
Study of Codon Usage Patterns and Influencing Factors in Rice Yellow Mottle Virus Based on Coding Sequence Data

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequences
2.2. Analysis of Nucleotide Composition
2.3. Analysis of Relative Synonymous Codon Usage (RSCU)
2.4. Analysis of Codon Adaptation Index (CAI)
2.5. Indices of Codon Usage
2.6. Correspondence Analysis (COA)
2.7. Analysis of Neutrality Plot
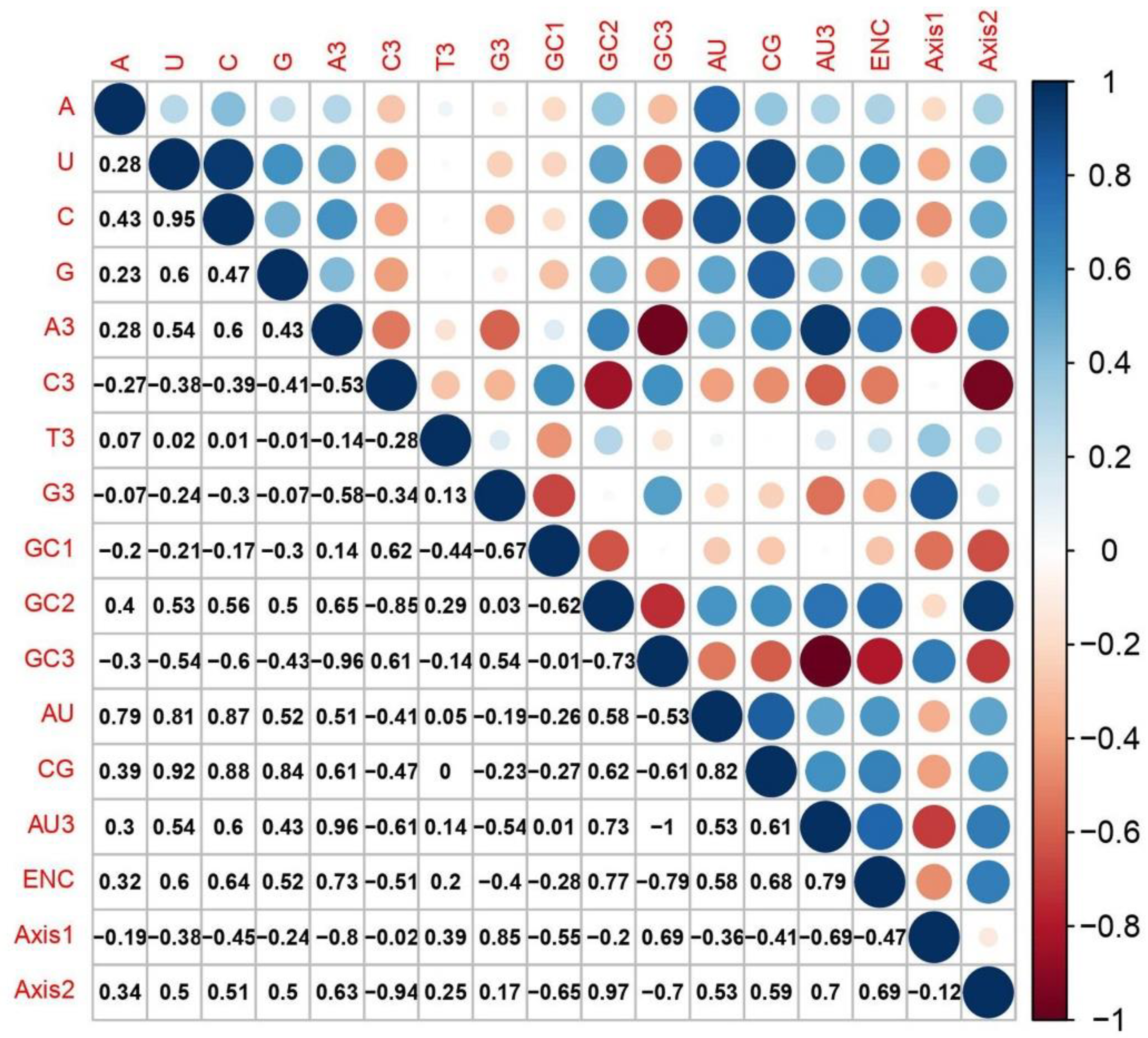
2.8. Correlation Analysis
3. Results
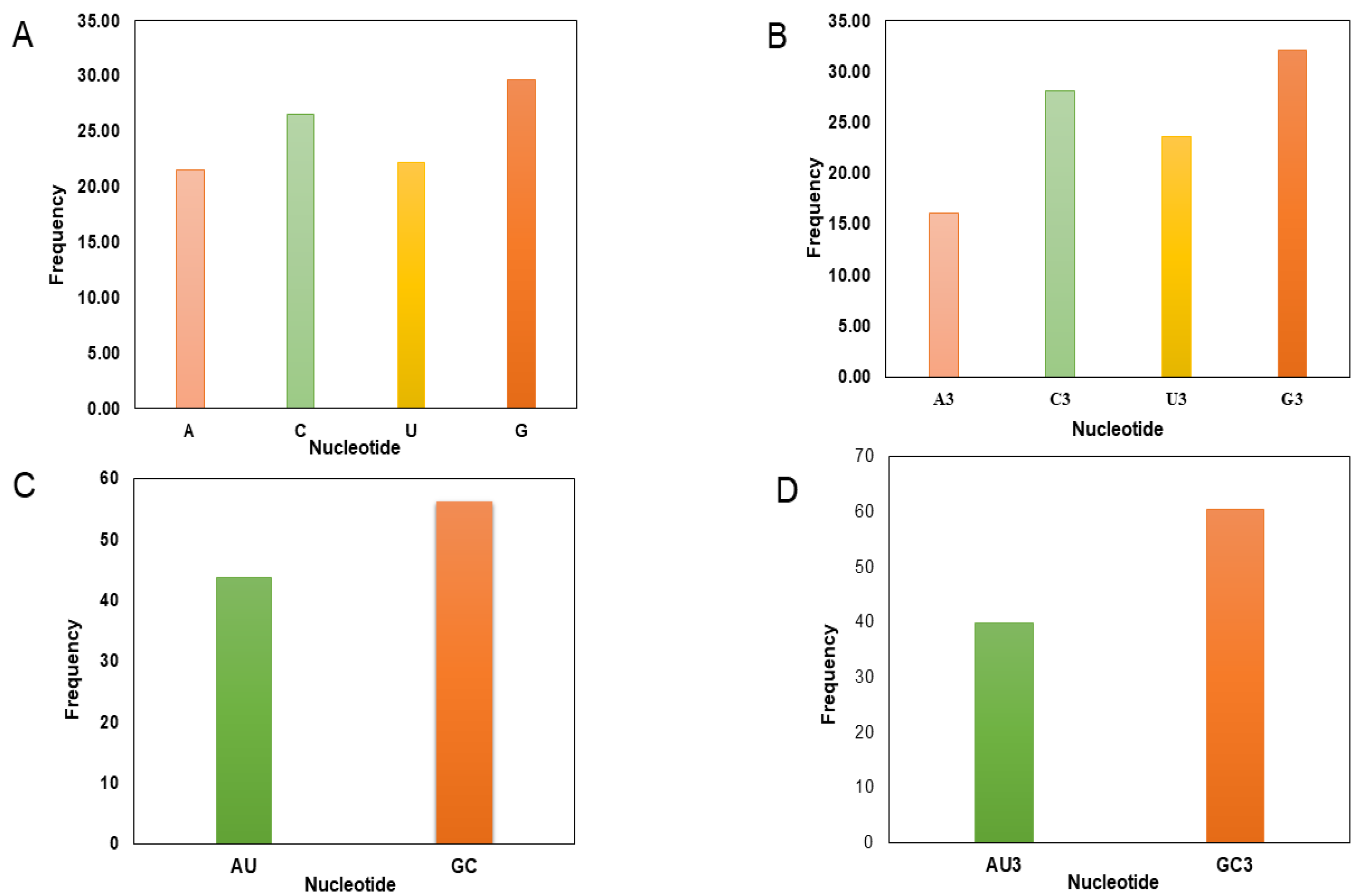
3.1. Base Compositional Analysis in RYMV Coding Sequences
3.2. Defining Codon Usage Patterns
3.3. Adaptation of RYMV to the Host Genome
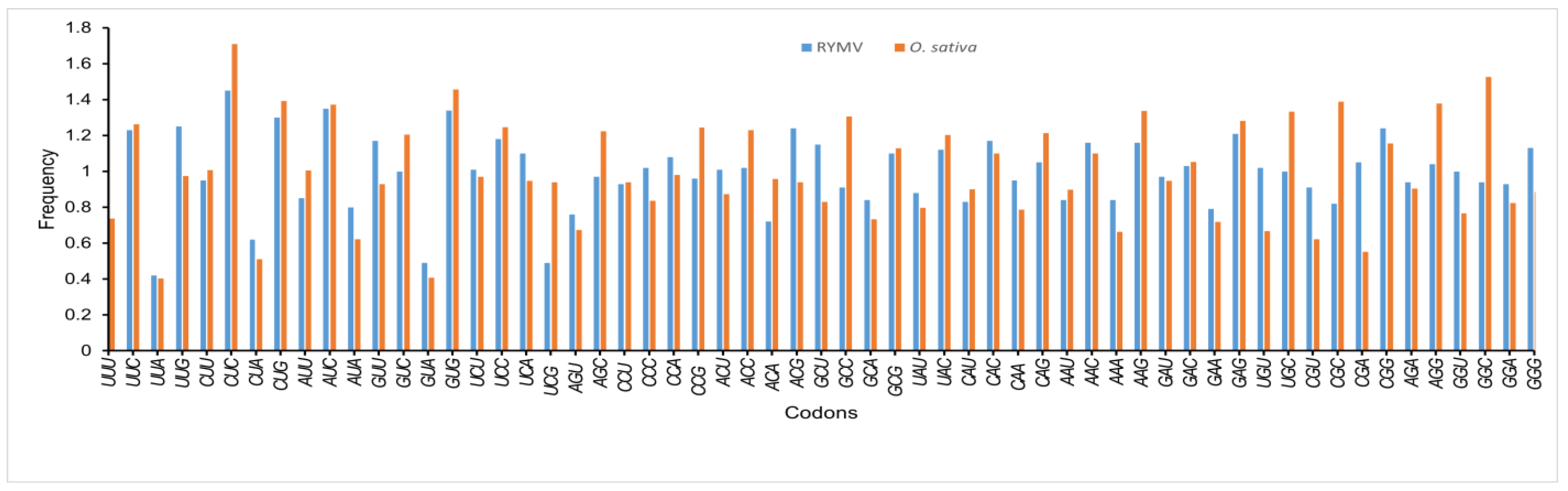
3.4. Use of Codons Biases in RYMV
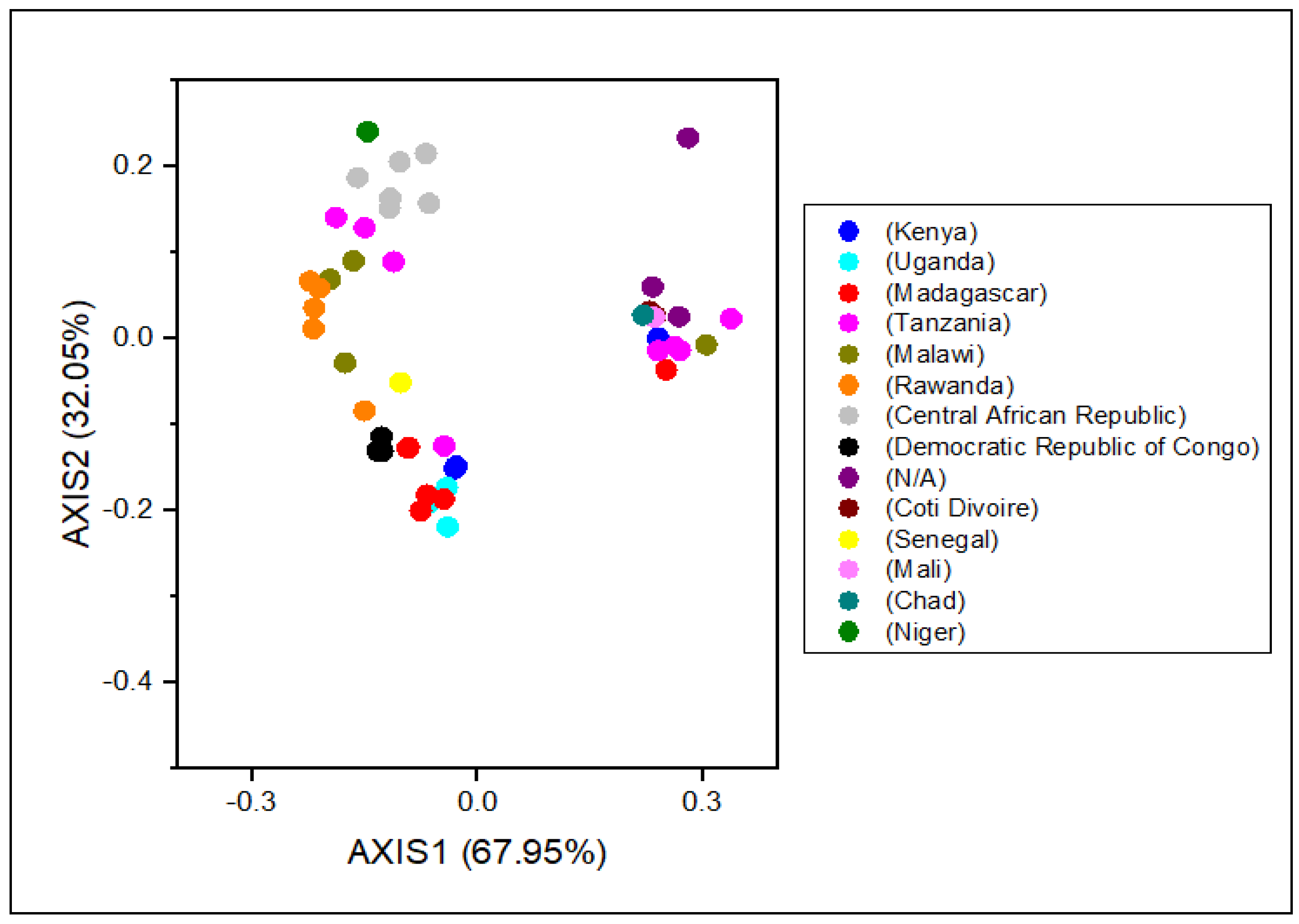
3.5. Discrepancy in the Usage of Codon among RYMVs
3.6. Neutrality Plot Analysis
3.7. Codon Usage Pattern Dominating Effects on RYMV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Rakotomalala, M.; Abera, B.; Rakotoarisoa, J.; Alemu, D.; Hébrard, E. Complete Genome Sequences of Rice Yellow Mottle Virus Isolates from the Federal Democratic Republic of Ethiopia. Microbiol. Resour. Announc. 2019, 8, e00589-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voinnet, O.; Pinto, Y.M.; Baulcombe, D.C. Suppression of Gene Silencing: A General Strategy Used by Diverse DNA and RNA Viruses of Plants. Proc. Natl. Acad. Sci. USA 1999, 96, 14147–14152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koudamiloro, A.; Nwilene, F.E.; Togola, A.; Akogbeto, M. Insect Vectors of Rice Yellow Mottle Virus. J. Insects 2015, 2015, 721751. [Google Scholar] [CrossRef] [Green Version]
- Fargette, D.; Abubakar, Z.; Traore, O.; Brugidou, C.; Choisy, M.; Se, Y.; Fauquet, C.; Konate, G. Inferring the Evolutionary History of Rice Yellow Mottle Virus from Genomic, Phylogenetic, and Phylogeographic Studies. J. Virol. 2004, 78, 3252–3261. [Google Scholar] [CrossRef] [Green Version]
- Bakker, W. Characterization and Ecological Aspects of Rice Yellow Mottle Virus in Kenya; Centre for Agricultural Publishing and Documentatio: Wageningen, The Netherlands, 1974. [Google Scholar]
- Kouassi, N.K.; N’Guessan, P.; Albar, L.; Fauquet, C.M.; Brugidou, C. Distribution and Characterization of Rice Yellow Mottle Virus: A Threat to African Farmers. Plant Dis. 2005, 89, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Konate, G.; Traore, O.; Coulibaly, M.M. Characterization of Rice Yellow Mottle Virus Isolates in Sudano-Sahelian Areas. Arch. Virol. 1997, 142, 1117–1124. [Google Scholar] [CrossRef]
- Sarra, S.; Peters, D. Rice Yellow Mottle Virus Is Transmitted by Cows, Donkeys, and Grass Rats in Irrigated Rice Crops. Plant Dis. 2003, 87, 804–808. [Google Scholar] [CrossRef] [Green Version]
- Konate, G.; Sarra, S.; Traore, O. Rice Yellow Mottle Virus Is Seed-Borne but Not Seed Transmitted in Rice Seeds. Eur. J. Plant Pathol. 2001, 107, 361–364. [Google Scholar] [CrossRef]
- Hubert, J.; Lyimo, H.J.F.; Luzi-kihupi, A. Geographical Variation, Distribution and Diversity of Rice Yellow Mottle Virus Phylotypes in Tanzania. Am. J. Plant Sci. 2017, 8, 1264–1284. [Google Scholar] [CrossRef]
- Ayaka, U.; Naswiru, T.; Keiko, T.N.; Nobuhito, S.; Nobuaki, O. Characterization of Rice Yellow Mottle Virus in North-Eastern Tanzania. J. Agric. Sci. 2015, 60, 116–126. [Google Scholar]
- N’Guessan, P.; Pinel, A.; Caruana, M.L.; Frutos, R.; Sy, A.; Ghesquière, A.; Fargette, D. Evidence of the Presence of Two Serotypes of Rice Yellow Mottle Sobemovirus in Côte d’Ivoire. Eur. J. Plant Pathol. 2000, 106, 167–178. [Google Scholar] [CrossRef]
- Rossel, H.W.; Ayotade, K.A.; Thottapilly, G.; Adeoti, A.A.; Alluri, K.; Alam, M.S.; Zan, K. A new record of rice yellow mottle virus disease in Badeggi, Nigeria. Int. Rice Comm. Newsl. 1982, 31, 23–24. [Google Scholar]
- Fauquet, C.; Thouvenel, J.C. Isolation of the rice yellow mottle virus in Ivory Coast. Plant Dis. Report. 1977, 61, 4–7. [Google Scholar]
- Raymundo, S.A.; Konteh, I. Distribution, importance, screening methods and varietal reaction to rice pale yellow mottle disease [in West Africa]. Int. Rice Comm. Newsl. 1980, 29, 51–53. [Google Scholar]
- John, V.T.; Thottappilly Ibadan, G.; Awoderu, V.A. Occurrence of Rice Yellow Mottle Virus in Some Sahelian Countries in West Africa. FAO Plant Prot. Bull. 1984, 32, 86–87. [Google Scholar]
- Reckhaus, P.M.; Andriamasintseheno, H.F. Rice Yellow Mottle Virus in Madagascar and Its Epidemiology in the Northwest of the Island/Rice Yellow Mottle Virus in Madagaskar Und Seine Epidemiologie Im Nordwesten Der Insel. Z. Pflanzenkrankh. Pflanzenschutz/J. Plant Dis. Prot. 1997, 104, 289–295. [Google Scholar]
- Traoré, O.; Pinel, A.; Fargette, D.; Konaté, G. First Report and Characterization of Rice Yellow Mottle Virus in Central Africa. Plant Dis. 2001, 85, 920. [Google Scholar] [CrossRef] [PubMed]
- Awoderu, V.A. The Rice Yellow Mottle Virus Situation in West Africa. J. Basic Microbiol. 1991, 31, 91–99. [Google Scholar] [CrossRef]
- Asante, M.D.; Amadu, B.; Traore, V.S.E.; Oppong, A.; Adebayo, M.A.; Aculey, P.; Marfo, E.A.; Kang, K.-H. Assessment of Korean Rice Lines for Their Reaction to Rice Yellow Mottle Virus in Ghana. Heliyon 2020, 6, e05551. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of Codon Usage in Zika Virus Genomes Is Host and Vector Speci Fi C. Emerg. Microbes Infect. 2016, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. TL-42. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, S.G.; Kurland, C.G. Codon Preferences in Free-Living Microorganisms. Microbiol. Rev. 1990, 54, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-Wide Analysis of Codon Usage Bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.U.; Mao, Y.; Tao, S. Codon Usage Bias and Evolutionary Analyses of Zika Virus Genomes. Genes Genom. 2017, 39, 855–866. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. Codon Usage in Regulatory Genes in Escherichia Coli Does Not Reflect Selection for “rare” Codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Nasrullah, I.; Tong, Y. Genome-Wide Analysis of Codon Usage and Influencing Factors in Chikungunya Viruses. PLoS ONE 2014, 9, e90905. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces That Influence the Evolution of Codon Bias. Philos. Trans. R. Soc. B 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.U.; Abdullah, M.; Khan, A.W.; Haq, M.I.U.; Haq, N.U.; Aziz, A.; Tao, S. A Detailed Comparative Analysis of Codon Usage Bias in Alongshan Virus. Virus Res. 2022, 308, 198646. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the Rate of Poliovirus Protein Synthesis through Large-Scale Codon Deoptimization Causes Attenuation of Viral Virulence by Lowering Specific Infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [Green Version]
- Costafreda, M.I.; Pérez-Rodriguez, F.J.; D’Andrea, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Hepatitis A Virus Adaptation to Cellular Shutoff Is Driven by Dynamic Adjustments of Codon Usage and Results in the Selection of Populations with Altered Capsids. J. Virol. 2014, 88, 5029–5041. [Google Scholar] [CrossRef] [Green Version]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of Poliovirus Replicative Fitness in HeLa Cells by Deoptimization of Synonymous Codon Usage in the Capsid Region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karumathil, S.; Raveendran, N.T.; Ganesh, D.; Kumar Ns, S.; Nair, R.R.; Dirisala, V.R. Evolution of Synonymous Codon Usage Bias in West African and Central African Strains of Monkeypox Virus. Evol. Bioinform. Online 2018, 14, 1176934318761368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odongo, P.J.; Onaga, G.; Ricardo, O.; Natsuaki, K.T.; Alicai, T.; Geuten, K. Insights Into Natural Genetic Resistance to Rice Yellow Mottle Virus and Implications on Breeding for Durable Resistance. Front. Plant Sci. 2021, 12, 671355. [Google Scholar] [CrossRef] [PubMed]
- Pidon, H.; Chéron, S.; Ghesquière, A.; Albar, L. Allele Mining Unlocks the Identification of RYMV Resistance Genes and Alleles in African Cultivated Rice. BMC Plant Biol. 2020, 20, 222. [Google Scholar] [CrossRef]
- Yao, X.; Fan, Q.; Yao, B.; Lu, P.; Rahman, S.U.; Chen, D.; Tao, S. Codon Usage Bias Analysis of Bluetongue Virus Causing Livestock Infection. Front. Microbiol. 2020, 11, 655. [Google Scholar] [CrossRef]
- Wong, E.H.M.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L.M. Codon Usage Bias and the Evolution of Influenza A Viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, D.; Ma, G.; Liu, M.; Wang, M.; Jia, R.; Chen, S.; Sun, K.; Yang, Q.; Wu, Y.; et al. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella Anatipestifer. Int. J. Mol. Sci. 2016, 17, 1304. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Uechi, T.; Higa, S.; Torihara, H.; Kenmochi, N. Loss of Ribosomal Protein L11 Affects Zebrafish Embryonic Development through a P53-Dependent Apoptotic Response. PLoS ONE 2009, 4, e4152. [Google Scholar] [CrossRef] [Green Version]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A Combined Set of Tools to Assess Codon Usage Adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The “effective Number of Codons” Used in a Gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Cristina, J.; Fajardo, A.; Sonora, M.; Moratorio, G.; Musto, H. A Detailed Comparative Analysis of Codon Usage Bias in Zika Virus. Virus Res. 2016, 223, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Greenacre, M. Theory and Applications of Correspondence Analysis; Academic Press: London, UK, 1984; ISBN 0122990501. [Google Scholar]
- Guan, D.L.; Ma, L.B.; Khan, M.S.; Zhang, X.X.; Xu, S.Q.; Xie, J.Y. Analysis of Codon Usage Patterns in Hirudinaria Manillensis Reveals a Preference for GC-Ending Codons Caused by Dominant Selection Constraints. BMC Genom. 2018, 19, 542. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional Mutation Pressure and Neutral Molecular Evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhao, D.; Tao, J. Analysis of Codon Usage Patterns in Herbaceous Peony (Paeonia Lactiflora Pall.) Based on Transcriptome Data. Genes 2015, 6, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.K.; Tyagi, A. A Detailed Analysis of Codon Usage Patterns and Influencing Factors in Zika Virus. Arch. Virol. 2017, 162, 1963–1973. [Google Scholar] [CrossRef]
- Hassan, S.; Mahalingam, V.; Kumar, V. Synonymous Codon Usage Analysis of Thirty Two Mycobacteriophage Genomes. Adv. Bioinform. 2009, 2009, 316936. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. An Evolutionary Perspective on Synonymous Codon Usage in Unicellular Organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Rahman, S.U.; Yao, X.; Li, X.; Chen, D.; Tao, S. Analysis of Codon Usage Bias of Crimean-Congo Hemorrhagic Fever Virus and Its Adaptation to Hosts. Infect. Genet. Evol. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, R.; Taylor, K.; Briley, A.; Caldwell, C.; Cheng, J. The Properties of Genome Conformation and Spatial Gene Interaction and Regulation Networks of Normal and Malignant Human Cell Types. PLoS ONE 2013, 8, e58793. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, G.M.; Holmes, E.C. The Extent of Codon Usage Bias in Human RNA Viruses and Its Evolutionary Origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-Wide Analysis of Codon Usage Bias in Four Sequenced Cotton Species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sablok, G.; Wu, X.; Kuo, J.; Nayak, K.C.; Baev, V.; Varotto, C.; Zhou, F. Combinational Effect of Mutational Bias and Translational Selection for Translation Efficiency in Tomato (Solanum Lycopersicum) Cv. Micro-Tom. Genomics 2013, 101, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis Thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, A.; Miyashita, N.T. Patterns of Codon Usage Bias in Three Dicot and Four Monocot Plant Species. Genes Genet. Syst. 2003, 78, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Wang, D.; Rahman, S.U.; Bai, H.; Yao, X.; Chen, D.; Tao, S. Analysis of Codon Usage Patterns and Influencing Factors in Rice Tungro Bacilliform Virus. Infect. Genet. Evol. 2021, 90, 104750. [Google Scholar] [CrossRef]
- Anwar, A.M.; Aljabri, M.; El-soda, M. Patterns of Genome-Wide Codon Usage Bias in Tobacco, Tomato and Potato. Biotechnol. Biotechnol. Equip. 2021, 35, 657–664. [Google Scholar] [CrossRef]
- Wei, L.; He, J.; Jia, X.; Qi, Q.; Liang, Z.; Zheng, H.; Ping, Y.; Liu, S.; Sun, J. Analysis of Codon Usage Bias of Mitochondrial Genome in Bombyx Moriand Its Relation to Evolution. BMC Evol. Biol. 2014, 14, 262. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, Y.; Zheng, C.; Huang, J.; Zhang, S. Genome-Wide Comparative Analysis of the Codon Usage Patterns in Plants. Genes Genom. 2016, 38, 723–731. [Google Scholar] [CrossRef]
- Cardinale, D.J.; Derosa, K.; Duffy, S. Base Composition and Translational Selection Are Insufficient to Explain Codon Usage Bias in Plant Viruses. Viruses 2013, 5, 162–181. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Gan, H.; Liang, X. Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, J.; Song, Q.; Zhang, Q.; Tian, P.; Nan, Z. Comprehensive Analysis of Codon Usage Bias in Seven Epichloë Species and Their Peramine-Coding Genes. Front. Microbiol. 2017, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hemert, F.; Berkhout, B. Nucleotide Composition of the Zika Virus RNA Genome and Its Codon Usage. Virol. J. 2016, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhao, Q.; Wang, Y.; Zhao, J.; Qiao, L.; Wu, B.; Yan, S.; Zheng, J.; Zheng, X. Comparative Analysis of Genomic and Transcriptome Sequences Reveals Divergent Patterns of Codon Bias in Wheat and Its Ancestor Species. Front. Genet. 2021, 12, 732432. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, R.; Zhang, H.; Huang, Y.; Tian, M.; Zhang, J. Analysis of Synonymous Codon Usage in Zea Mays. Mol. Biol. Rep. 2010, 37, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Rup Sarkar, R. Data in Support of Large Scale Comparative Codon Usage Analysis in Leishmania and Trypanosomatids. Data Br. 2015, 4, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Vasanthi, S.; Dass, J.F.P. Comparative Genome-Wide Analysis of Codon Usage of Different Bacterial Species Infecting Oryza Sativa. J. Cell. Biochem. 2018, 119, 9346–9356. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Welti, R.; Wang, X. Quantitative Analysis of Major Plant Hormones in Crude Plant Extracts by High-Performance Liquid Chromatography–Mass Spectrometry. Nat. Protoc. 2010, 5, 986–992. [Google Scholar] [CrossRef]
- Jia, J.; Xue, Q. Codon Usage Biases of Transposable Elements and Host Nuclear Genes in Arabidopsis Thaliana and Oryza Sativa. Genom. Proteom. Bioinform. 2009, 7, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Qing, Y.; Cheng, L.; Li, R.; Liu, G.; Zhang, Y.; Tang, X.; Wang, J.; Liu, H.; Qin, Y. Potential Antibacterial Mechanism of Silver Nanoparticles and the Optimization of Orthopedic Implants by Advanced Modification Technologies. Int. J. Nanomed. 2018, 13, 3311–3327. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.P.; Liu, Z.X.; Hao, L.; Ma, J.Y.; Liang, Z.L.; Li, Y.G.; Ke, H. Analysing Codon Usage Bias of Cyprinid Herpesvirus 3 and Adaptation of This Virus to the Hosts. J. Fish Dis. 2015, 38, 665–673. [Google Scholar] [CrossRef]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic Analysis of Codon Usage Shows Influence of Mutation Pressure, Natural Selection, and Host Features on Marburg Virus Evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Bera, B.C.; Greenbaum, B.D.; Bhatia, S.; Sood, R.; Selvaraj, P.; Anand, T.; Tripathi, B.N.; Virmani, N. Revelation of Influencing Factors in Overall Codon Usage Bias of Equine Influenza Viruses. PLoS ONE 2016, 11, e0154376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, J.; Cook, N.; Lloyd, G.; Mioulet, V.; Tolley, H.; Hewson, R. Co-Evolutionary Patterns of Variation in Small and Large RNA Segments of Crimean-Congo Hemorrhagic Fever Virus. J. Gen. Virol. 2005, 86, 3337–3341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, Z.; Meng, X.; Zhang, L.; Liu, Z.; Liu, M.; Zhang, F.; Zhao, J. Codon Usage Patterns across Seven Rosales Species. BMC Plant Biol. 2022, 22, 65. [Google Scholar] [CrossRef]
- Hewson, R.; Gmyl, A.; Gmyl, L.; Smirnova, S.E.; Karganova, G.; Jamil, B.; Hasan, R.; Chamberlain, J.; Clegg, C. Evidence of Segment Reassortment in Crimean-Congo Haemorrhagic Fever Virus. J. Gen. Virol. 2004, 85, 3059–3070. [Google Scholar] [CrossRef]
- Cheng, X.; Wu, X.; Wang, H.; Sun, Y.; Qian, Y.; Luo, L. High Codon Adaptation in Citrus Tristeza Virus to Its Citrus Host. Virol. J. 2012, 9, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Bhattacharyya, T.K.; Ghosh, T.C. Synonymous Codon Usage in Lactococcus Lactis: Mutational Bias versus Translational Selection. J. Biomol. Struct. Dyn. 2004, 21, 527–536. [Google Scholar] [CrossRef]
- Biswas, K.K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.K.; Ghosh, D.K.; Debnath, P.; Ramadugu, C.; Keremane, M.L.; Khetarpal, R.K.; Lee, R.F. Codon Usage Bias Analysis of Citrus Tristeza Virus: Higher Codon Adaptation to Citrus Reticulata Host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Alam, C.M.; Kumar, R.V.; Gnanasekaran, P.; Chakraborty, S. Potential Linkage between Compound Microsatellites and Recombination in Geminiviruses: Evidence from Comparative Analysis. Virology 2015, 482, 41–50. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Gnanasekaran, P.; Jain, S.K.; Chakraborty, S. Genome Wide Survey and Analysis of Small Repetitive Sequences in Caulimoviruses. Infect. Genet. Evol. 2014, 27, 15–24. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | A | C | U | G | GC | AU | A3 | C3 | U3 | G3 | GC1 | GC2 | GC3 | AU3 | GC3s | ENC | Gravy | Aromo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21.99 | 25.66 | 23.72 | 28.63 | 54.29 | 45.71 | 19.82 | 23.32 | 22.22 | 34.63 | 47.45 | 57.46 | 57.96 | 42.04 | 56.41 | 57.45 | −0.3005 | 0.092184 |
| 2 | 21.57 | 26.13 | 23.20 | 29.10 | 55.23 | 44.77 | 16.50 | 30.90 | 21.90 | 30.70 | 57.60 | 46.50 | 61.60 | 38.40 | 60.23 | 56.82 | −0.2919 | 0.092092 |
| 3 | 21.67 | 26.18 | 23.76 | 28.38 | 54.57 | 45.43 | 15.68 | 27.56 | 24.75 | 32.01 | 55.12 | 49.01 | 59.57 | 40.43 | 58.85 | 57.58 | −0.3021 | 0.07438 |
| 4 | 21.73 | 26.29 | 23.71 | 28.27 | 54.57 | 45.43 | 15.84 | 27.72 | 24.59 | 31.85 | 54.95 | 49.17 | 59.57 | 40.43 | 58.78 | 57.32 | −0.2988 | 0.071074 |
| 5 | 22.03 | 25.87 | 23.40 | 28.70 | 54.57 | 45.43 | 17.50 | 30.40 | 22.50 | 29.60 | 57.20 | 46.50 | 60.00 | 40.00 | 58.52 | 57.33 | −0.2933 | 0.092092 |
| 6 | 22.17 | 26.46 | 23.21 | 28.16 | 54.62 | 45.38 | 17.33 | 27.72 | 23.27 | 31.68 | 54.95 | 49.50 | 59.41 | 40.59 | 58.75 | 57.61 | −0.2925 | 0.07438 |
| 7 | 21.57 | 26.43 | 22.93 | 29.07 | 55.50 | 45.50 | 17.20 | 31.60 | 21.30 | 29.90 | 57.70 | 47.30 | 61.50 | 38.50 | 60.12 | 56.63 | −0.2891 | 0.089089 |
| 8 | 21.53 | 26.13 | 23.33 | 29.00 | 55.13 | 44.87 | 16.90 | 30.80 | 22.00 | 30.30 | 57.40 | 46.90 | 61.10 | 38.90 | 59.67 | 57.83 | −0.2876 | 0.091091 |
| 9 | 21.67 | 25.90 | 23.40 | 29.03 | 54.93 | 45.07 | 16.90 | 30.30 | 22.40 | 30.40 | 57.10 | 47.00 | 60.70 | 39.30 | 59.21 | 57.80 | −0.3038 | 0.093093 |
| 10 | 21.47 | 26.27 | 23.13 | 29.13 | 55.40 | 44.60 | 17.00 | 31.50 | 21.60 | 29.90 | 57.80 | 47.00 | 61.40 | 38.60 | 59.98 | 57.24 | −0.2798 | 0.091091 |
| 11 | 21.51 | 26.18 | 23.76 | 28.55 | 54.73 | 45.27 | 15.02 | 27.72 | 24.26 | 33.00 | 54.46 | 49.01 | 60.73 | 39.27 | 60.03 | 56.10 | −0.3044 | 0.069421 |
| 12 | 21.62 | 26.57 | 23.43 | 28.38 | 54.95 | 45.05 | 15.84 | 27.72 | 24.09 | 32.34 | 55.12 | 49.67 | 60.07 | 39.93 | 59.36 | 55.92 | −0.3173 | 0.069421 |
| 13 | 21.84 | 26.40 | 23.54 | 28.22 | 54.62 | 45.38 | 15.84 | 27.72 | 24.42 | 32.01 | 54.79 | 49.34 | 59.74 | 40.26 | 58.95 | 57.36 | −0.3292 | 0.069421 |
| 14 | 21.83 | 26.30 | 23.22 | 28.66 | 54.96 | 45.04 | 27.58 | 25.42 | 24.14 | 22.86 | 60.28 | 56.30 | 48.28 | 51.72 | 48.47 | 59.60 | −0.4022 | 0.074859 |
| 15 | 21.92 | 25.93 | 23.49 | 28.66 | 54.59 | 45.41 | 17.02 | 29.73 | 23.02 | 30.23 | 56.86 | 46.95 | 59.96 | 40.04 | 58.52 | 57.33 | −0.2975 | 0.089178 |
| 16 | 21.66 | 25.57 | 23.73 | 29.04 | 54.61 | 45.39 | 18.15 | 25.98 | 24.22 | 31.65 | 51.08 | 55.13 | 57.62 | 42.38 | 56.61 | 58.32 | −0.2354 | 0.081211 |
| 17 | 21.68 | 25.78 | 23.55 | 28.99 | 54.77 | 45.23 | 18.42 | 25.84 | 24.22 | 31.51 | 51.55 | 55.40 | 57.35 | 42.65 | 56.35 | 58.58 | −0.2448 | 0.079725 |
| 18 | 21.66 | 25.73 | 23.55 | 29.06 | 54.79 | 45.21 | 18.42 | 25.78 | 24.29 | 31.51 | 51.62 | 55.47 | 57.29 | 42.71 | 56.28 | 58.51 | −0.2439 | 0.079092 |
| 19 | 22.22 | 25.66 | 23.66 | 28.45 | 54.12 | 45.88 | 22.60 | 28.34 | 23.14 | 25.91 | 55.60 | 52.50 | 54.25 | 45.75 | 53.61 | 59.27 | −0.3880 | 0.088687 |
| 20 | 21.95 | 26.07 | 23.23 | 28.74 | 54.81 | 45.19 | 19.84 | 27.33 | 23.48 | 29.35 | 53.10 | 54.66 | 56.68 | 43.32 | 55.79 | 59.73 | −0.3074 | 0.074534 |
| 21 | 21.93 | 25.91 | 23.44 | 28.72 | 54.63 | 45.37 | 20.04 | 26.79 | 24.02 | 29.15 | 53.24 | 54.72 | 55.94 | 44.06 | 55.01 | 59.64 | −0.2971 | 0.071923 |
| 22 | 21.61 | 25.62 | 23.82 | 28.95 | 54.57 | 45.43 | 20.78 | 29.42 | 24.29 | 25.51 | 56.95 | 51.82 | 54.93 | 45.07 | 54.13 | 58.86 | −0.4036 | 0.089013 |
| 23 | 21.79 | 25.78 | 23.55 | 28.88 | 54.66 | 45.34 | 18.96 | 25.84 | 24.16 | 31.04 | 51.42 | 55.67 | 56.88 | 43.12 | 55.96 | 58.64 | −0.2448 | 0.07777 |
| 24 | 21.82 | 25.71 | 23.57 | 28.90 | 54.61 | 45.39 | 18.89 | 25.71 | 24.29 | 31.11 | 51.42 | 55.60 | 56.82 | 43.18 | 55.89 | 58.69 | −0.2486 | 0.07777 |
| 25 | 22.29 | 25.96 | 23.17 | 28.59 | 54.54 | 45.46 | 22.20 | 27.73 | 23.55 | 26.52 | 56.55 | 52.83 | 54.25 | 45.75 | 53.59 | 59.21 | −0.4560 | 0.084203 |
| 26 | 21.69 | 26.39 | 23.17 | 28.75 | 55.14 | 44.86 | 21.85 | 28.86 | 22.39 | 26.90 | 57.11 | 53.53 | 55.77 | 44.23 | 55.22 | 58.25 | −0.3994 | 0.086171 |
| 27 | 22.14 | 26.34 | 22.95 | 28.57 | 54.91 | 45.09 | 22.52 | 29.06 | 22.72 | 25.69 | 57.18 | 52.80 | 54.75 | 45.25 | 54.43 | 58.93 | −0.4301 | 0.082696 |
| 28 | 22.13 | 26.09 | 23.19 | 28.59 | 54.68 | 45.32 | 20.11 | 27.19 | 23.41 | 29.28 | 53.04 | 54.52 | 56.48 | 43.52 | 55.58 | 59.61 | −0.3165 | 0.074586 |
| 29 | 21.94 | 25.87 | 23.62 | 28.57 | 54.44 | 45.56 | 25.02 | 21.38 | 23.47 | 30.14 | 53.61 | 58.19 | 51.52 | 48.48 | 50.40 | 59.71 | −0.2559 | 0.079132 |
| 30 | 21.66 | 26.23 | 23.17 | 28.95 | 55.17 | 44.83 | 22.13 | 28.61 | 22.47 | 26.79 | 56.82 | 53.31 | 55.40 | 44.60 | 54.94 | 58.74 | −0.3946 | 0.083043 |
| 31 | 22.29 | 25.91 | 23.23 | 28.57 | 54.48 | 45.52 | 22.20 | 27.73 | 23.62 | 26.45 | 56.68 | 52.56 | 54.18 | 45.82 | 53.52 | 59.23 | −0.4475 | 0.084899 |
| 32 | 21.81 | 26.27 | 23.09 | 28.83 | 55.10 | 44.90 | 24.58 | 21.74 | 22.89 | 30.79 | 54.42 | 58.34 | 52.53 | 47.47 | 51.51 | 59.24 | −0.2734 | 0.068966 |
| 33 | 22.12 | 25.85 | 23.32 | 28.71 | 54.56 | 45.44 | 25.47 | 21.83 | 22.10 | 30.59 | 53.37 | 57.88 | 52.43 | 47.57 | 51.65 | 59.07 | −0.2822 | 0.078762 |
| 34 | 22.27 | 25.69 | 23.60 | 28.43 | 54.12 | 45.88 | 25.56 | 21.04 | 22.59 | 30.82 | 53.14 | 57.38 | 51.85 | 48.15 | 50.77 | 58.95 | −0.2796 | 0.082691 |
| 35 | 22.25 | 25.62 | 23.65 | 28.48 | 54.10 | 45.90 | 22.59 | 27.51 | 23.20 | 26.70 | 56.17 | 51.92 | 54.21 | 45.79 | 53.67 | 59.14 | −0.3769 | 0.085834 |
| 36 | 21.86 | 25.86 | 23.50 | 28.78 | 54.64 | 45.36 | 21.48 | 29.97 | 23.30 | 25.25 | 57.17 | 51.52 | 55.22 | 44.78 | 54.35 | 59.00 | −0.4505 | 0.089708 |
| 37 | 21.97 | 26.07 | 23.30 | 28.65 | 54.72 | 45.28 | 19.97 | 27.26 | 23.68 | 29.08 | 53.04 | 54.79 | 56.34 | 43.66 | 55.55 | 59.93 | −0.2964 | 0.070491 |
| 38 | 21.79 | 26.19 | 23.18 | 28.84 | 55.03 | 44.97 | 23.52 | 29.45 | 23.05 | 23.99 | 59.03 | 52.63 | 53.44 | 46.56 | 53.17 | 59.06 | −0.4666 | 0.080724 |
| 39 | 21.83 | 26.05 | 23.40 | 28.73 | 54.78 | 45.22 | 22.39 | 28.12 | 22.99 | 26.50 | 57.05 | 52.66 | 54.62 | 45.38 | 53.84 | 59.20 | −0.3835 | 0.087561 |
| 40 | 21.69 | 26.21 | 23.24 | 28.86 | 55.07 | 44.93 | 22.05 | 28.66 | 22.52 | 26.77 | 57.18 | 52.60 | 55.43 | 44.57 | 54.92 | 58.50 | −0.3786 | 0.086866 |
| 41 | 21.66 | 25.78 | 23.59 | 28.97 | 54.75 | 45.25 | 18.69 | 25.64 | 24.43 | 31.24 | 51.42 | 55.94 | 56.88 | 43.12 | 56.02 | 58.83 | −0.2484 | 0.077029 |
| 42 | 21.75 | 25.71 | 23.62 | 28.92 | 54.63 | 45.37 | 18.69 | 25.64 | 24.36 | 31.31 | 51.28 | 55.67 | 56.95 | 43.05 | 56.06 | 58.69 | −0.2461 | 0.07777 |
| 43 | 21.71 | 26.30 | 23.06 | 28.93 | 55.23 | 44.77 | 22.39 | 28.32 | 22.93 | 26.37 | 57.99 | 53.00 | 54.69 | 45.31 | 54.30 | 59.30 | −0.4041 | 0.082754 |
| 44 | 21.92 | 26.12 | 23.29 | 28.68 | 54.80 | 45.20 | 22.25 | 28.46 | 23.20 | 26.10 | 57.05 | 52.80 | 54.55 | 45.45 | 53.90 | 58.95 | −0.4244 | 0.085595 |
| 45 | 22.12 | 26.41 | 22.95 | 28.52 | 54.93 | 45.07 | 22.52 | 29.20 | 22.72 | 25.56 | 56.91 | 53.14 | 54.75 | 45.25 | 54.48 | 58.84 | −0.4297 | 0.081533 |
| 46 | 21.92 | 26.25 | 22.97 | 28.86 | 55.11 | 44.89 | 24.81 | 21.71 | 22.52 | 30.95 | 54.75 | 57.92 | 52.66 | 47.34 | 51.90 | 59.57 | −0.2966 | 0.070827 |
| 47 | 21.58 | 26.39 | 23.13 | 28.91 | 55.29 | 44.71 | 21.51 | 24.07 | 22.52 | 31.89 | 48.21 | 61.70 | 55.97 | 44.03 | 54.85 | 59.80 | −0.2944 | 0.069376 |
| 48 | 22.09 | 25.84 | 23.55 | 28.52 | 54.36 | 45.64 | 22.13 | 28.21 | 23.35 | 26.32 | 55.67 | 52.90 | 54.52 | 45.48 | 54.12 | 59.26 | −0.4056 | 0.085434 |
| 49 | 22.18 | 25.92 | 23.42 | 28.48 | 54.39 | 45.61 | 19.76 | 26.57 | 23.74 | 29.94 | 52.73 | 53.94 | 56.51 | 43.49 | 55.28 | 59.64 | −0.3229 | 0.07687 |
| 50 | 21.73 | 25.62 | 23.73 | 28.92 | 54.54 | 45.46 | 18.89 | 25.71 | 24.29 | 31.11 | 51.42 | 55.40 | 56.82 | 43.18 | 55.96 | 58.74 | −0.2293 | 0.07989 |
| Mean | 21.86 | 26.03 | 23.39 | 28.73 | 54.75 | 45.27 | 20.27 | 27.22 | 23.29 | 29.22 | 54.88 | 52.97 | 56.44 | 43.56 | 55.59 | 58.55 | −0.32791 | 0.08092 |
| SD | 0.23 | 0.27 | 0.25 | 0.24 | 0.32 | 0.31 | 3.04 | 2.62 | 0.87 | 2.67 | 2.76 | 3.63 | 2.97 | 2.97 | 2.76 | 1.00 | 0.067628 | 0.007244 |
| RSCU | RSCU | ||||||
|---|---|---|---|---|---|---|---|
| AA | Codon | RYMV | O. sativa | AA | Codon | RYMV | O. sativa |
| Phe | UUU | 0.77 | 0.738 | Ala | GCU | 1.15 | 0.831 |
| UUC | 1.23 | 1.262 | GCC | 0.91 | 1.306 | ||
| Leu | UUA | 0.42 | 0.404 | GCA | 0.84 | 0.734 | |
| UUG | 1.25 | 0.975 | GCG | 1.10 | 1.128 | ||
| CUU | 0.95 | 1.008 | Tyr | UAU | 0.88 | 0.797 | |
| CUC | 1.45 | 1.71 | UAC | 1.12 | 1.203 | ||
| CUA | 0.62 | 0.51 | His | CAU | 0.83 | 0.900 | |
| CUG | 1.30 | 1.392 | CAC | 1.17 | 1.100 | ||
| Ile | AUU | 0.85 | 1.005 | Gln | CAA | 0.95 | 0.787 |
| AUC | 1.35 | 1.373 | CAG | 1.05 | 1.213 | ||
| AUA | 0.80 | 0.623 | Asn | AAU | 0.84 | 0.899 | |
| Val | GUU | 1.17 | 0.93 | AAC | 1.16 | 1.101 | |
| GUC | 1.00 | 1.205 | Lys | AAA | 0.84 | 0.663 | |
| GUA | 0.49 | 0.408 | AAG | 1.16 | 1.337 | ||
| GUG | 1.34 | 1.457 | Asp | GAU | 0.97 | 0.948 | |
| Ser | UCU | 1.01 | 0.971 | GAC | 1.03 | 1.052 | |
| UCC | 1.18 | 1.246 | Glu | GAA | 0.79 | 0.718 | |
| UCA | 1.10 | 0.948 | GAG | 1.21 | 1.282 | ||
| UCG | 0.49 | 0.94 | Cys | UGU | 1.02 | 0.667 | |
| AGU | 0.76 | 0.673 | UGC | 1.00 | 1.333 | ||
| AGC | 0.97 | 1.223 | Arg | CGU | 0.91 | 0.621 | |
| CGC | 0.82 | 1.388 | |||||
| Pro | CCU | 0.93 | 0.94 | CGA | 1.05 | 0.552 | |
| CCC | 1.02 | 0.836 | CGG | 1.24 | 1.155 | ||
| CCA | 1.08 | 0.981 | AGA | 0.94 | 0.905 | ||
| CCG | 0.96 | 1.244 | AGG | 1.04 | 1.379 | ||
| Thr | ACU | 1.01 | 0.874 | Gly | GGU | 1.00 | 0.766 |
| ACC | 1.02 | 1.229 | GGC | 0.94 | 1.527 | ||
| ACA | 0.72 | 0.957 | GGA | 0.93 | 0.823 | ||
| ACG | 1.24 | 0.94 | GGG | 1.13 | 0.885 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.U.; Nawaz, S.; Ullah, S.; Rahman, I.U.; Haq, M.I.U.; Khan, M.A.; Al-Ghamdi, A.A.; Al-Hemaid, F.M.; Elshikh, M.S.; Aljowaie, R.M.; et al. Study of Codon Usage Patterns and Influencing Factors in Rice Yellow Mottle Virus Based on Coding Sequence Data. Agronomy 2022, 12, 1990. https://doi.org/10.3390/agronomy12091990
Rahman SU, Nawaz S, Ullah S, Rahman IU, Haq MIU, Khan MA, Al-Ghamdi AA, Al-Hemaid FM, Elshikh MS, Aljowaie RM, et al. Study of Codon Usage Patterns and Influencing Factors in Rice Yellow Mottle Virus Based on Coding Sequence Data. Agronomy. 2022; 12(9):1990. https://doi.org/10.3390/agronomy12091990
Chicago/Turabian StyleRahman, Siddiq Ur, Sajid Nawaz, Sifat Ullah, Inayat Ur Rahman, Muhammad Inam Ul Haq, Muazzam Ali Khan, Abdullah Ahmed Al-Ghamdi, Fahad M. Al-Hemaid, Mohamed S. Elshikh, Reem M. Aljowaie, and et al. 2022. "Study of Codon Usage Patterns and Influencing Factors in Rice Yellow Mottle Virus Based on Coding Sequence Data" Agronomy 12, no. 9: 1990. https://doi.org/10.3390/agronomy12091990
APA StyleRahman, S. U., Nawaz, S., Ullah, S., Rahman, I. U., Haq, M. I. U., Khan, M. A., Al-Ghamdi, A. A., Al-Hemaid, F. M., Elshikh, M. S., Aljowaie, R. M., & Eltayb, W. A. (2022). Study of Codon Usage Patterns and Influencing Factors in Rice Yellow Mottle Virus Based on Coding Sequence Data. Agronomy, 12(9), 1990. https://doi.org/10.3390/agronomy12091990