Color in Sweet Potato" Agronomy 12, no. 5: 991. https://doi.org/10.3390/agronomy12050991
APA StyleLiu, Y., Pan, R., Zhang, W., Lei, J., Wang, L., Chai, S., Jin, X., Jiao, C., & Yang, X. (2022). Integrating Genome-Wide Association Study with Transcriptomic Analysis to Predict Candidate Genes Controlling Storage Root Flesh Color in Sweet Potato. Agronomy, 12(5), 991. https://doi.org/10.3390/agronomy12050991