
Genetic Diversity in Oilseed and Vegetable Mustard (Brassica juncea L.) Accessions Revealed by Nuclear and Mitochondrial Molecular Markers



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Samples
2.2. Genomic DNA Extraction
2.3. Sequence Related Amplified Polymorphism (SRAP) Analysis
2.4. Simple Sequence Repeats (SSR) Analysis
2.5. Intron-Exon Splice Junction (ISJ) Analysis
2.6. Analysis of Differentiation of Mitochondrial Genomes
2.7. Data Collection and Analysis
3. Results
3.1. Level of Polymorphism
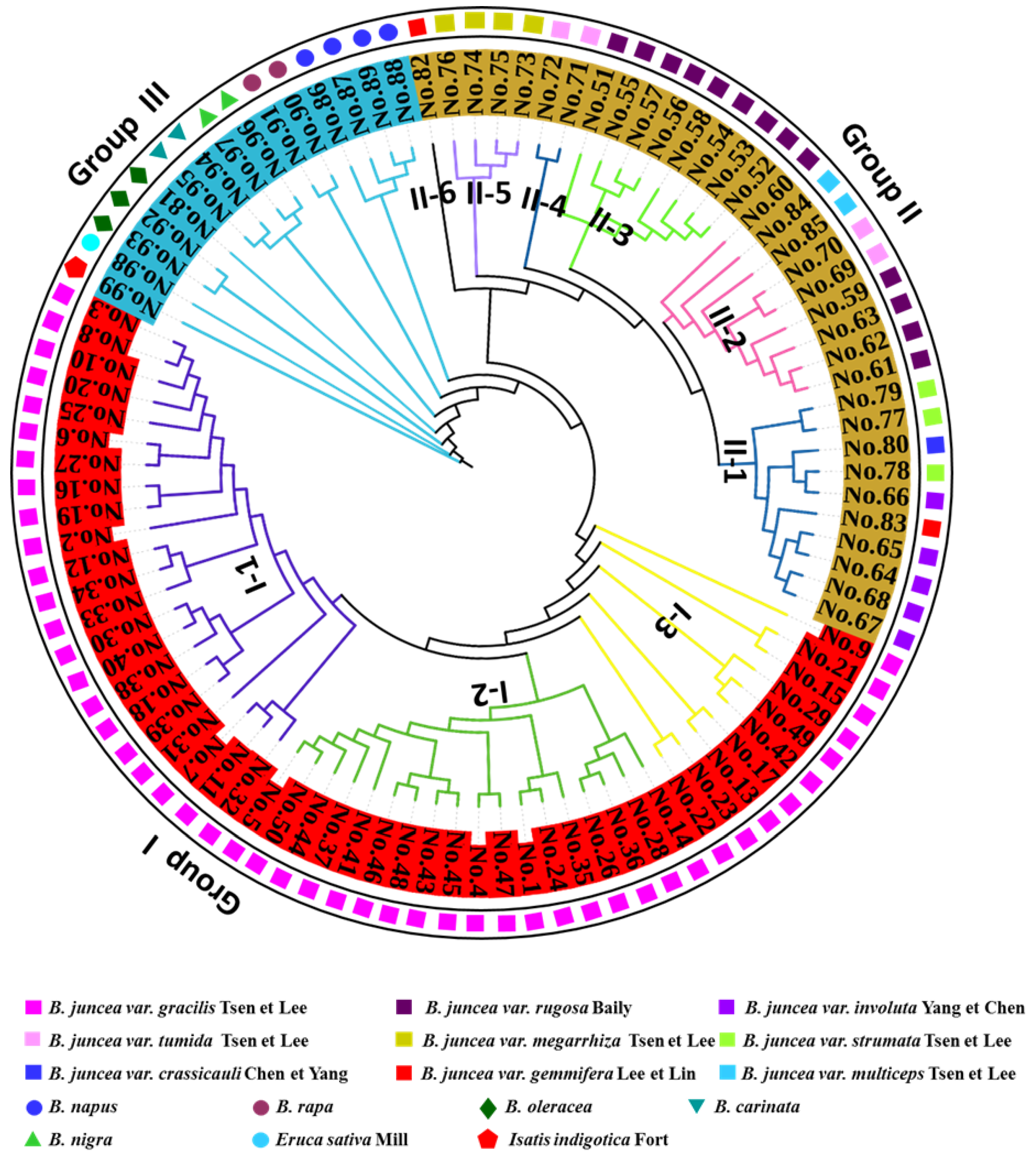
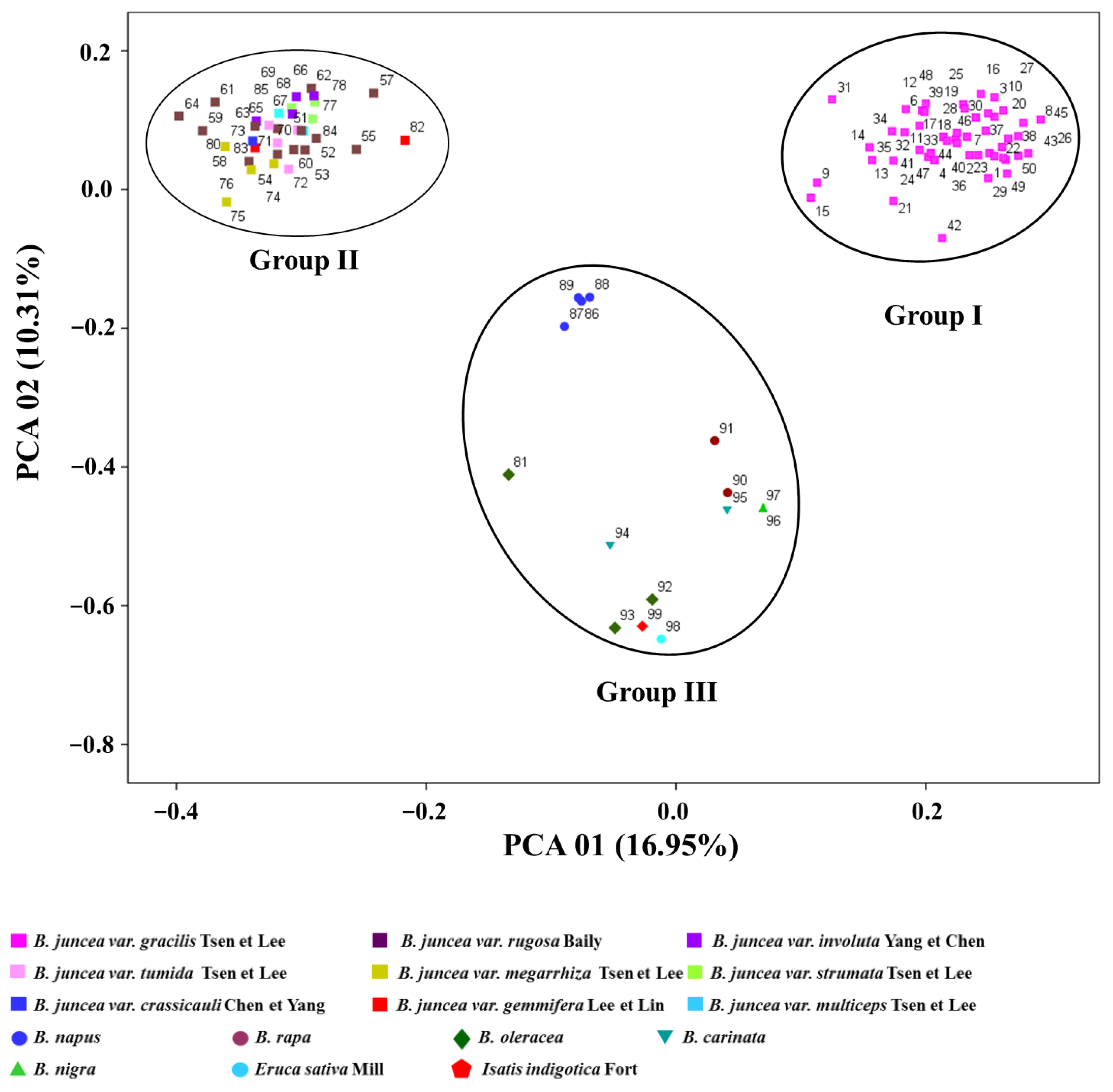
3.2. Cluster Analysis
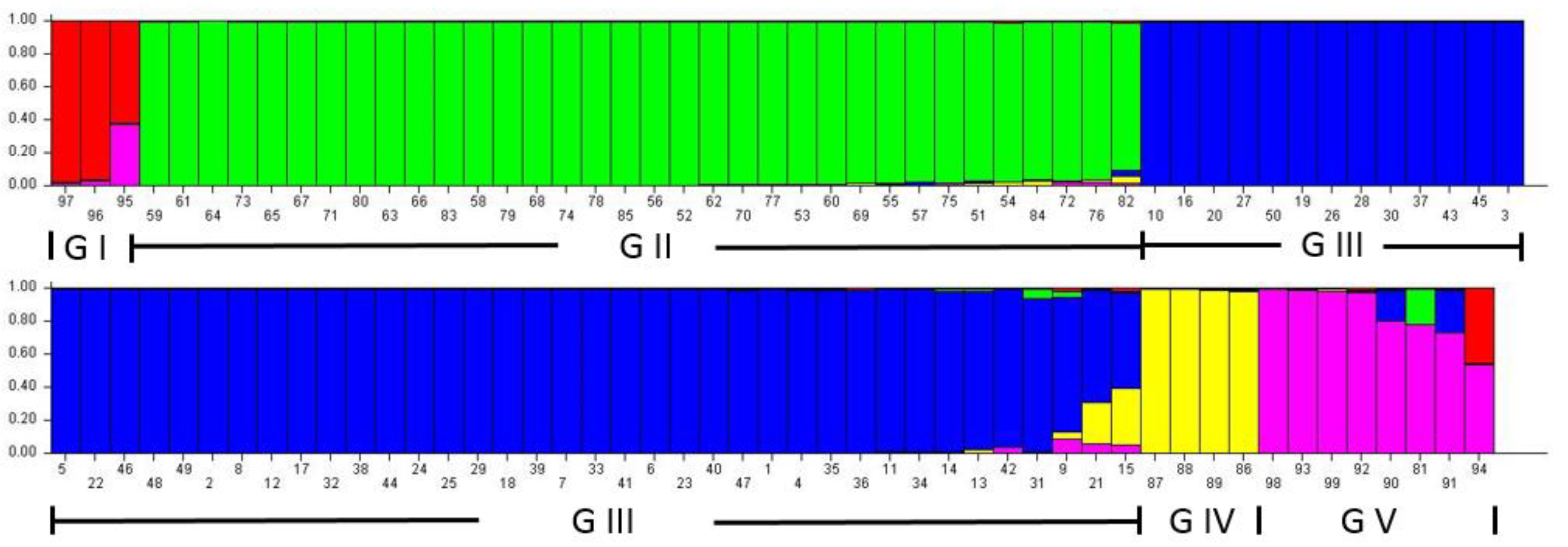
3.3. Population Structure Analysis
3.4. Analysis of Molecular Variance
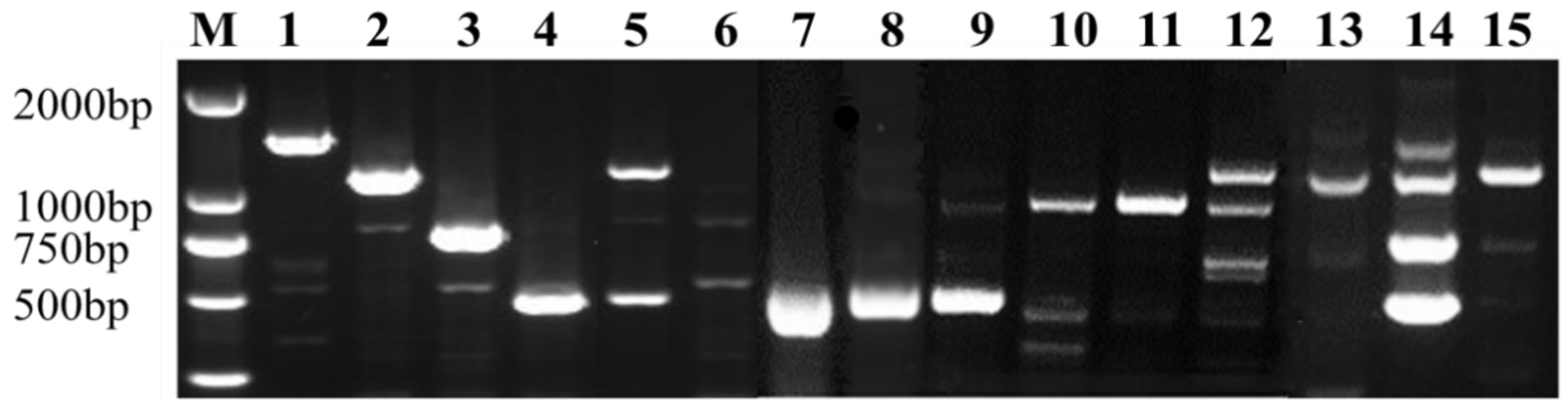
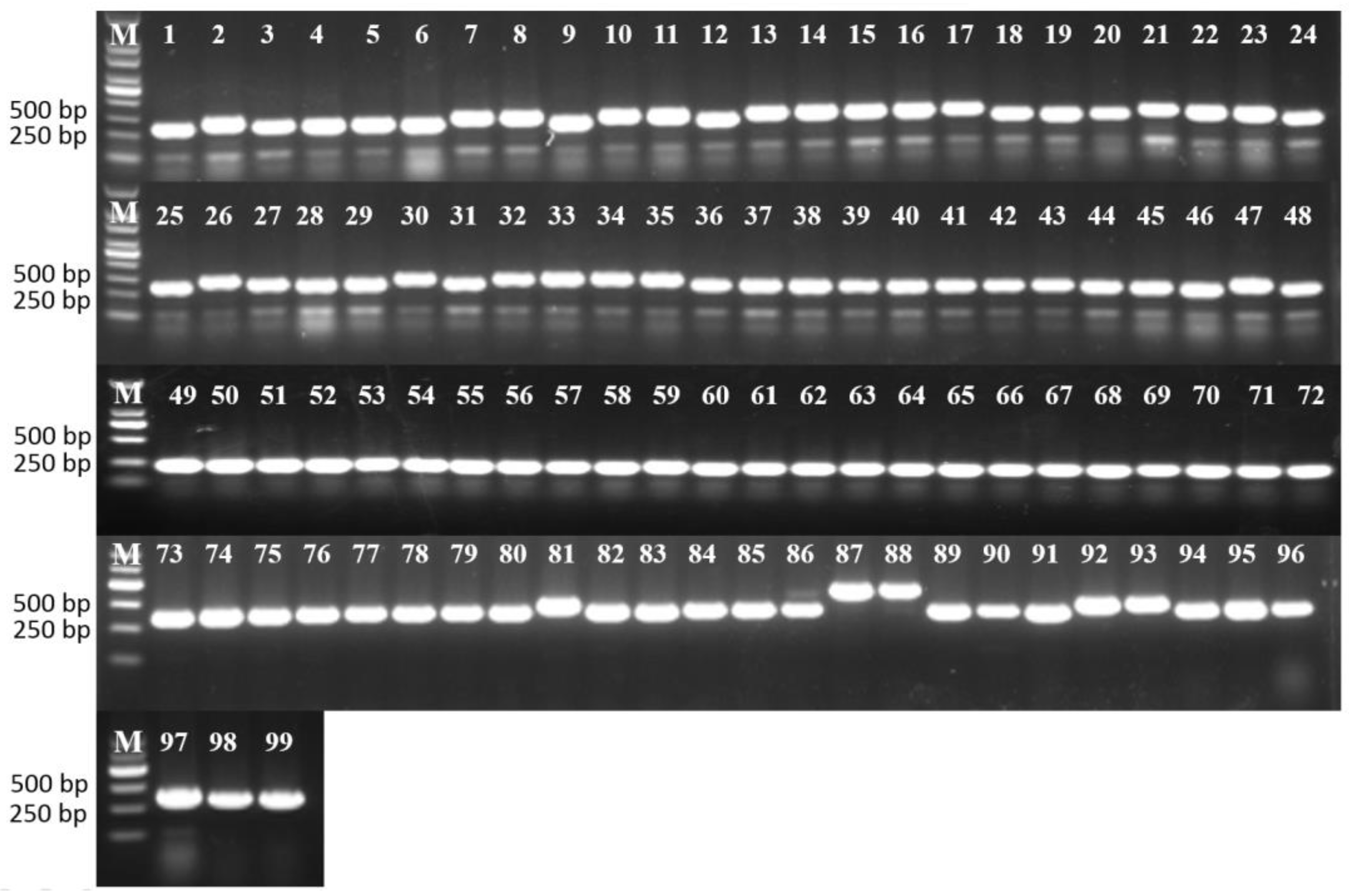
3.5. Analysis of the Cytoplasm Types by Mitochondrial Markers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Pradhan, A.K.; Pental, D. Genetics of Brassica juncea. In Genetics and Genomics of the Brassicaceae, Plant Genetics and Genomics: Crops and Models; Schmidt, R., Bancroft, I., Eds.; Springer Science + Business Media: Berlin, Germany, 2011; pp. 323–345. [Google Scholar]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, C.; Zhao, N.; Zhang, L.; Hu, Z.; Chen, S.; Zhang, M. Chinese root-type mustard provides phylogenomic insights into the evolution of the multi-use diversified allopolyploid Brassica juncea. Mol. Plant 2018, 11, 512–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.G.; Liu, N.C.; Chen, X.Q.; Chen, C.L.; Zhou, G.F.; Liao, K.L. A study on classification of mustard. Acta. Hortic. Sin. 1989, 16, 114–121. [Google Scholar]
- Gladis, T.; Hammer, K. The Brassica collection in gatersleben: Brassica juncea, Brassica napus, Brassica nigra and Brassica rapa. Feddes Rep. 1992, 103, 469–507. [Google Scholar] [CrossRef]
- Chen, C.L.; Zhou, G.F.; Yang, Y.G.; Chen, X.Q.; Liu, N.C. Distribution of Chinese mustard (Brassica juncea (L.) Coss). Southwest China J. Agric. Sci. 1990, 3, 17–21. [Google Scholar]
- Kang, L.; Qian, L.; Zheng, M.; Chen, L.; Chen, H.; Yang, L.; You, L.; Bin Yang, B.; Yan, M.; Gu, Y.; et al. Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 2021, 53, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Paritosh, K.; Yadava, S.K.; Singh, P.; Bhayana, L.; Mukhopadhyay, A.; Gupta, V.; Bisht, N.C.; Zhang, J.; Kudrna, D.A.; Copetti, D.; et al. A chromosome-scale assembly of allotetraploid Brassica juncea (AABB) elucidates comparative architecture of the A and B genomes. Plant Biotechnol. J. 2021, 19, 602–614. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, H.Z.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.-K.; et al. A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 2020, 6, 929–941. [Google Scholar] [CrossRef]
- Erickson, L.R.; Straus, N.A.; Beversdorf, W.D. Restriction patterns reveal origins of chloroplast genomes in Brassica amphiploids. Theor. Appl. Genet. 1983, 65, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Shields, C.R.; Cohen, D.B.; Orton, T.J. Chloroplast DNA evolution and the origin of amphiploid Brassica species. Theor. Appl. Genet. 1983, 65, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, S.; Li, F.; Zhang, S.; Zhang, H.; Wang, X.; Sun, R.; Bonnema, G.; Borm, T.J.A. A phylogenetic analysis of chloroplast genomes elucidates the relationships of the six economically important Brassica species comprising the triangle of U. Front. Plant Sci. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, J.G.; Hemingway, J.S.; Schofield, H.J. Contributions to a study of variation in Brassica juncea Coss. & Czern. J. Linn. Soc. 1963, 58, 435–447. [Google Scholar]
- Song, K.M.; Osborn, T.C.; Williams, P.H. Brassica taxonomy based on nuclear restriction fragment length polymorphism (RFLPs). Theor. Appl. Genet. 1988, 76, 593–600. [Google Scholar] [CrossRef]
- Chen, S.; Wan, Z.; Nelson, M.N.; Chauhan, J.S.; Redden, R.; Burton, W.A.; Lin, P.; Salisbury, P.A.; Fu, T.; Cowling, W.A. Evidence from genome-wide simple sequence repeat markers for a polyphyletic origin and secondary centers of genetic diversity of Brassica juncea in China and India. J. Hered. 2013, 104, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Banga, S.; Kumar, N.; Gupta, S.; Akhatar, J.; Banga, S.S. Polyphyletic origin of Brassica juncea with B. rapa and B. nigra (Brassicaceae) participating as cytoplasm donor parents in independent hybridization events. Am. J. Bot. 2014, 101, 1157–1166. [Google Scholar] [CrossRef]
- Vavilov, N.I. Phytogeographic basis of plant breeding. Chron. Bot. 1951, 13, 14–56. [Google Scholar]
- Saroj, R.; Soumya, S.L.; Singh, S.; Sankar, S.M.; Chaudhary, R.; Yashpal; Saini, N.; Vasudev, S.; Yadava, D.K. Unraveling the relationship between seed yield and yield-related traits in a diversity panel of Brassica juncea using multi-traits mixed model. Front. Plant. Sci. 2021, 12, 651936. [Google Scholar] [CrossRef]
- Nanjundan, J.; Aravind, J.; Radhamani, J.; Singh, K.H.; Kumar, A.; Thakur, A.K.; Singh, K.; Meena, K.N.; Tyagi, R.K.; Singh, D. Development of Indian mustard [Brassica juncea (L.) Czern.] core collection based on agro-morphological traits. Genet. Resour. Crop Evol. 2022, 69, 145–162. [Google Scholar] [CrossRef]
- Jain, A.; Bhatia, S.; Banga, S.S.; Prakash, S.; Lakshmikumaran, M. Potential use of random amplified polymorphic DNA (RAPD) technique to study the genetic diversity in Indian mustard (Brassica juncea) and its relationship to heterosis. Theor. Appl. Genet. 1994, 88, 116–122. [Google Scholar] [CrossRef]
- Rabbani, M.A.; Iwabuchi, A.; Murakami, Y.; Suzuki, T.; Takayanagi, K. Genetic diversity in mustard (Brassica juncea L.) germplasm from Pakistan as determined by RAPDs. Euphytica 1998, 103, 235–242. [Google Scholar] [CrossRef]
- Srivastava, A.; Gupta, V.; Pental, D.; Pradhan, A.K. AFLP-based genetic diversity assessment amongst agronomically important natural and some newly synthesized lines of Brassica juncea. Theor. Appl. Genet. 2001, 102, 193–199. [Google Scholar] [CrossRef]
- Qi, X.H.; Yang, J.H.; Zhang, M.F. AFLP-based genetic diversity assessment among Chinese vegetable mustards (Brassica juncea (L.) Czern.). Genet. Resour. Crop Evol. 2008, 55, 705–711. [Google Scholar] [CrossRef]
- Wu, X.-M.; Chen, B.-Y.; Lu, G.; Wang, H.-Z.; Xu, K.; Guizhan, G.; Song, Y. Genetic diversity in oil and vegetable mustard (Brassica juncea) landraces revealed by SRAP markers. Genet. Resour. Crop Evol. 2009, 56, 1011–1022. [Google Scholar] [CrossRef]
- Liu, H.H.; Hu, S.W. Genetic diversity of twenty-three Brassica juncea accessions based on isozyme markers. Acta. Agric. Boreali-occidentalis Sin. 2010, 19, 82–85, 106. [Google Scholar]
- Yao, Q.-L.; Chen, F.-B.; Fang, P.; Zhou, G.-F.; Fan, Y.-H.; Zhang, Z.-R. Genetic diversity of Chinese vegetable mustard (Brassica juncea Coss) landraces based on SSR data. Biochem. Syst. Ecol. 2012, 45, 41–48. [Google Scholar] [CrossRef]
- Singh, V.K.; Avtar, R.; Mahavir, K.N.; Kumar, R. Assessment of genetic diversity and population structure in Indian mustard (Brassica juncea L.) using SSR markers. J. Environ. Biol. 2021, 42, 396–405. [Google Scholar]
- Khan, M.A.; Rabbani, M.A.; Munir, M.; Ajmal, S.K.; Malik, M.A. Assessment of genetic variation with in Indian mustard (Brassica juncea) germplasm using random amplified polymorphic DNA markers. J. Integr. Plant Biol. 2008, 50, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Li, R.G.; Zhu, L.; Wu, N.F.; Fan, Y.L.; Wu, X.M.; Qian, X.Z. Genetic diversity among oilseed cultivars of Brassica juncea Czern. et Coss in China. Biotechnol. Bull. 1997, 5, 26–31. [Google Scholar]
- An, X.H.; Chen, B.Y.; Fu, T.D.; Liu, H.L. Genetic diversity of Chinese landraces in Brassica napus was analysed by RAPD markers. J. Huazhong Agric. Univ. 1999, 18, 524–527. [Google Scholar]
- Pu, X.B.; Wang, M.L.; Li, L. Genetic diversity analysis of Brassica juncea L. and races in southwest China. Sci. Agric. Sin. 2007, 40, 1610–1621. [Google Scholar]
- Xu, A.X.; Ma, C.Z.; Xiao, E.S. Genetic diversity of Brassica juncea from western China. Acta. Agron Sin. 2008, 34, 754–763. [Google Scholar] [CrossRef]
- Song, W.L.; Xu, K.; Li, F.; Chen, B.; Cai, M.; Wu, J.; Wu, X. Genetic diversity of Brassica juncea from Tibet autonomous region of China. Chinese J. Oil Crop Sci. 2013, 35, 153–161. [Google Scholar]
- Qiao, A.M.; Liu, P.Y.; Lei, J.J. RAPD analysis of sixteen varieties of mustard. Acta. Bot. Sin. 1998, 40, 915–921. [Google Scholar]
- Fu, J.; Zhang, M.F.; Qi, X.H. Genetic diversity of traditional Chinese mustard crops Brassica juncea as revealed by phenotypic differences and RAPD markers. Genet. Resour. Crop Evol. 2006, 53, 1513–1519. [Google Scholar] [CrossRef]
- Song, M.; Liu, T.; Tang, Q.; Wang, Z.; Yang, Y.; Zhang, M. Genetic diversity analysis of mustard germplasm based on RAPD and ISSR. Acta. Hortic. Sin. 2009, 36, 835–842. [Google Scholar]
- Li, N.; Si, J.; Ren, X.; Song, H.; Li, C. SRAP analysis of mustard germplasm genetic diversity and genetic relationship. China Veg. 2014, 5, 26–33. [Google Scholar]
- Fang, P.; Chen, F.-B.; Yao, Q.-L.; Yang, K.-C.; Zhou, G.-F.; Fan, Y.-H.; Zhang, Z.-R.; Shen, J.-J.; Zhang, H. Analysis of genetic diversity in the tuber mustard (Brassica juncea var. tumida Tsen et Lee) in the Yangtze river basin of China. Genet. Resour. Crop Evol. 2013, 60, 129–143. [Google Scholar] [CrossRef]
- Sharma, D.; Nanjundan, J.; Singh, L.; Singh, S.P.; Parmar, N.; Kumar, M.S.S.; Singh, K.H.; Mishra, A.K.; Singh, R.; Verma, K.S.; et al. Genetic diversity in leafy mustard (Brassica juncea var. rugosa) as revealed by agro-morphological traits and SSR markers. Physiol Mol. Biol. Plants 2020, 26, 2005–2018. [Google Scholar] [CrossRef]
- Zhao, H.X.; Li, Z.J.; Hu, S.W.; Sun, G.L.; Chang, J.J.; Zhang, Z.H. Identification of cytoplasm types in rapeseed (Brassica napus L.) accessions by a multiplex PCR assay. Theor. Appl. Genet. 2010, 121, 643–650. [Google Scholar] [CrossRef]
- Heng, S.; Chen, F.; Wei, C.; Hu, K.; Yang, Z.; Wen, J.; Yi, B.; Ma, C.; Tu, J.; Si, P.; et al. Identification of different cytoplasms based on newly developed mitotype-specific markers for marker-assisted selection breeding in Brassica napus L. Plant Cell Rep. 2017, 36, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, H.; Channa, S.A.; Zhang, Y.; Guo, Y.; Klima, M.; Yu, F.; Hu, S. Genetic diversity in Chinese and exotic Brassica rapa L. accessions revealed by SSR and SRAP markers. Braz. J. Bot. 2017, 40, 973–982. [Google Scholar] [CrossRef]
- Song, W.N.; Langridge, P. Identification and mapping of polymorphisms in cereals based on the polymerase chain reaction. Theor. Appl. Genet. 1991, 82, 209–216. [Google Scholar]
- Bonhomme, S.; Budar, F.; Lancelin, D.; Small, I.; Defrance, M.-C.; Pelletier, G. Sequence and transcript analysis of the Nco2.5 ogura-specific fragment correlated with cytoplasmic male sterility in Brassica cybrids. Mol. Gen. Genet. 1992, 235, 340–348. [Google Scholar] [CrossRef]
- Grelon, M.; Budar, F.; Bonhomme, S.; Pelletier, G. Ogura cytoplasmic male-sterility (CMS)-associated orf138 is translated into a mitochondrial membrane polypeptide in male-sterility Brassica cybrids. Mol. Gen. Genet. 1994, 243, 540–547. [Google Scholar] [CrossRef]
- L’Homme, Y.; Stahl, R.J.; Li, X.-Q.; Hameed, A.; Brown, G.G. Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS-associated orf224 gene. Curr. Genet. 1997, 31, 325–335. [Google Scholar] [CrossRef]
- Singh, M.; Brown, G.G. Suppression of cytoplasmic male sterility by nuclear genes alters expression of a novel mitochondrial gene region. Plant cell 1991, 3, 1349–1362. [Google Scholar]
- L’Homme, Y.; Brown, G.G. Organizational differences between cytoplasmic male-sterile and male-fertile Brassica mitochondrial genomes are confined to a single transposed locus. Nucleic Acids Res. 1993, 21, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- Jing, B.; Heng, S.; Tong, D.; Wan, Z.; Fu, T.; Tu, J.; Ma, C.; Yi, B.; Wen, J.; Shen, J. A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J. Exp. Bot. 2012, 63, 1285–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatono, S.; Nishimura, K.; Murakami, Y.; Tsujimura, M.; Yamagishi, H. Complete mitochondrial genome sequences of Brassica rapa (Chinese cabbage and mizuna), and intraspecific differentiation of cytoplasm in B. rapa and Brassica juncea. Breed. Sci. 2017, 67, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokal, R.R.; Michener, C.D. A statistical method for evaluating systematic relationships. Sci. Bull. 1958, 28, 1409–1438. [Google Scholar]
- Tang, Q.Y.; Zhang, C.X. Data processing system (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Li, Y.L.; Han, G.M.; He, S.E.; Zhang, Z.J. A new strategy for construction of phylogenetic tree based on DNA molecular marker data. China J. Bioinform. 2008, 4, 168–170. [Google Scholar]
- Rohlf, F.J. NTSYS-pc, Numerical Taxonomy and Multivariate Analysis System, version 2.02g; Exeter Software: New York, NY, USA, 1998. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under linux and windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE Version 1.32: Microsoft Window-Based Freeware for Population Genetic Analysis; Molecular Biology and Biotechnology Center, University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Zhang, R.J.; Hu, S.W.; Yan, J.Q.; Sun, G.L. Cytoplasmic diversity in Brassica rapa L. investigated by mitochondrial markers. Genet. Resour. Crop Evol. 2013, 60, 967–974. [Google Scholar] [CrossRef]
- You, L.; Yang, L.; Liu, F.Y.; Kang, L.; Chen, H.; Yang, B.; Yang, Q.; Liu, Z.S. Assembly and marker analysis of mitochondrial genomes provide insights into origin, evolution and spread of Brassica juncea (L.) Czern. et Coss. Crop J. 2022, 10, 611–616. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oilseed Mustard | Vegetable Mustard | Other Brassicaceae | |
|---|---|---|---|
| Oilseed mustard | 59.62041 | ||
| Vegetable mustard | 32.13425 | 52.81462 | |
| Other Brassicaceae | 31.54465 | 35.97280 | 105.84762 |
| Subspecies a | Variety b | Mitochondrial Type c | ||
|---|---|---|---|---|
| MT1 | MT2 | MT3 | ||
| juncea | gracilis | 14 | 20 | 16 |
| integrifolia | rugosa | 4 | 0 | 9 |
| crassicauli | 0 | 0 | 1 | |
| gemmifera | 0 | 0 | 2 | |
| involuta | 0 | 0 | 5 | |
| strumata | 0 | 0 | 3 | |
| multiceps | 0 | 0 | 2 | |
| napiformis | megarrhiza | 4 | 0 | 0 |
| Tumida | tumida | 0 | 0 | 4 |
| Total | 22 | 20 | 42 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Yu, H.; Gao, L.; Wang, J.; Dong, H.; Guo, Y.; Hu, S. Genetic Diversity in Oilseed and Vegetable Mustard (Brassica juncea L.) Accessions Revealed by Nuclear and Mitochondrial Molecular Markers. Agronomy 2023, 13, 919. https://doi.org/10.3390/agronomy13030919
Zhang D, Yu H, Gao L, Wang J, Dong H, Guo Y, Hu S. Genetic Diversity in Oilseed and Vegetable Mustard (Brassica juncea L.) Accessions Revealed by Nuclear and Mitochondrial Molecular Markers. Agronomy. 2023; 13(3):919. https://doi.org/10.3390/agronomy13030919
Chicago/Turabian StyleZhang, Dongsuo, Haibo Yu, Lianliang Gao, Jing Wang, Hui Dong, Yuan Guo, and Shengwu Hu. 2023. "Genetic Diversity in Oilseed and Vegetable Mustard (Brassica juncea L.) Accessions Revealed by Nuclear and Mitochondrial Molecular Markers" Agronomy 13, no. 3: 919. https://doi.org/10.3390/agronomy13030919
APA StyleZhang, D., Yu, H., Gao, L., Wang, J., Dong, H., Guo, Y., & Hu, S. (2023). Genetic Diversity in Oilseed and Vegetable Mustard (Brassica juncea L.) Accessions Revealed by Nuclear and Mitochondrial Molecular Markers. Agronomy, 13(3), 919. https://doi.org/10.3390/agronomy13030919