A Dual Role of Heme Oxygenase-1 in Angiotensin II-Induced Abdominal Aortic Aneurysm in the Normolipidemic Mice

 , , ,
, , , 
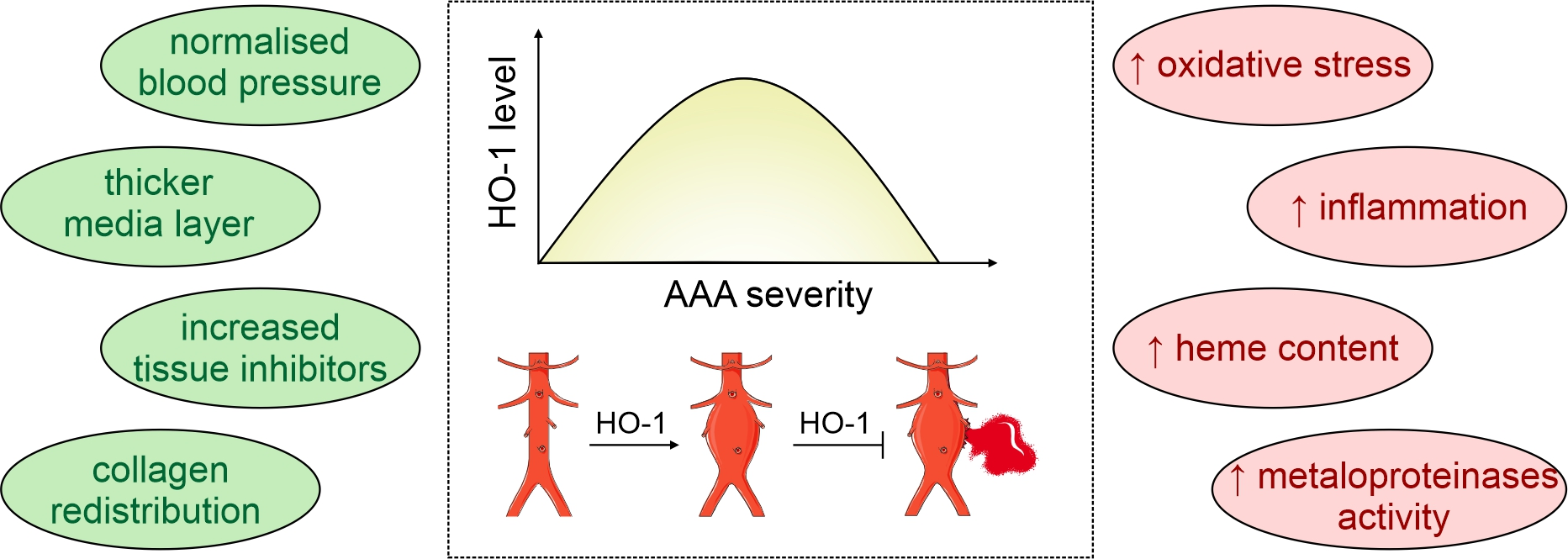
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Development of Abdominal Aortic Aneurysm
2.3. Blood Pressure Monitoring
2.4. In Vivo Ultrasound Imaging of Abdominal Aorta and Analysis
2.5. Blood Cell Count
2.6. Total RNA Isolation, Reverse Transcription and Quantitative PCR
2.7. Histological and Immunofluorescent Stainings
2.8. Heme Level Measurement
2.9. In Situ Gelatin Zymography (ISZ)
2.10. In Vitro Assays
2.11. Statistical Analysis
3. Results
3.1. Heme Oxygenase 1 Is Upregulated During the Angiotensin II-Induced Formation of AAA in Non-Hypercholesterolemic Mice
3.2. Lack of Ho-1 Protects Against Induction of AAA in Non-Hyperlipidemic Mice; However, If the Aneurysms Form, They Are Unstable and Prone to Rupture
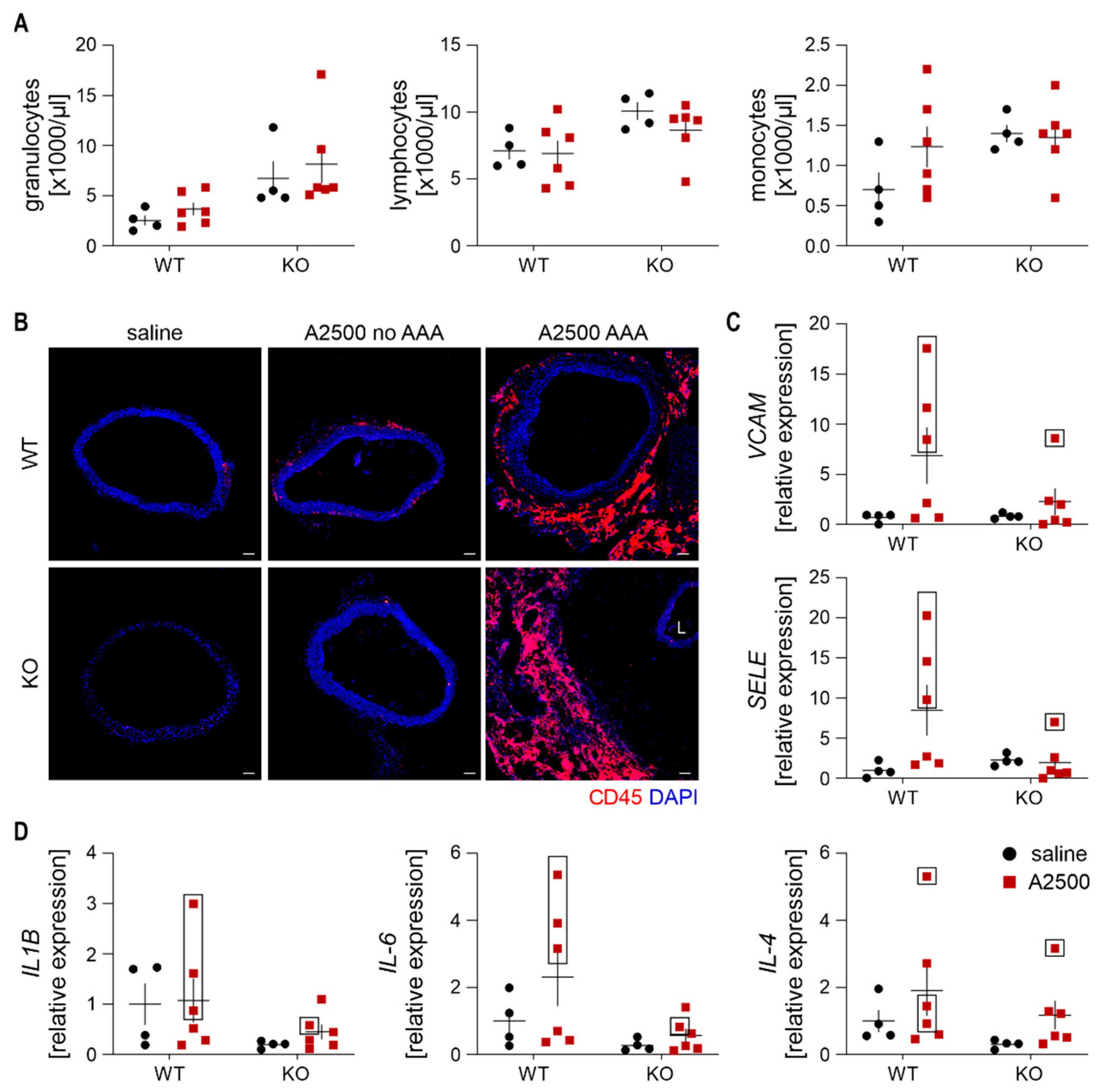
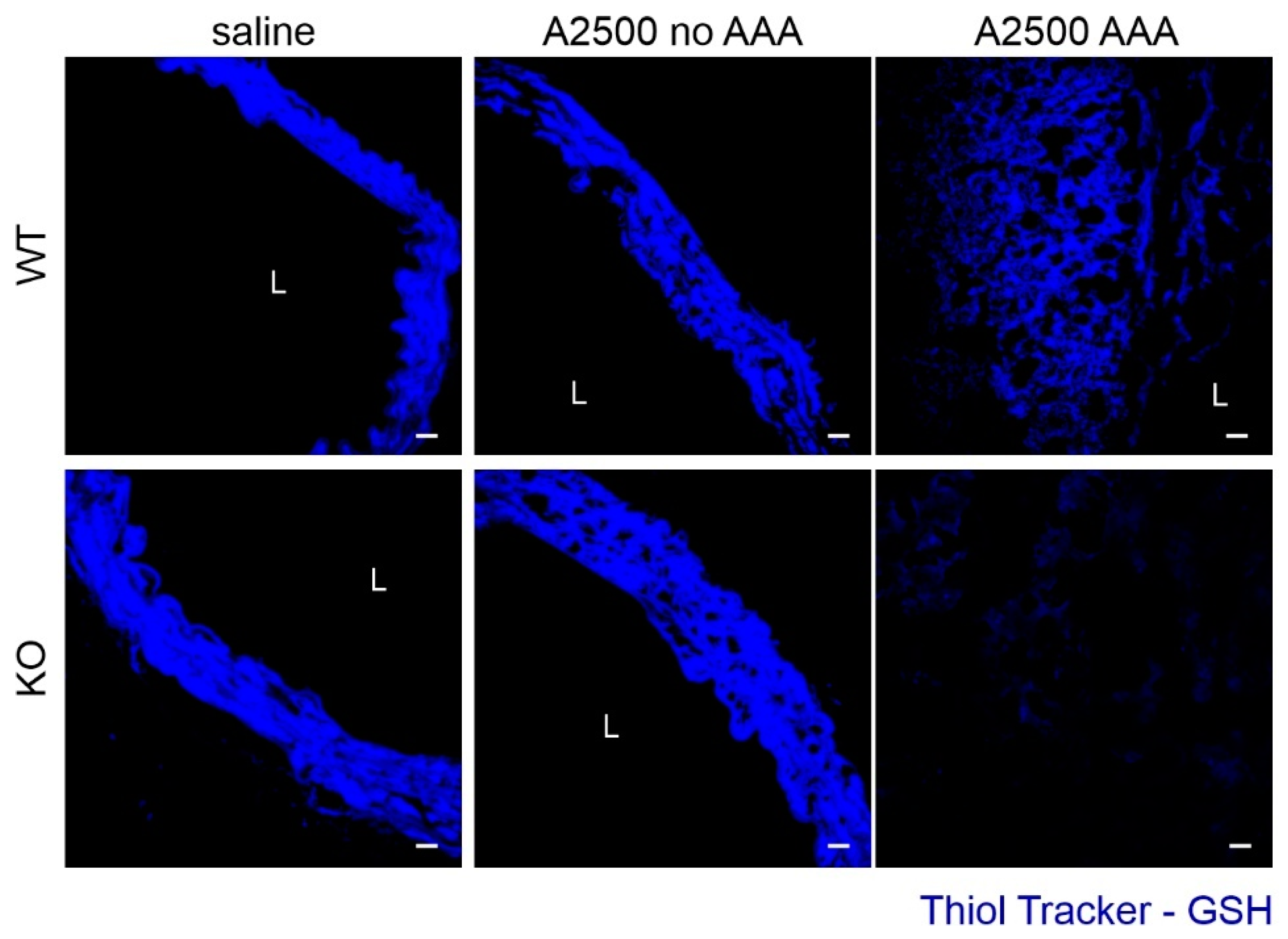
3.3. Enhanced Immune Cell Infiltration and Oxidative Stress Are Present Only in the Formed Aneurysms
3.4. Ho-1 Deficiency Delays Angii-Induced Blood Pressure Rise, With Concomitant Changes in at2r Expression
3.5. AngII Infusion Increases Total Heme Content Especially in AAA
3.6. AngII Infusion Leads to the Thickening of Aortic Media in Ho-1 Ko Mice
3.7. Differential Effect on the Collagens Upon AngIIInfusion between the Genotypes
3.8. MMP2 and TIMP2 are Strongly Upregulated Upon AngII in HO-1-Deficient Mice
4. Discussion
Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sakalihasan, N.; Michel, J.-B.; Katsargyris, A.; Kuivaniemi, H.; Defraigne, J.-O.; Nchimi, A.; Powell, J.T.; Yoshimura, K.; Hultgren, R. Abdominal Aortic Aneurysms. Nat. Rev. Dis. Primer 2018, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Mix, D.S.; Yang, L.; Johnson, C.C.; Couper, N.; Zarras, B.; Arabadjis, I.; Trakimas, L.E.; Stoner, M.C.; Day, S.W.; Richards, M.S. Detecting Regional Stiffness Changes in Aortic Aneurysmal Geometries Using Pressure-Normalized Strain. Ultrasound Med. Biol. 2017, 43, 2372–2394. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Jozkowicz, A.; Nowak, W.; Eilenberg, W.; Neumayer, C.; Malinski, T.; Huk, I.; Brostjan, C. The Abdominal Aortic Aneurysm and Intraluminal Thrombus: Current Concepts of Development and Treatment. Front. Cardiovasc. Med. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Choi, A.M.K. Targeting Heme Oxygenase-1/Carbon Monoxide for Therapeutic Modulation of Inflammation. Transl. Res. J. Lab. Clin. Med. 2016, 167, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Taha, H.; Skrzypek, K.; Guevara, I.; Nigisch, A.; Mustafa, S.; Grochot-Przeczek, A.; Ferdek, P.; Was, H.; Kotlinowski, J.; Kozakowska, M.; et al. Role of Heme Oxygenase-1 in Human Endothelial Cells: Lesson from the Promoter Allelic Variants. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Piechota-Polanczyk, A.; Kopacz, A.; Kloska, D.; Zagrapan, B.; Neumayer, C.; Grochot-Przeczek, A.; Huk, I.; Brostjan, C.; Dulak, J.; Jozkowicz, A. Simvastatin Treatment Upregulates HO-1 in Patients with Abdominal Aortic Aneurysm but Independently of Nrf2. Oxid. Med. Cell. Longev. 2018, 2018, 2028936. [Google Scholar] [CrossRef] [Green Version]
- Kloska, D.; Kopacz, A.; Piechota-Polanczyk, A.; Neumayer, C.; Huk, I.; Dulak, J.; Jozkowicz, A.; Grochot-Przeczek, A. Biliverdin Reductase Deficiency Triggers an Endothelial-to-Mesenchymal Transition in Human Endothelial Cells. Arch. Biochem. Biophys. 2019, 108182. [Google Scholar] [CrossRef]
- Anja, H.; Margarete, M.; Steffen, W.; David, P.; Coy, B.; Henning, M. Reeps Christian Abstract 416: Upregulation of Heme Oxygenase-1 Is Associated With Inflammation in Patients With Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2020, 40, A416. [Google Scholar] [CrossRef]
- Schillinger, M.; Exner, M.; Mlekusch, W.; Domanovits, H.; Huber, K.; Mannhalter, C.; Wagner, O.; Minar, E. Heme Oxygenase-1 Gene Promoter Polymorphism Is Associated with Abdominal Aortic Aneurysm. Thromb. Res. 2002, 106, 131–136. [Google Scholar] [CrossRef]
- Gregorek, A.C.; Gornik, K.C.; Polancec, D.S.; Dabelic, S. GT Microsatellite Repeats in the Heme Oxygenase-1 Gene Promoter Associated with Abdominal Aortic Aneurysm in Croatian Patients. Biochem. Genet. 2013, 51, 482–492. [Google Scholar] [CrossRef]
- Azuma, J.; Wong, R.J.; Morisawa, T.; Hsu, M.; Maegdefessel, L.; Zhao, H.; Kalish, F.; Kayama, Y.; Wallenstein, M.B.; Deng, A.C.; et al. Heme Oxygenase-1 Expression Affects Murine Abdominal Aortic Aneurysm Progression. PLoS ONE 2016, 11, e0149288. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Wu, M.-L.; Gung, P.-Y.; Chen, C.-H.; Kuo, C.-C.; Yet, S.-F. Heme Oxygenase-1 Deficiency Exacerbates Angiotensin II-Induced Aortic Aneurysm in Mice. Oncotarget 2016, 7, 67760–67776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, A.; Cassis, L.A. Mouse Models of Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, S.H.; Forsdahl, S.H.; Singh, K.; Jacobsen, B.K. Atherosclerosis in Abdominal Aortic Aneurysms: A Causal Event or a Process Running in Parallel? The Tromsø Study. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadière, C.; Rénia, L.; et al. TGF-β Activity Protects against Inflammatory Aortic Aneurysm Progression and Complications in Angiotensin II–Infused Mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Mulorz, J.; Spin, J.M.; Beck, H.C.; Tha Thi, M.L.; Wagenhäuser, M.U.; Rasmussen, L.M.; Lindholt, J.S.; Tsao, P.S.C.; Steffensen, L.B. Hyperlipidemia Does Not Affect Development of Elastase-Induced Abdominal Aortic Aneurysm in Mice. Atherosclerosis 2020, 311, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Werner, E.; Kloska, D.; Hajduk, K.; Fichna, J.; Jozkowicz, A.; Piechota-Polanczyk, A. Nrf2 Transcriptional Activity in the Mouse Affects the Physiological Response to Tribromoethanol. Biomed. Pharmacother. 2020, 128, 110317. [Google Scholar] [CrossRef]
- Kopacz, A.; Werner, E.; Grochot-Przęczek, A.; Klóska, D.; Hajduk, K.; Neumayer, C.; Józkowicz, A.; Piechota-Polanczyk, A. Simvastatin Attenuates Abdominal Aortic Aneurysm Formation Favoured by Lack of Nrf2 Transcriptional Activity. Oxid. Med. Cell. Longev. 2020, 2020, 6340190. [Google Scholar] [CrossRef]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin-II Mediates ACE2 Internalization and Degradation through an Angiotensin-II Type I Receptor-Dependent Mechanism. Hypertens. Dallas Tex 1979 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Watanabe-Matsui, M. Wearing Red for Signaling: The Heme-Bach Axis in Heme Metabolism, Oxidative Stress Response and Iron Immunology. Tohoku J. Exp. Med. 2014, 232, 229–253. [Google Scholar] [CrossRef] [Green Version]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genomics Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hussien, H.; Soekhoe, R.G.V.; Weber, E.; von der Thüsen, J.H.; Kleemann, R.; Mulder, A.; van Bockel, J.H.; Hanemaaijer, R.; Lindeman, J.H.N. Collagen Degradation in the Abdominal Aneurysm. Am. J. Pathol. 2007, 170, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeman, J.H.N.; Ashcroft, B.A.; Beenakker, J.-W.M.; van Es, M.; Koekkoek, N.B.R.; Prins, F.A.; Tielemans, J.F.; Abdul-Hussien, H.; Bank, R.A.; Oosterkamp, T.H. Distinct Defects in Collagen Microarchitecture Underlie Vessel-Wall Failure in Advanced Abdominal Aneurysms and Aneurysms in Marfan Syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharm. Basel Switz. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.G.; Martin-McNulty, B.; Sukovich, D.A.; Freay, A.; Halks-Miller, M.; Thinnes, T.; Loskutoff, D.J.; Carmeliet, P.; Dole, W.P.; Wang, Y.X. Urokinase-Type Plasminogen Activator Plays a Critical Role in Angiotensin II–Induced Abdominal Aortic Aneurysm. Circ. Res. 2003, 92, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Lysgaard Poulsen, J.; Stubbe, J.; Lindholt, J.S. Animal Models Used to Explore Abdominal Aortic Aneurysms: A Systematic Review. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2016, 52, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Szade, A.; Nowak, W.N.; Szade, K.; Gese, A.; Czypicki, R.; Waś, H.; Dulak, J.; Józkowicz, A. Effect of Crossing C57BL/6 and FVB Mouse Strains on Basal Cytokine Expression. Mediat. Inflamm. 2015, 2015, 762419. [Google Scholar] [CrossRef] [Green Version]
- Trachet, B.; Aslanidou, L.; Piersigilli, A.; Fraga-Silva, R.A.; Sordet-Dessimoz, J.; Villanueva-Perez, P.; Stampanoni, M.F.M.; Stergiopulos, N.; Segers, P. Angiotensin II Infusion into ApoE-/- Mice: A Model for Aortic Dissection Rather than Abdominal Aortic Aneurysm? Cardiovasc. Res. 2017, 113, 1230–1242. [Google Scholar] [CrossRef]
- Jiang, W.-C.; Chen, C.-M.; Hamdin, C.D.; Orekhov, A.N.; Sobenin, I.A.; Layne, M.D.; Yet, S.-F. Therapeutic Potential of Heme Oxygenase-1 in Aneurysmal Diseases. Antioxidants 2020, 9, 1150. [Google Scholar] [CrossRef]
- Budbazar, E.; Rodriguez, F.; Sanchez, J.M.; Seta, F. The Role of Sirtuin-1 in the Vasculature: Focus on Aortic Aneurysm. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- King, V.L.; Lin, A.Y.; Kristo, F.; Anderson, T.J.T.; Ahluwalia, N.; Hardy, G.J.; Owens, A.P.; Howatt, D.A.; Shen, D.; Tager, A.M.; et al. Interferon-Gamma and the Interferon-Inducible Chemokine CXCL10 Protect against Aneurysm Formation and Rupture. Circulation 2009, 119, 426–435. [Google Scholar] [CrossRef]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-Derived Netrin-1 Promotes Abdominal Aortic Aneurysm Formation by Activating MMP3 in Vascular Smooth Muscle Cells. Nat. Commun. 2018, 9, 5022. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Upchurch, G.R.; Kuivaniemi, H.; Thompson, R.W.; Tilson, M.D. Challenges and Opportunities in Abdominal Aortic Aneurysm Research. J. Vasc. Surg. 2007, 45, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T Cell in the Genesis of Angiotensin II Induced Hypertension and Vascular Dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Moore, J.P.; Sakkal, S.; Bullen, M.L.; Kemp-Harper, B.K.; Ricardo, S.D.; Sobey, C.G.; Drummond, G.R. A Flow Cytometric Method for the Analysis of Macrophages in the Vascular Wall. J. Immunol. Methods 2013, 396, 33–43. [Google Scholar] [CrossRef]
- Navar, L.G.; Mitchell, K.D.; Harrison-Bernard, L.M.; Kobori, H.; Nishiyama, A. Review: Intrarenal Angiotensin II Levels in Normal and Hypertensive States. J. Renin-Angiotensin-Aldosterone Syst. 2001, 2, S176–S184. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Villalobos, R.A.; Satou, R.; Seth, D.M.; Semprun-Prieto, L.C.; Katsurada, A.; Kobori, H.; Navar, L.G. Angiotensin-Converting Enzyme-Derived Angiotensin II Formation during Angiotensin II-Induced Hypertension. Hypertension 2009, 53, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Intengan Hope, D.; Schiffrin Ernesto, L. Vascular Remodeling in Hypertension. Hypertension 2001, 38, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Sassa, S. Why Heme Needs to Be Degraded to Iron, Biliverdin IXα, and Carbon Monoxide? Antioxid. Redox Signal. 2004, 6, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.W.; Geraghty, P.J.; Lee, J.K. Abdominal Aortic Aneurysms: Basic Mechanisms and Clinical Implications. Curr. Probl. Surg. 2002, 39, 110–230. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.K.; Soini, Y.; Melkko, J.; Satta, J.; Risteli, L.; Risteli, J. Increased Amount of Type III PN-Collagen in Human Abdominal Aortic Aneurysms: Evidence for Impaired Type III Collagen Fibrillogenesis. J. Vasc. Surg. 2000, 32, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, H.A.; Pajala, A.; Leppilahti, J.; Risteli, J. Increased Content of Type III Collagen at the Rupture Site of Human Achilles Tendon. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2002, 20, 1352–1357. [Google Scholar] [CrossRef]
- Saito, K.; Nakatomi, M.; Ida-Yonemochi, H.; Ohshima, H. Osteopontin Is Essential for Type I Collagen Secretion in Reparative Dentin. J. Dent. Res. 2016, 95, 1034–1041. [Google Scholar] [CrossRef]
- Shen Ying, H.; LeMaire Scott, A.; Webb Nancy, R.; Cassis Lisa, A.; Daugherty, A.; Lu Hong, S. Aortic Aneurysms and Dissections Series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef]
- Cheema, T.A.; Kim, G.M.; Lee, C.Y.; Hong, J.G.; Kwak, M.K.; Park, C.W. Characteristics of Blood Vessel Wall Deformation with Porous Wall Conditions in an Aortic Arch. Appl. Rheol. 2014, 24, 17–24. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Spinetti, G.; Jiang, L.-Q.; Monticone, R.; Zhao, D.; Cheng, L.; Krawczyk, M.; Talan, M.; Pintus, G.; et al. Angiotensin II Activates Matrix Metalloproteinase Type II and Mimics Age-Associated Carotid Arterial Remodeling in Young Rats. Am. J. Pathol. 2005, 167, 1429–1442. [Google Scholar] [CrossRef] [Green Version]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix Metalloproteinases 2 and 9 Work in Concert to Produce Aortic Aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Carmeliet, P. Proteinases in Cardiovascular Aneurysms and Rupture: Targets for Therapy? J. Clin. Investig. 2000, 105, 1519–1520. [Google Scholar] [CrossRef] [Green Version]
- Ottl, J.; Gabriel, D.; Murphy, G.; Knäuper, V.; Tominaga, Y.; Nagase, H.; Kröger, M.; Tschesche, H.; Bode, W.; Moroder, L. Recognition and Catabolism of Synthetic Heterotrimeric Collagen Peptides by Matrix Metalloproteinases. Chem. Biol. 2000, 7, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Van Doren, S.R. Matrix Metalloproteinase Interactions with Collagen and Elastin. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 44–46, 224–231. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Tsai, H.-L.; Chiang, M.-T.; Chau, L.-Y. Carbon Monoxide-Induced Early Thrombolysis Contributes to Heme Oxygenase-1-Mediated Inhibition of Neointimal Growth after Vascular Injury in Hypercholesterolemic Mice. J. Biomed. Sci. 2006, 13, 721–730. [Google Scholar] [CrossRef]
- Qian, H.S.; Gu, J.-M.; Liu, P.; Kauser, K.; Halks-Miller, M.; Vergona, R.; Sullivan, M.E.; Dole, W.P.; Deng, G.G. Overexpression of PAI-1 Prevents the Development of Abdominal Aortic Aneurysm in Mice. Gene Ther. 2008, 15, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Defawe, O.D.; Colige, A.; Lambert, C.A.; Munaut, C.; Delvenne, P.; Lapière, C.M.; Limet, R.; Nusgens, B.V.; Sakalihasan, N. TIMP-2 and PAI-1 MRNA Levels Are Lower in Aneurysmal as Compared to Athero-Occlusive Abdominal Aortas. Cardiovasc. Res. 2003, 60, 205–213. [Google Scholar] [CrossRef] [Green Version]
- DiMusto, P.D.; Lu, G.; Ghosh, A.; Roelofs, K.J.; Su, G.; Zhao, Y.; Lau, C.L.; Sadiq, O.; McEvoy, B.; Laser, A.; et al. Increased PAI-1 in Females Compared with Males Is Protective for Abdominal Aortic Aneurysm Formation in a Rodent Model. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1378–H1386. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Shen, M.; Parajuli, N.; Oudit, G.Y.; McMurtry, M.S.; Kassiri, Z. Gender-Dependent Aortic Remodelling in Patients with Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm. J. Mol. Med. 2014, 92, 939–949. [Google Scholar] [CrossRef]
- Guala, A.; Camporeale, C.; Ridolfi, L. Compensatory Effect between Aortic Stiffening and Remodelling during Ageing. PLoS ONE 2015, 10, e0139211. [Google Scholar] [CrossRef] [Green Version]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the Heme Recycling System in Heme Oxygenase 1-Deficient Mice: Effects on Macrophage Viability and Tissue Iron Distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [Green Version]
- Szade, K.; Zukowska, M.; Szade, A.; Nowak, W.; Skulimowska, I.; Ciesla, M.; Bukowska-Strakova, K.; Gulati, G.S.; Kachamakova-Trojanowska, N.; Kusienicka, A.; et al. Heme Oxygenase-1 Deficiency Triggers Exhaustion of Hematopoietic Stem Cells. EMBO Rep. 2020, 21, e47895. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopacz, A.; Klóska, D.; Werner, E.; Hajduk, K.; Grochot-Przęczek, A.; Józkowicz, A.; Piechota-Polańczyk, A. A Dual Role of Heme Oxygenase-1 in Angiotensin II-Induced Abdominal Aortic Aneurysm in the Normolipidemic Mice. Cells 2021, 10, 163. https://doi.org/10.3390/cells10010163
Kopacz A, Klóska D, Werner E, Hajduk K, Grochot-Przęczek A, Józkowicz A, Piechota-Polańczyk A. A Dual Role of Heme Oxygenase-1 in Angiotensin II-Induced Abdominal Aortic Aneurysm in the Normolipidemic Mice. Cells. 2021; 10(1):163. https://doi.org/10.3390/cells10010163
Chicago/Turabian StyleKopacz, Aleksandra, Damian Klóska, Ewa Werner, Karolina Hajduk, Anna Grochot-Przęczek, Alicja Józkowicz, and Aleksandra Piechota-Polańczyk. 2021. "A Dual Role of Heme Oxygenase-1 in Angiotensin II-Induced Abdominal Aortic Aneurysm in the Normolipidemic Mice" Cells 10, no. 1: 163. https://doi.org/10.3390/cells10010163
APA StyleKopacz, A., Klóska, D., Werner, E., Hajduk, K., Grochot-Przęczek, A., Józkowicz, A., & Piechota-Polańczyk, A. (2021). A Dual Role of Heme Oxygenase-1 in Angiotensin II-Induced Abdominal Aortic Aneurysm in the Normolipidemic Mice. Cells, 10(1), 163. https://doi.org/10.3390/cells10010163