





Inflorescence Trait Diversity and Genotypic Differentiation as Influenced by the Environment in Elymus nutans Griseb. from Qinghai–Tibet Plateau


Abstract
:1. Introduction
2. Materials and Methods
2.1. Transcriptome Sequencing and EST-SSR Design
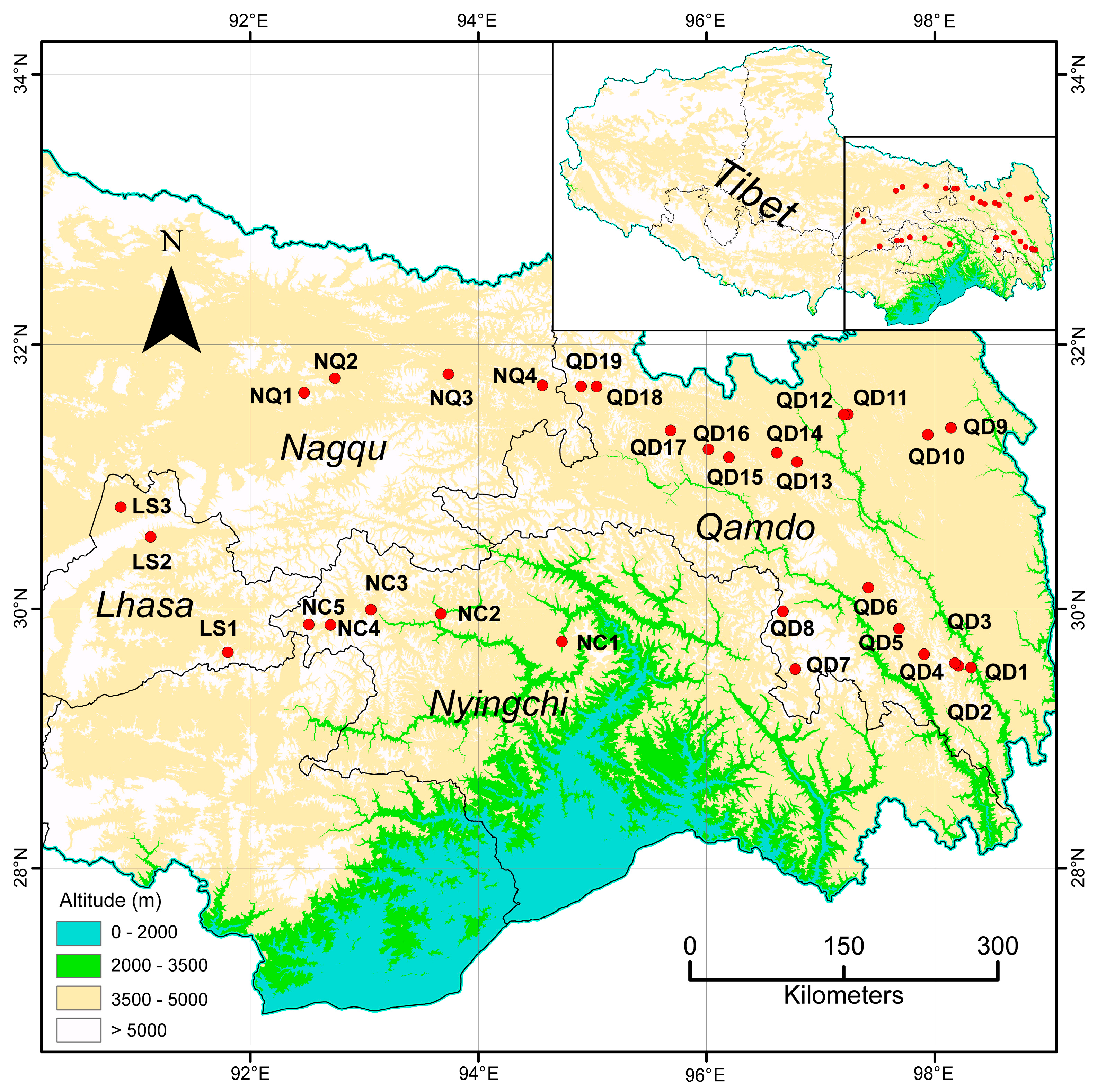
2.2. Plant Materials and DNA Extraction
2.3. Genetic Diversity Analysis
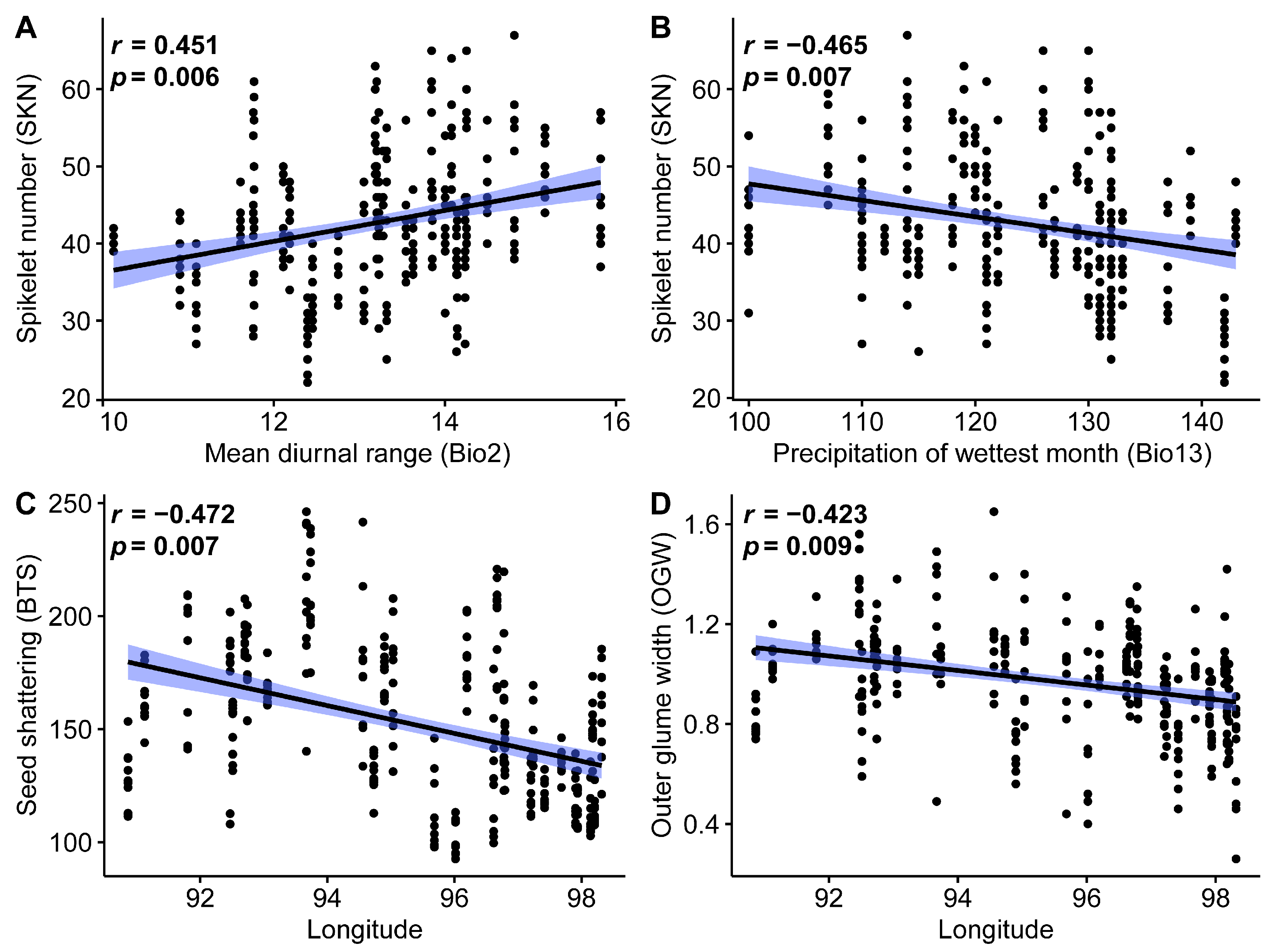
2.4. Phenotypic Variation and Association Analysis
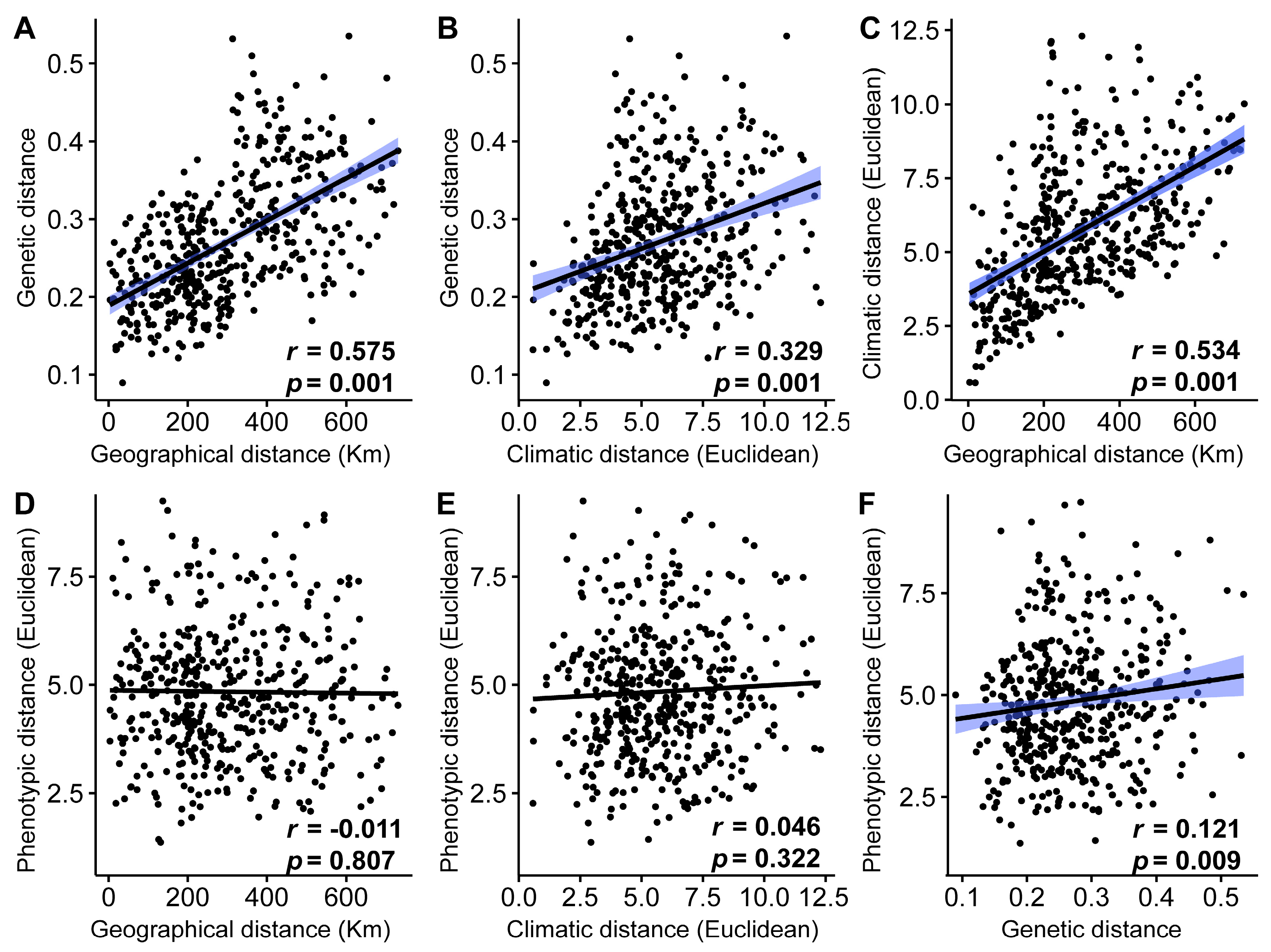
2.5. Genetic and Phenotypic Differentiation Analysis
3. Results
3.1. De Novo Assembly and Distribution of SSR Repeats
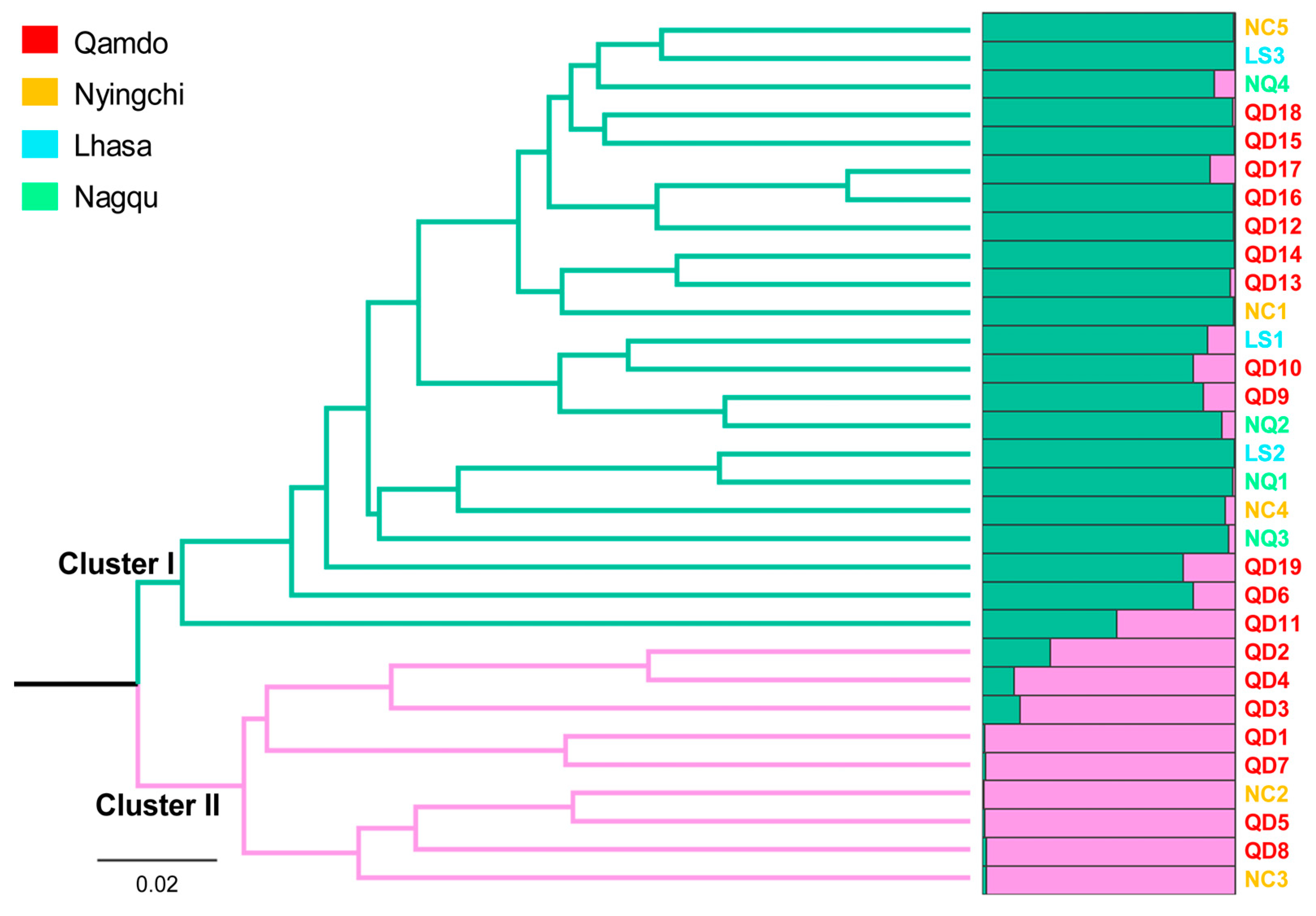
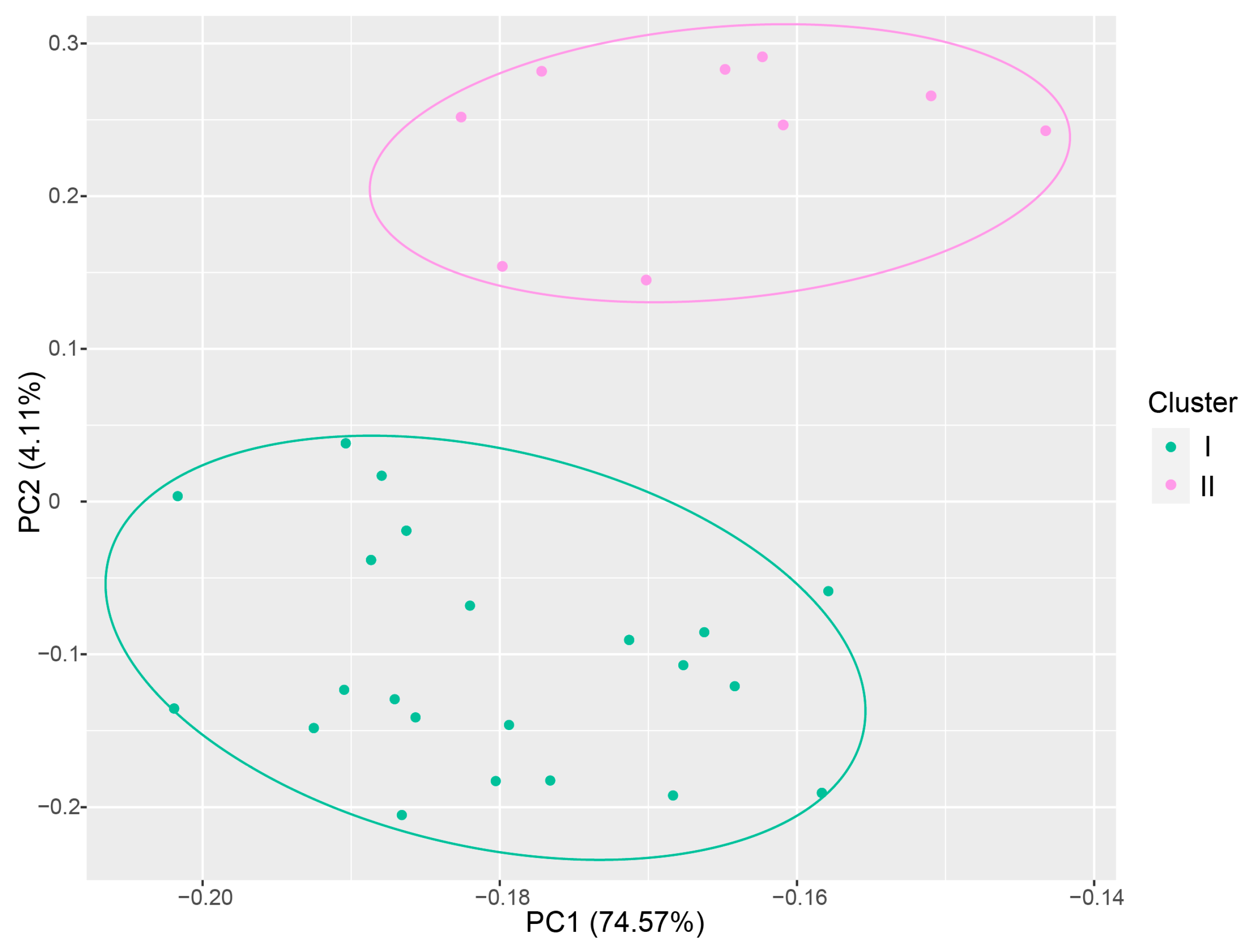
3.2. Genetic Relationship Analysis
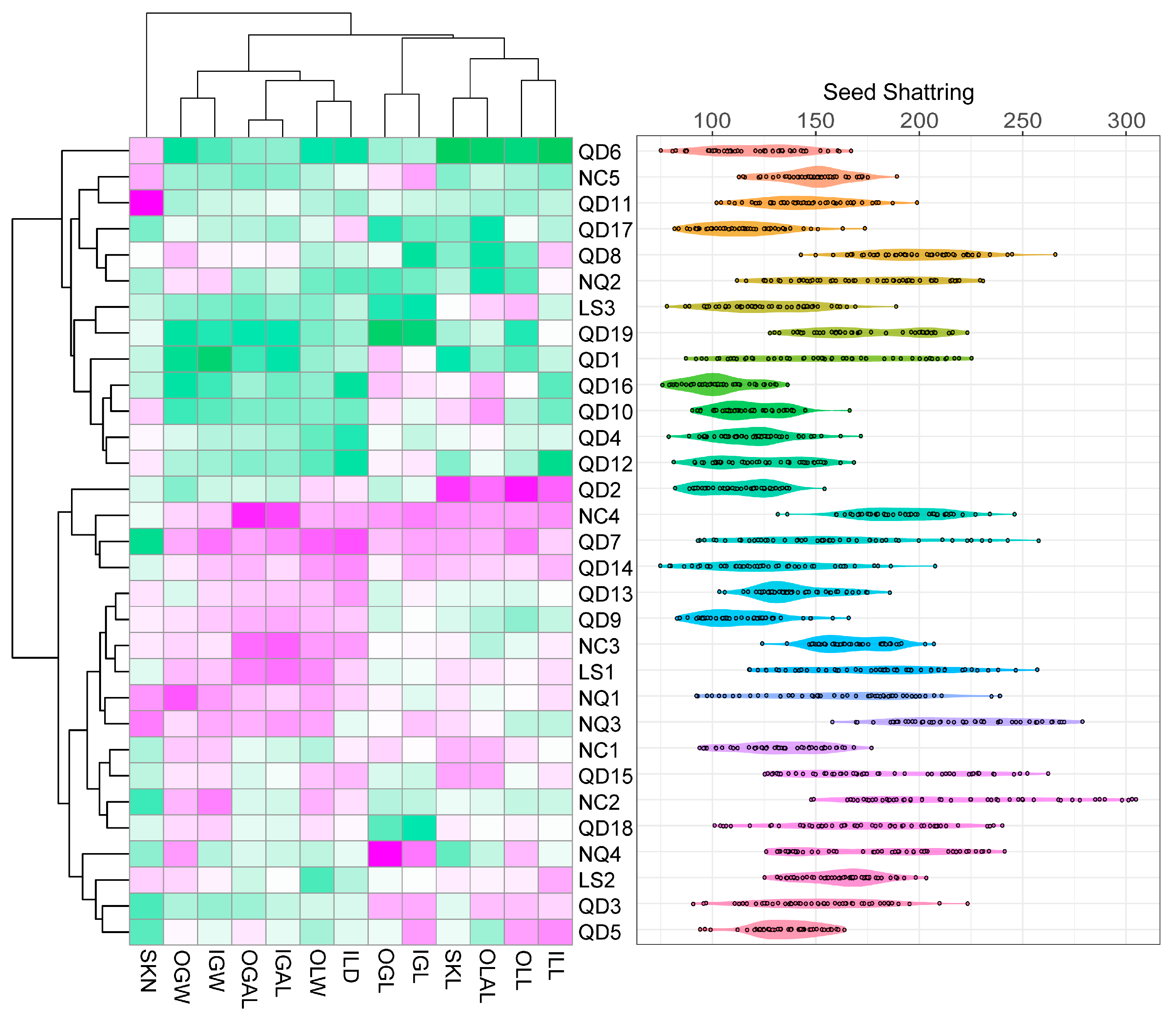
3.3. Inflorescence Phenotypic Characteristics, Shattering, and Tassel Analysis
3.4. Associations between Genetic, Phylogenetic, Geographical, and Climatic Distance
4. Discussion
4.1. Characterization of E. nutans Transcriptome and EST-SSR Distribution
4.2. SSR Validation and Genetic Analysis
4.3. Phenotypic Variation and Association Analysis
4.4. How Genotypic and Phenotypic Differentiation Is Affected by Geography and Environment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Löve, A. Conspectuse of Triticeae. Feddes Repert. 1984, 95, 425–524. [Google Scholar]
- Lu, B.R. Meiotic studies of Elymus nutans and Elymus jacquemontii (Poaceae, Triticeae) and their hybrids with Pseudoroegneria spicata and seventeen Elymus species. Plant Syst. Evol. 1993, 186, 193–211. [Google Scholar] [CrossRef]
- Yen, C.; Yang, J.L.; Baum, B. Synopsis of Leymus Hochst. (Triticeae: Poaceae). J. Syst. Evol. 2009, 47, 67–86. [Google Scholar] [CrossRef]
- Ferdinandez, Y.; Somers, D.J.; Coulman, B.E. Estimating the genetic relationship of hybrid bromegrass to smooth bromegrass and meadow bromegrass using RAPD markers. Plant Breed. 2010, 120, 149–153. [Google Scholar] [CrossRef]
- Wang, R.R.C.; von Bothmer, R.; Dvorak, J.; Fedak, G.; Linde-Laursen, I.; Muramatsu, M. Genome symbols in the Triticeae (Poaceae). In Proceedings of 2nd International Triticeae Symposium; Wang, R.R.C., Ed.; Utah State University Publications on Design and Production: Logan, UT, USA, 1994; pp. 29–34. [Google Scholar]
- Bor, N.L. The Grasses of Burma, Ceylon, India and Pakistan; Pergamon Press: Oxford, UK, 1960. [Google Scholar]
- Clayton, W.D.; Harman, K.T.; Williamson, H. GrassBase–The Online World Grass Flora. 2006. Available online: http://www.kew.org/data/grasses-db.html (accessed on 15 May 2020.).
- Chen, S.L.; Zhu, G.H. Elymus L. Flora of China (Poaceae); Science Press: Beijing, China; Missouri Botanical Garden: Louis, MO, USA, 2006. [Google Scholar]
- Sun, J.; Fu, B.J.; Zhao, W.W.; Liu, S.L.; Liu, G.H.; Zhou, H.K.; Shao, X.Q.; Chen, Y.C.; Zhang, Y. Optimizing grazing exclusion practices to achieve goal 15 of the sustainable development goals in the Tibetan plateau. Sci. Bull. 2021, 66, 1493–1496. [Google Scholar] [CrossRef]
- Lei, Y.T.; Zhao, Y.Y.; Yu, F.; Li, Y.; Dou, Q.W. Development and characterization of 53 polymorphic genomic-SSR markers in Siberian wildrye (Elymus sibiricus L.). Conserv. Genet. Resour. 2014, 6, 861–864. [Google Scholar] [CrossRef]
- Shi, N.; Naudiyal, N.; Wang, J.N.; Gaire, N.P.; Wu, Y.; Wei, Y.Q.; He, J.L.; Wang, C.Y. Assessing the impact of climate change on potential distribution of Meconopsis punicea and its influence on ecosystem services supply in the southeastern margin of Qinghai-Tibet plateau. Front. Plant Sci. 2022, 12, 830119. [Google Scholar] [CrossRef]
- Kou, Y.X.; Wu, Y.X.; Jia, D.R.; Li, Z.H.; Wang, Y.J. Rang expansion, genetic differentiation, and phenotypic adaption of Hippophaë neurocarpa (Elaeagnaceae) on the Qinghai-Tibet Plateau. J. Syst. Evol. 2014, 52, 303–312. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Ma, X.; Huang, L. Assessment of genetic diversity and differentiation of Elymus nutans indigenous to Qinghai-Tibet Plateau using simple sequence repeats markers. Can. J. Plant. Sci. 2013, 93, 1089–1096. [Google Scholar] [CrossRef]
- Chen, S.Y.; Ma, X.; Zhang, X.Q.; Chen, Z.H. Genetic variation and geographical divergence in Elymus nutans Griseb. (Poaceae: Triticeae) from West China. Biochem. Syst. Ecol. 2009, 37, 716–722. [Google Scholar] [CrossRef]
- Miao, J.M.; Zhang, X.Q.; Chen, S.Y.; Ma, X.; Chen, Z.H.; Zhong, J.C.; Bai, S.Q. Gliadin analysis of Elymus nutans Griseb. from the Qinghai-Tibetan Plateau and Xinjiang, China. Grassl. Sci. 2011, 57, 127–134. [Google Scholar] [CrossRef]
- Yan, X.B.; Guo, Y.X.; Liu, F.Y.; Zhao, C.; Liu, Q.L.; Lu, B.R. Population structure affected by excess gene flow in self-pollinating Elymus nutans and E. burchan-buddae (Triticeae: Poaceae). Popul. Ecol. 2010, 52, 233–241. [Google Scholar] [CrossRef]
- Karan, M.; Evans, D.S.; Reilly, D.; Schulte, K.; Wright, C.; Innes, D.; Holton, T.A.; Nikles, D.G.; Dickinson, G.R. Rapid microsatellite marker development for African mahogany (Khaya senegalensis, Meliaceae) using next-generation sequencing and assessment of its intra-specific genetic diversity. Mol. Ecol. Resour. 2012, 12, 344–353. [Google Scholar] [CrossRef]
- Soler, S.; Gramazio, P.; Figàs, M.R.; Vilanova, S.; Rosa, E.; Llosa, E.R.; Borras, D.; Plazas, M.; Prohens, J. Genetic structure of Cannabis sativa var. indica cultivars based on genomic SSR (gSSR) markers: Implications for breeding and germplasm management. Ind. Crops Prod. 2017, 104, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xie, W.; Zhao, Y.; Zhang, J.; Wang, N.; Ntakirutimana, F.; Yan, J.J.; Wang, Y.R. EST-SSR marker development based on RNA-sequencing of E. sibiricus and its application for phylogenetic relationships analysis of seventeen Elymus species. BMC Plant Biol. 2019, 19, 235. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Yang, J.; Xiong, Y.; Zhao, J.; Liu, L.; Liu, W.; Sha, L.N.; Zhou, J.Q.; You, M.H.; Li, D.X.; et al. Full-length transcriptome sequencing analysis and characterization, development and validation of microsatellite markers in Kengyilia melanthera. Front. Plant Sci. 2022, 13, 959042. [Google Scholar] [CrossRef]
- Sun, M.; Dong, Z.X.; Yang, J.; Wu, W.D.; Zhang, C.L.; Zhang, J.B.; Zhao, J.M.; Xiong, Y.; Jia, S.G.; Ma, X. Transcriptomic resources for prairie grass (Bromus catharticus): Expressed transcripts, tissue-specific genes, and identification and validation of EST-SSR markers. BMC Plant Biol. 2021, 21, 264. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, Q.; Ma, L.C.; Xie, W.G.; Wang, Y.R.; Hu, X.W.; Liu, Z.P. Novel polymorphic expressed-sequence tag–simple-sequence repeat markers in Campeiostachys nutans for genetic diversity analyses. Crop Sci. 2015, 55, 2712–2718. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, J.; Zhang, Z.; Xie, W. Elymus nutans genes for seed shattering and candidate gene-derived EST-SSR markers for germplasm evaluation. BMC Plant Biol. 2019, 19, 102. [Google Scholar] [CrossRef] [Green Version]
- Kamali, M.; Samsampour, D.; Bagheri, A.; Mehrafarin, A.; Homaei, A. Association analysis and evaluation of genetic diversity of Teucrium stocksianum Boiss. populations using ISSR markers. Genet. Resour. Crop Evol. 2023, 70, 691–709. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, Y.; Zeng, B.; Yin, G.; Zhang, X.; Ji, Y.; Huang, L.K.; Jiang, X.M.; Liu, X.C.; Peng, Y.; et al. Genetic diversity and association of EST-SSR and SCoT markers with rust traits in Orchardgrass (Dactylis glomerata L.). Molecules 2016, 21, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, S.; Sajjad, M.; Khan, S.H.; Shahid, M.; Zubair, M.; Awan, F.S.; Khan, A.I.; Mubarak, M.S.; Tahir, A.; Umer, M.; et al. Genome-wide association analysis for stripe rust resistance in spring wheat (Triticum aestivum L.) germplasm. J. Integr. Agric. 2020, 19, 2035–2043. [Google Scholar] [CrossRef]
- Bourne, E.C.; Bocedi, G.; Travis, J.M.J.; Pakeman, R.J.; Brooker, R.W.; Schiffers, K. Between migration load and evolutionary rescue: Dispersal, adaptation and the response of spatially structured populations to environmental change. Proc. R. Soc. B Biol. Sci. 2014, 281, 20132795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.J.; Summers, K. Genetic structure is correlated with phenotypic divergence rather than geographic isolation in the highly polymorphic strawberry poison-dart frog. Mol. Ecol. 2010, 19, 447–458. [Google Scholar] [CrossRef]
- Noguerales, V.; Cordero, P.J.; Ortego, J. Hierarchical genetic structure shaped by topography in a narrow-endemic montane grasshopper. BMC Evol. Biol. 2016, 16, 96. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Isolation by distance under diverse systems of mating. Genetics 1945, 30, 571–572. [Google Scholar] [CrossRef]
- Lichstein, J. Multiple regression on distance matrices: A multivariate spatial analysis tool. Plant Ecol. 2007, 188, 117–131. [Google Scholar] [CrossRef]
- Li, J.; Ma, S.; Jiang, K.; Zhang, C.; Liu, W.; Chen, S. Drivers of population divergence and genetic variation in Elymus breviaristatus (Keng) Keng f. (Poaceae: Triticeae), an endemic perennial herb of the Qinghai-Tibet plateau. Front. Ecol. Evol. 2022, 10, 1068739. [Google Scholar] [CrossRef]
- Marchese, C. Biodiversity hotspots: A shortcut for a more complicated concept. Glob. Ecol. Conserv. 2015, 3, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Yeh, F.C.; Boyle, T.J.B. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg. J. Bot. 1997, 129, 157–163. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes. 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, R.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Liu, K.; Xu, H.; Liu, G.; Guan, P.; Zhou, X.; Peng, H.; Yao, Y.; Ni, Z.; Sun, Q.; Du, J. QTL mapping of fag leaf-related traits in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2018, 131, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.B.; Guo, Y.X.; Zhou, H.; Lu, B.R.; Wang, K. Genetic patterns of ten Elymus species from the Tibetan and Inner Mongolian plateaus of China. Grass Forage Sci. 2006, 61, 398–404. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, W.; Zhang, J.; Zhao, X.; Zhao, Y.; Wang, Y. Phenotype- and SSR-based estimates of genetic variation between and within two important Elymus species in western and northern China. Genes 2018, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Jun, T.H.; Van, K.; Kim, M.Y.; Lee, S.H.; Walker, D.R. Association analysis using SSR markers to find QTL for seed protein content in soybean. Euphytica 2008, 162, 179–185. [Google Scholar] [CrossRef]
- Azmach, G.; Gedil, M.; Menkir, A.; Spillane, C. Marker-trait association analysis of functional gene markers for provitamin A levels across diverse tropical yellow maize inbred lines. BMC Plant Biol. 2013, 13, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, G.L.; Liu, S.; Hall, M.D.; Brooks, W.S.; Chao, S.; Muehlbauer, G.J.; Baik, B.K.; Steffenson, B.; Griffey, C.A. Marker-trait associations in Virginia Tech winter barley identified using genome-wide mapping. Theor. Appl. Genet. 2013, 126, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Castilla, A.R.; Méndez-Vigo, B.; Marcer, A.; Martínez-Minaya, J.; Conesa, D.; Picó, F.X.; Alonso-Blanco, C. Ecological, genetic and evolutionary drivers of regional genetic differentiation in Arabidopsis thaliana. BMC Evol. Biol. 2020, 20, 71. [Google Scholar] [CrossRef]
- Sexton, J.P.; Hangartner, S.B.; Hoffmann, A.A. Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution 2014, 68, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ghalambor, C.K.; McKay, J.K.; Carroll, S.P.; Reznick, D.N. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Funct. Ecol. 2007, 21, 394–407. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range (bp) | Transcripts | Unigene |
|---|---|---|
| 200–500 | 205,785 | 88,679 |
| 500–1000 | 123,056 | 111,828 |
| 1000–2000 | 83,850 | 82,173 |
| >2000 | 21,973 | 21,874 |
| Total Number | 434,637 | 304,554 |
| Total Length | 327,236,286 | 283,614,840 |
| N50 Length | 1062 | 1194 |
| N90 Length | 337 | 478 |
| Mean Length | 753 | 931 |
| Number of Repeat Units | Mono- | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Percentage (%) |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 12,830 | 860 | 198 | 79 | 13,967 | 28.82 |
| 6 | 0 | 3733 | 4731 | 272 | 11 | 23 | 8770 | 18.10 |
| 7 | 0 | 1784 | 2002 | 31 | 9 | 7 | 3833 | 7.91 |
| 8 | 0 | 1131 | 870 | 17 | 2 | 7 | 2027 | 4.18 |
| 9 | 0 | 742 | 158 | 6 | 0 | 1 | 907 | 1.87 |
| 10 | 6017 | 501 | 112 | 4 | 0 | 0 | 6634 | 13.69 |
| 11 | 2665 | 476 | 63 | 0 | 0 | 0 | 3204 | 6.61 |
| 12 | 1501 | 511 | 39 | 0 | 0 | 0 | 2051 | 4.23 |
| 13 | 821 | 219 | 19 | 1 | 0 | 0 | 1060 | 2.19 |
| 14 | 707 | 210 | 16 | 0 | 0 | 0 | 933 | 1.93 |
| 15 | 506 | 195 | 9 | 0 | 0 | 0 | 710 | 1.47 |
| >15 | 3550 | 793 | 18 | 0 | 0 | 0 | 4361 | 9.00 |
| Total | 15,767 | 10,295 | 20,867 | 1191 | 220 | 117 | 48,457 | 100.00 |
| Percentage (%) | 32.54 | 21.25 | 43.06 | 2.46 | 0.45 | 0.24 | 100.00 |
| Quantitative Traits | Mean | Max | Min | SD | Variation Coefficient (%) | Genetic Diversity Index |
|---|---|---|---|---|---|---|
| SKN | 42.99 | 68.80 | 27.70 | 7.20 | 16.74 | 1.94 |
| SKL (mm) | 27.97 | 40.16 | 14.16 | 4.92 | 17.61 | 1.88 |
| OGL (mm) | 4.62 | 7.37 | 3.04 | 0.74 | 16.03 | 1.77 |
| OGW (mm) | 0.95 | 1.30 | 0.62 | 0.16 | 16.94 | 1.82 |
| OGAL (mm) | 1.51 | 3.07 | 0.30 | 0.54 | 35.50 | 1.94 |
| IGL (mm) | 3.66 | 5.90 | 1.53 | 0.69 | 19.01 | 1.92 |
| IGW (mm) | 0.79 | 1.14 | 0.35 | 0.17 | 21.76 | 2.03 |
| IGAL (mm) | 1.23 | 2.37 | 0.33 | 0.45 | 36.48 | 2.03 |
| OLL (mm) | 9.42 | 11.46 | 7.67 | 0.69 | 7.34 | 1.94 |
| OLW (mm) | 2.18 | 3.10 | 1.60 | 0.43 | 19.73 | 1.63 |
| OLAL (mm) | 14.55 | 21.27 | 5.43 | 3.55 | 24.37 | 1.97 |
| ILL (mm) | 8.75 | 10.16 | 6.77 | 0.71 | 8.17 | 1.96 |
| ILW (mm) | 1.81 | 2.37 | 1.35 | 0.24 | 13.15 | 2.04 |
| Trait | Marker | p | Marker_R2 | Trait | Marker | p | Marker_R2 |
|---|---|---|---|---|---|---|---|
| BTS | EN48-260 bp | 0.00305 | 0.28356 | OGL | EN57-183 bp | 0.00084 | 0.33773 |
| IGAL | EN5-256 bp | 0.00019 | 0.40221 | OGL | EN99-150 bp | 0.00153 | 0.31079 |
| IGAL | EN55-256 bp | 0.00368 | 0.26985 | OGW | EN5-256 bp | 0.00126 | 0.32361 |
| IGL | EN57-183 bp | 0.00975 | 0.21398 | OGW | EN58-243 bp | 0.00898 | 0.22701 |
| IGW | EN5-256 bp | 0.00018 | 0.40794 | OLAL | EN5-235 bp | 0.00079 | 0.32793 |
| IGW | EN58-243 bp | 0.00967 | 0.22345 | OLL | EN5-235 bp | 0.00583 | 0.23654 |
| ILL | EN35-207 bp | 0.00252 | 0.26241 | OLW | EN55-208 bp | 0.00888 | 0.22983 |
| ILL | EN91-239 bp | 0.00746 | 0.21351 | SKL | EN5-235 bp | 0.00293 | 0.27998 |
| OGAL | EN5-256 bp | 0.00063 | 0.35446 | SKN | EN90-228 bp | 0.00567 | 0.19378 |
| OGAL | EN55-208 bp | 0.00594 | 0.24807 | SKN | EN99-164 bp | 0.00611 | 0.19025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Tian, H.; Ji, W.; Zhang, C.; Chen, S. Inflorescence Trait Diversity and Genotypic Differentiation as Influenced by the Environment in Elymus nutans Griseb. from Qinghai–Tibet Plateau. Agronomy 2023, 13, 1004. https://doi.org/10.3390/agronomy13041004
Li J, Tian H, Ji W, Zhang C, Chen S. Inflorescence Trait Diversity and Genotypic Differentiation as Influenced by the Environment in Elymus nutans Griseb. from Qinghai–Tibet Plateau. Agronomy. 2023; 13(4):1004. https://doi.org/10.3390/agronomy13041004
Chicago/Turabian StyleLi, Jin, Haoqi Tian, Wenqin Ji, Changbing Zhang, and Shiyong Chen. 2023. "Inflorescence Trait Diversity and Genotypic Differentiation as Influenced by the Environment in Elymus nutans Griseb. from Qinghai–Tibet Plateau" Agronomy 13, no. 4: 1004. https://doi.org/10.3390/agronomy13041004
APA StyleLi, J., Tian, H., Ji, W., Zhang, C., & Chen, S. (2023). Inflorescence Trait Diversity and Genotypic Differentiation as Influenced by the Environment in Elymus nutans Griseb. from Qinghai–Tibet Plateau. Agronomy, 13(4), 1004. https://doi.org/10.3390/agronomy13041004