Autophagy During Vertebrate Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
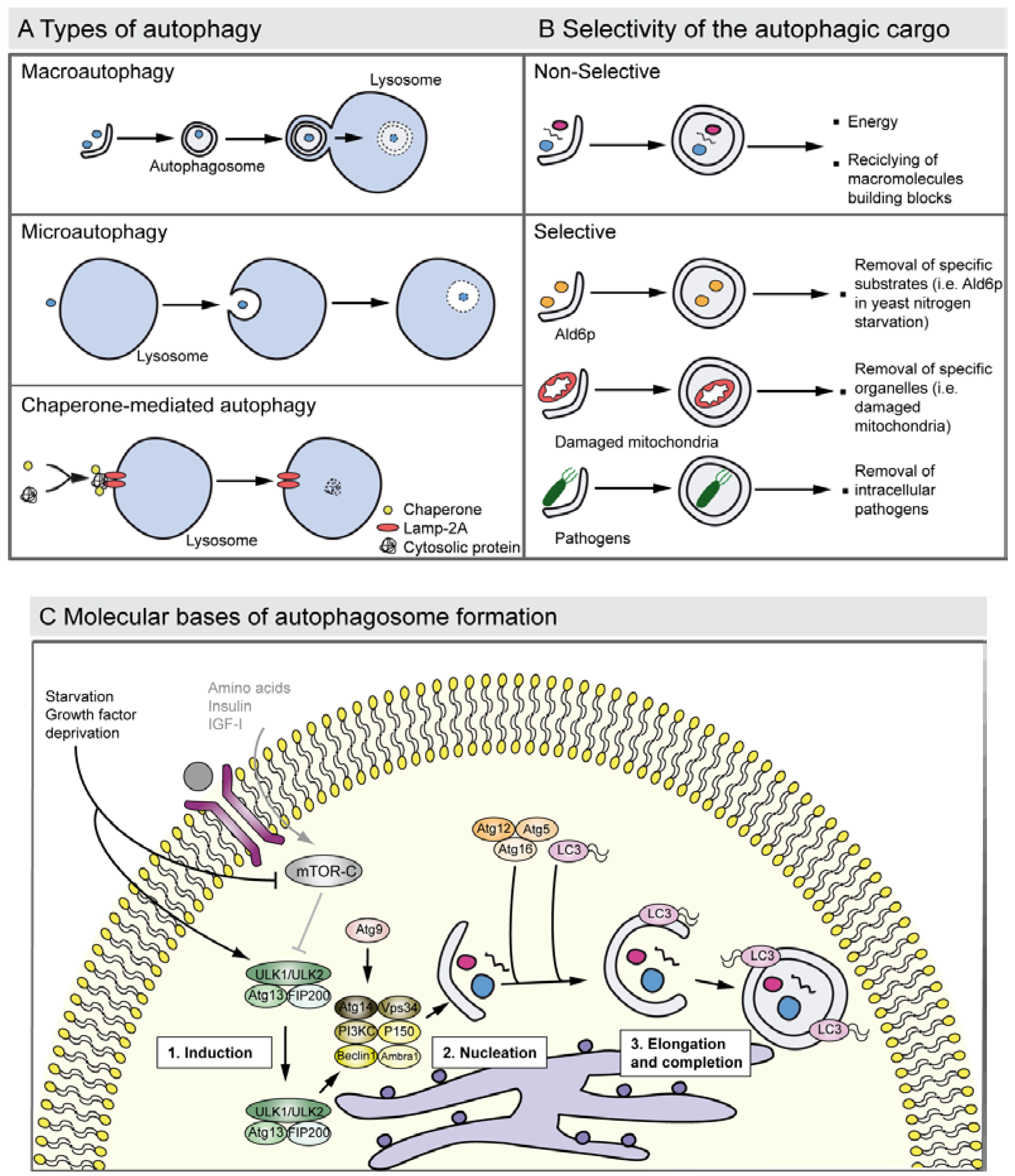
:1. Introduction


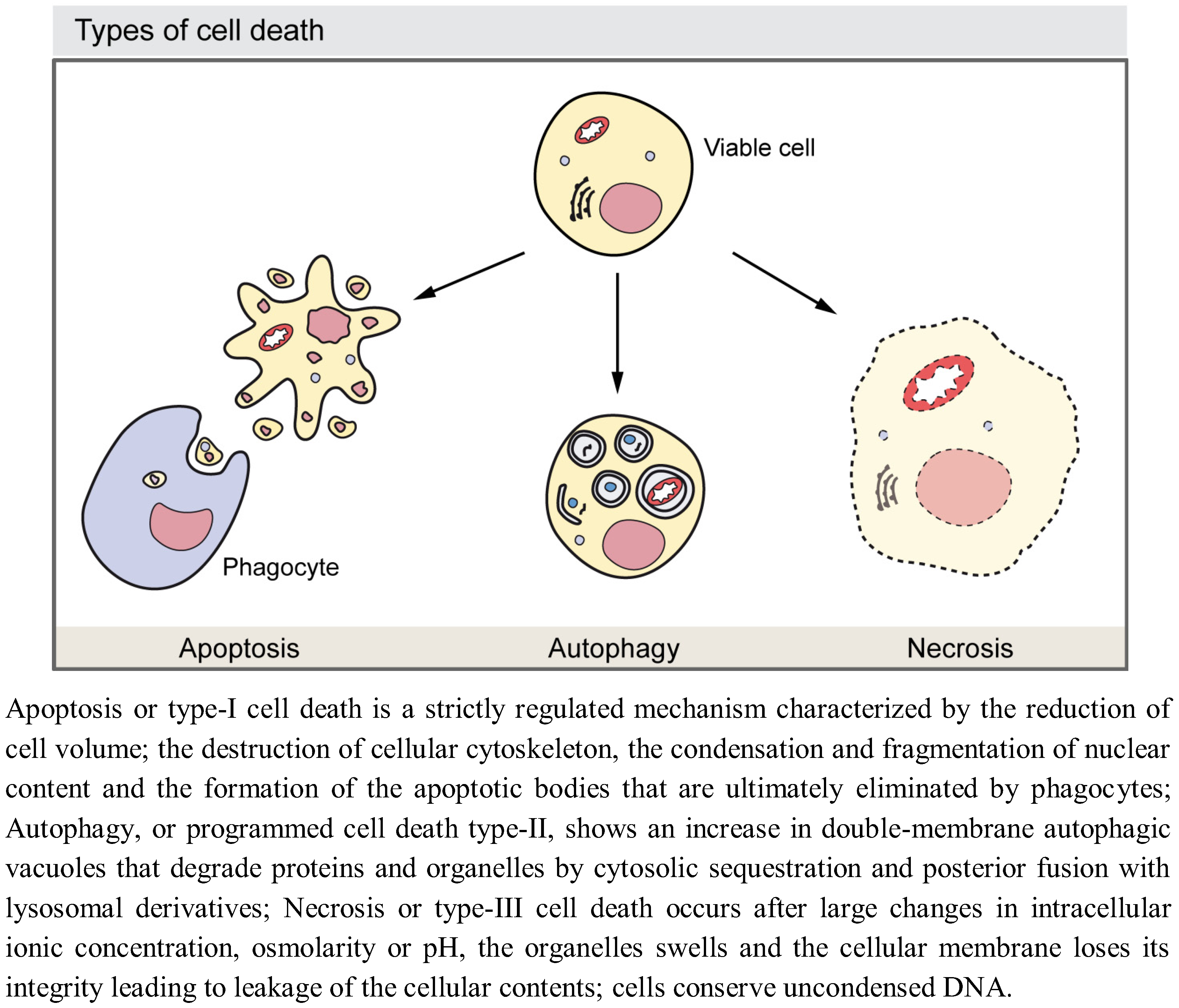
2. Autophagy as a Cell Death Mechanism

3. Autophagy in Cell Cycle Regulation
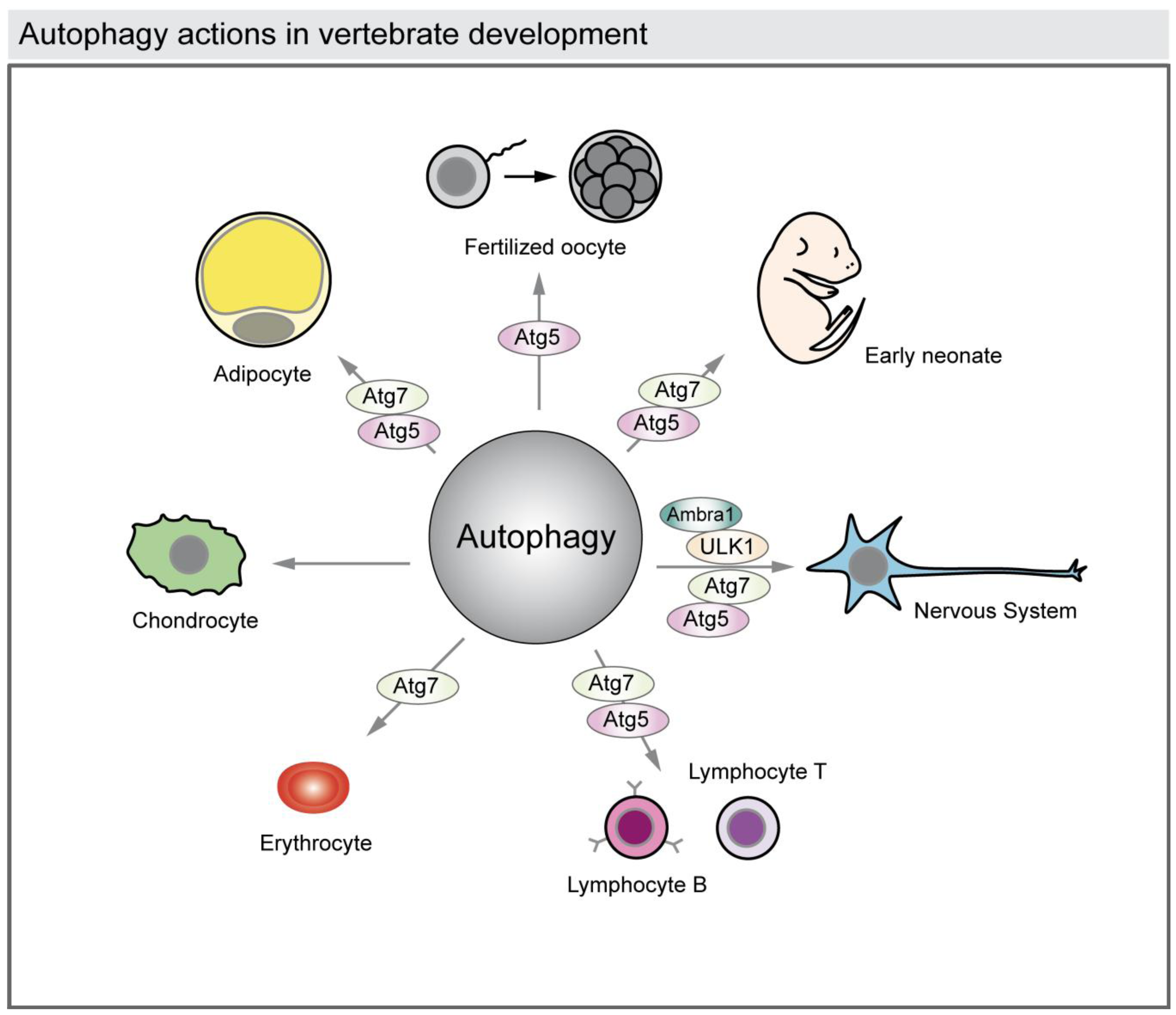
4. Autophagy in Cell Differentiation and Development

4.1. Autophagy Facilitates Remodeling at Specific Times of Development
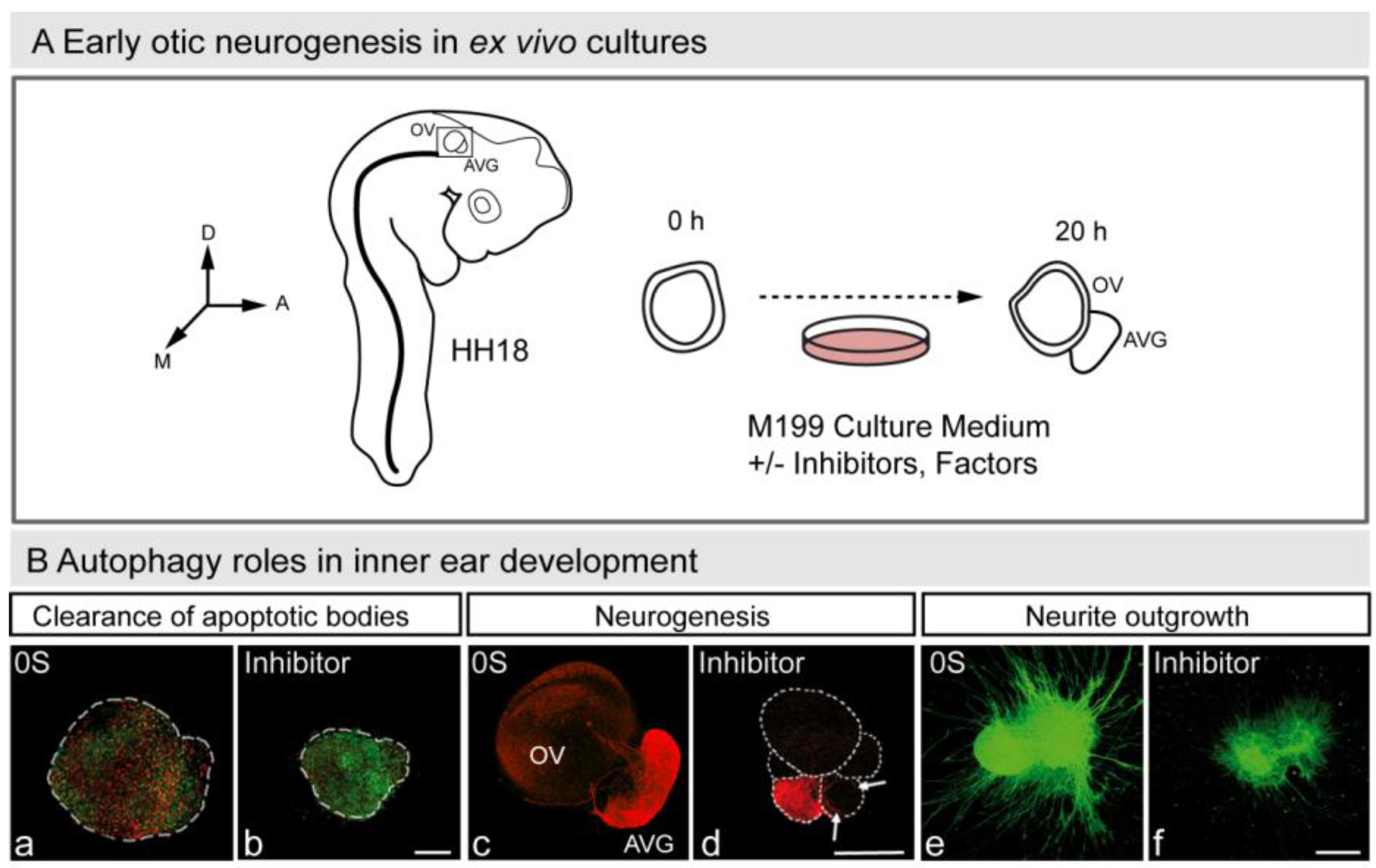
4.2. Autophagy in the Developing Nervous System
4.3. Autophagy During Hematopoiesis: Lymphocytes and Erythrocytes
4.4. Autophagy in Developing Osseous Tissue
4.5. Autophagy in Developing Adipose Tissue
5. Autophagy in Tissue Homeostasis and Aging
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Hafen, E.; Stocker, H. How are the sizes of cells, organs, and bodies controlled? PLoS Bio. 2003, 1, E86. [Google Scholar]
- Conlon, I.; Raff, M. Size control in animal development. Cell 1999, 96, 235–244. [Google Scholar]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar]
- Yuan, J.; Kroemer, G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010, 24, 2592–2602. [Google Scholar] [CrossRef]
- Zakeri, Z.; Lockshin, R.A. Cell death: History and future. Adv. Exp. Med. Biol. 2008, 615, 1–11. [Google Scholar]
- Penaloza, C.; Lin, L.; Lockshin, R.A.; Zakeri, Z. Cell death in development: Shaping the embryo. Histochem. Cell Biol. 2006, 126, 149–158. [Google Scholar] [CrossRef]
- Denton, D.; Shravage, B.; Simin, R.; Mills, K.; Berry, D.L.; Baehrecke, E.H.; Kumar, S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 2009, 19, 1741–1746. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Jo, E.-K.; Shin, D.-M.; Choi, A.M.K. Autophagy: Cellular defense to excessive inflammation. Microbes Infect. 2012, 14, 119–125. [Google Scholar] [CrossRef]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef]
- Mukaiyama, H.; Kajiwara, S.; Hosomi, A.; Giga-Hama, Y.; Tanaka, N.; Nakamura, T.; Takegawa, K. Autophagy-deficient Schizosaccharomyces pombe mutants undergo partial sporulation during nitrogen starvation. Microbiology 2009, 155, 3816–3826. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Baehrecke, E.H. Distinct death mechanisms in Drosophila development. Curr. Opin. Cell Biol. 2010, 22, 889–895. [Google Scholar] [CrossRef]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.-L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar]
- Tsukamoto, S.; Kuma, A.; Murakami, M.; Kishi, C.; Yamamoto, A.; Mizushima, N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008, 321, 117–120. [Google Scholar]
- Montero, J.A.; Lorda-Diez, C.I.; Certal, A.C.; Moreno, N.; Rodriguez-Leon, J.; Torriglia, A.; Hurle, J.M. Coordinated and sequential activation of neutral and acidic DNases during interdigital cell death in the embryonic limb. Apoptosis 2010, 15, 1197–1210. [Google Scholar]
- Mellén, M.A.; de la Rosa, E.J.; Boya, P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008, 15, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar]
- Onodera, J.; Ohsumi, Y. Ald6p is a preferred target for autophagy in yeast, Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 16071–16076. [Google Scholar] [CrossRef]
- Yu, L.; Strandberg, L.; Lenardo, M.J. The selectivity of autophagy and its role in cell death and survival. Autophagy 2008, 4, 567–573. [Google Scholar]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Deas, E.; Wood, N.W.; Plun-Favreau, H. Mitophagy and Parkinson’s disease: The PINK1-parkin link. Biochim. Biophys. Acta 2011, 1813, 623–633. [Google Scholar]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef]
- Zirin, J.; Perrimon, N. Drosophila as a model system to study autophagy. Semin. Immunopathol. 2010, 32, 363–372. [Google Scholar] [CrossRef]
- Fleming, A.; Rubinsztein, D.C. Zebrafish as a model to understand autophagy and its role in neurological disease. Biochim. Biophys. Acta 2011, 1812, 520–526. [Google Scholar]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, in press. [Google Scholar]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; Ohsumi, Y. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. The incredible ULKs. Cell Commun. Signal. 2012, 10, 7. [Google Scholar] [CrossRef]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar]
- Takahashi, Y.; Meyerkord, C.L.; Hori, T.; Runkle, K.; Fox, T.E.; Kester, M.; Loughran, T.P.; Wang, H.-G. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 2011, 7, 61–73. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Tanida, I.; Mizushima, N.; Kiyooka, M.; Ohsumi, M.; Ueno, T.; Ohsumi, Y.; Kominami, E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 1999, 10, 1367–1379. [Google Scholar]
- Ohsumi, Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohsumi, Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007, 581, 2156–2161. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar]
- Kim, J.; Huang, W.-P.; Stromhaug, P.E.; Klionsky, D.J. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J. Biol. Chem. 2002, 277, 763–773. [Google Scholar]
- Lang, T.; Schaeffeler, E.; Bernreuther, D.; Bredschneider, M.; Wolf, D.H.; Thumm, M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998, 17, 3597–3607. [Google Scholar] [CrossRef]
- Cann, G.M.; Guignabert, C.; Ying, L.; Deshpande, N.; Bekker, J.M.; Wang, L.; Zhou, B.; Rabinovitch, M. Developmental expression of LC3alpha and beta: Absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev. Dynam. 2008, 237, 187–195. [Google Scholar] [CrossRef]
- Mariño, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; López-Otín, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 1–3. [Google Scholar] [CrossRef]
- Scarlatti, F.; Granata, R.; Meijer, A.J.; Codogno, P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009, 16, 12–20. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: that is the autophagic question. Curr. Mol. Med. 2008, 8, 78–91. [Google Scholar] [CrossRef]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar]
- Lam, D.; Kosta, A.; Luciani, M.-F.; Golstein, P. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 2008, 19, 691–700. [Google Scholar]
- Berry, D.L.; Baehrecke, E.H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef]
- Rusten, T.E.; Lindmo, K.; Juhász, G.; Sass, M.; Seglen, P.O.; Brech, A.; Stenmark, H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell 2004, 7, 179–192. [Google Scholar] [CrossRef]
- Denton, D.; Shravage, B.; Simin, R.; Baehrecke, E.H.; Kumar, S. Larval midgut destruction in Drosophila: not dependent on caspases but suppressed by the loss of autophagy. Autophagy 2010, 6, 163–165. [Google Scholar] [CrossRef]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Juhász, G.; Erdi, B.; Sass, M.; Neufeld, T.P. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007, 21, 3061–3066. [Google Scholar] [CrossRef] [Green Version]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Ullman, E.; Fan, Y.; Stawowczyk, M.; Chen, H.-M.; Yue, Z.; Zong, W.-X. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. 2008, 15, 422–425. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Cho, D.-H.; Jo, Y.K.; Hwang, J.J.; Lee, Y.M.; Roh, S.A.; Kim, J.C. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 2009, 274, 95–100. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef]
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390. [Google Scholar] [CrossRef]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef]
- Li, W.; Zou, W.; Yang, Y.; Chai, Y.; Chen, B.; Cheng, S.; Tian, D.; Wang, X.; Vale, R.D.; Ou, G. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J. Cell Biol. 2012, 197, 27–35. [Google Scholar] [CrossRef]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef]
- Magariños, M.; Aburto, M.R.; Sánchez-Calderón, H.; Muñoz-Agudo, C.; Rapp, U.R.; Varela-Nieto, I. RAF kinase activity regulates neuroepithelial cell proliferation and neuronal progenitor cell differentiation during early inner ear development. PLoS ONE 2010, 5, e14435. [Google Scholar]
- Magariños, M.; Contreras, J.; Aburto, M.R.; Varela-Nieto, I. Early development of the vertebrate inner ear. Anat. Rec. 2012, in press. [Google Scholar]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; Pledger, W.J.; Wang, H.-G. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA: Degradation of paternal mitochondria by allogeneic organelle autophagy, allophagy. Autophagy 2012, in press. [Google Scholar]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar]
- Stitzel, M.L.; Seydoux, G. Regulation of the oocyte-to-zygote transition. Science 2007, 316, 407–408. [Google Scholar] [CrossRef]
- Schier, A.F. The maternal-zygotic transition: death and birth of RNAs. Science 2007, 316, 406–407. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar]
- Sou, Y.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef]
- Baerga, R.; Zhang, Y.; Chen, P.-H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef]
- Tomoda, T.; Bhatt, R.S.; Kuroyanagi, H.; Shirasawa, T.; Hatten, M.E. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 1999, 24, 833–846. [Google Scholar] [CrossRef]
- Mochizuki, H.; Toda, H.; Ando, M.; Kurusu, M.; Tomoda, T.; Furukubo-Tokunaga, K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS ONE 2011, 6, e19632. [Google Scholar]
- Ogura, K.; Wicky, C.; Magnenat, L.; Tobler, H.; Mori, I.; Müller, F.; Ohshima, Y. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994, 8, 2389–2400. [Google Scholar] [CrossRef]
- Mariño, G.; Fernández, A.F.; Cabrera, S.; Lundberg, Y.W.; Cabanillas, R.; Rodríguez, F.; Salvador-Montoliu, N.; Vega, J.A.; Germanà, A.; Fueyo, A.; et al. Autophagy is essential for mouse sense of balance. J. Clin. Invest. 2010, 120, 2331–2344. [Google Scholar] [CrossRef]
- Sanchez-Calderon, H.; Rodriguez-de la Rosa, L.; Milo, M.; Pichel, J.G.; Holley, M.; Varela-Nieto, I. RNA microarray analysis in prenatal mouse cochlea reveals novel IGF-I target genes: implication of MEF2 and FOXM1 transcription factors. PLoS ONE 2010, 5, e8699. [Google Scholar]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.-W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef]
- Mortensen, M.; Simon, A.K. Nonredundant role of Atg7 in mitochondrial clearance during erythroid development. Autophagy 2010, 6, 423–425. [Google Scholar] [CrossRef]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.-W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055. [Google Scholar] [CrossRef]
- Srinivas, V.; Shapiro, I.M. Chondrocytes embedded in the epiphyseal growth plates of long bones undergo autophagy prior to the induction of osteogenesis. Autophagy 2006, 2, 215–216. [Google Scholar]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrin. Met. 2011, 22, 234–240. [Google Scholar] [CrossRef]
- Goldman, S.J.; Zhang, Y.; Jin, S. Autophagic degradation of mitochondria in white adipose tissue differentiation. Antioxid. Redox Sign. 2011, 14, 1971–1978. [Google Scholar] [CrossRef]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 2009, 119, 3329–3339. [Google Scholar]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar]
- Langin, D. Recruitment of brown fat and conversion of white into brown adipocytes: strategies to fight the metabolic complications of obesity? Biochim. Biophys. Acta 2010, 1801, 372–376. [Google Scholar]
- Meng, Q.; Cai, D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J. Biol. Chem. 2011, 286, 32324–32332. [Google Scholar] [CrossRef]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Rubinsztein, D.C. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Meth. Enzymol. 2009, 453, 83–110. [Google Scholar]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef]
- Iwata, J.; Ezaki, J.; Komatsu, M.; Yokota, S.; Ueno, T.; Tanida, I.; Chiba, T.; Tanaka, K.; Kominami, E. Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 2006, 281, 4035–4041. [Google Scholar]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aburto, M.R.; Hurlé, J.M.; Varela-Nieto, I.; Magariños, M. Autophagy During Vertebrate Development. Cells 2012, 1, 428-448. https://doi.org/10.3390/cells1030428
Aburto MR, Hurlé JM, Varela-Nieto I, Magariños M. Autophagy During Vertebrate Development. Cells. 2012; 1(3):428-448. https://doi.org/10.3390/cells1030428
Chicago/Turabian StyleAburto, María R., Juan M. Hurlé, Isabel Varela-Nieto, and Marta Magariños. 2012. "Autophagy During Vertebrate Development" Cells 1, no. 3: 428-448. https://doi.org/10.3390/cells1030428
APA StyleAburto, M. R., Hurlé, J. M., Varela-Nieto, I., & Magariños, M. (2012). Autophagy During Vertebrate Development. Cells, 1(3), 428-448. https://doi.org/10.3390/cells1030428