Myeloid Cells in Glioblastoma Microenvironment


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor-Associated Macrophages (TAMs)
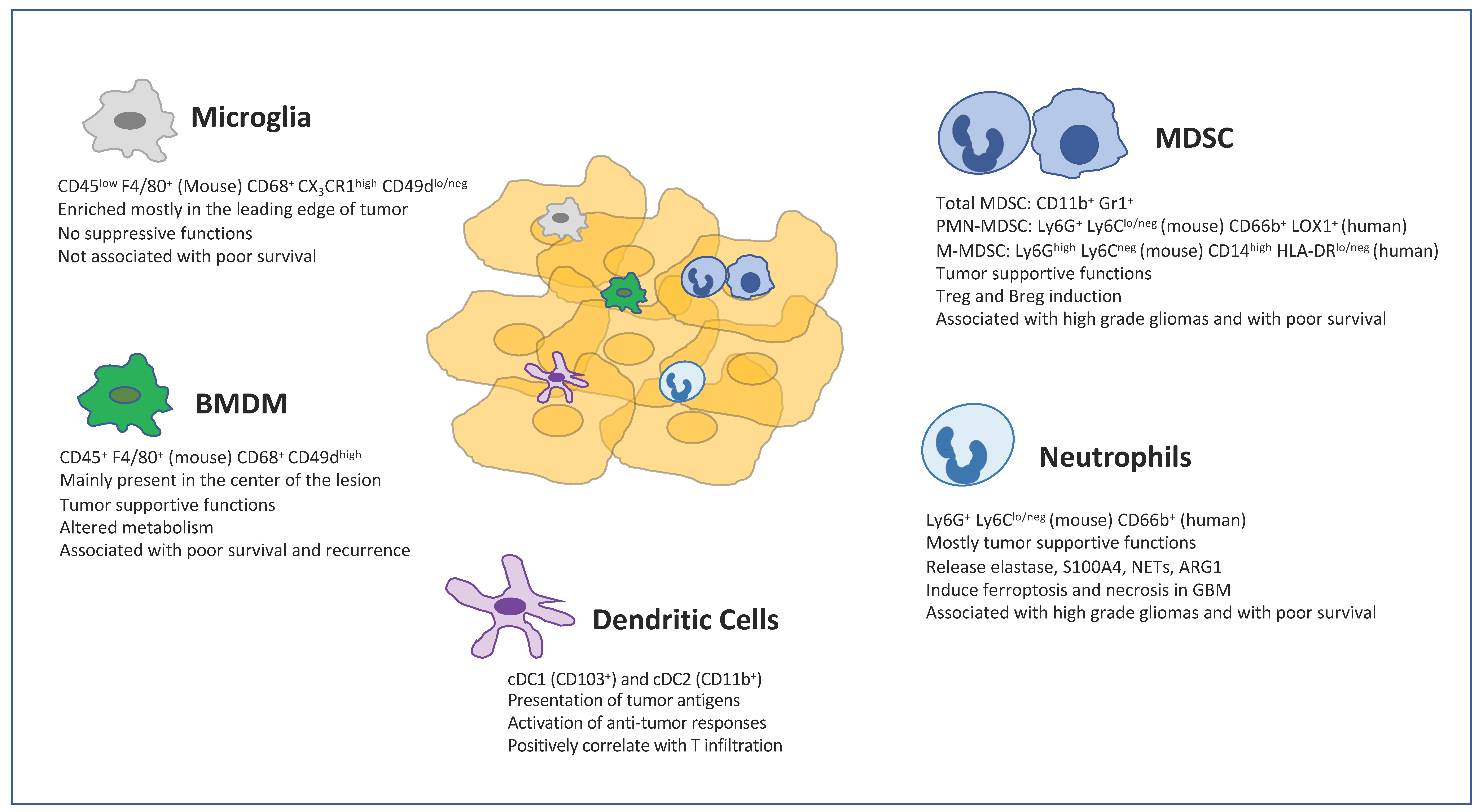
3. Distinctive Features of MG and BMDMs
4. Localization of MG and BMDMs
5. Epigenetic and Transcriptional Features of MG and BMDMs
6. Metabolic Profile of MG and BMDMs
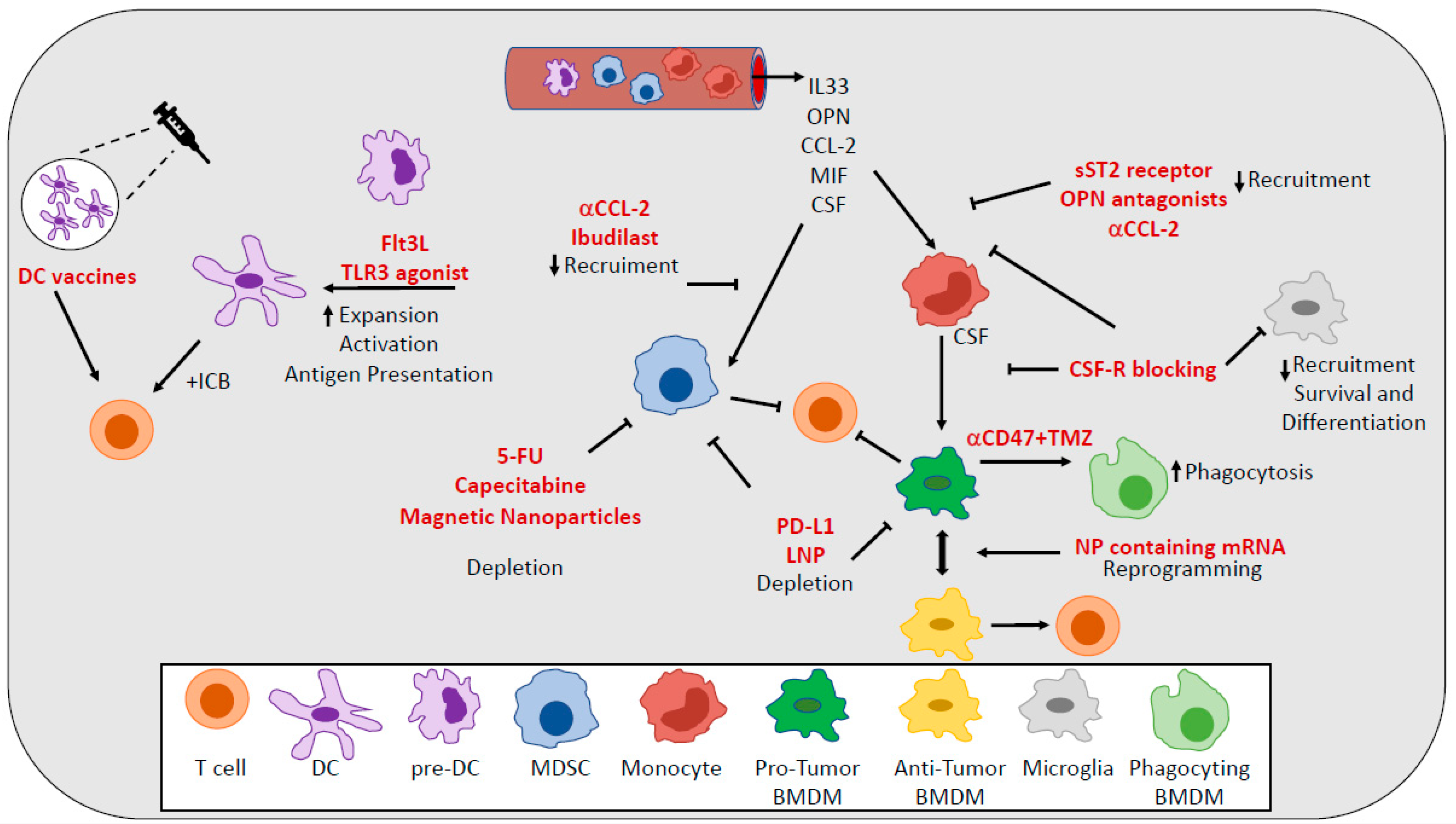
7. Therapeutic Targeting of TAMs
8. Myeloid Derived Suppressor Cells (MDSCs) in GBM
9. Therapeutic Targeting of MDSCs
10. Neutrophils in GBM
11. Neutrophils and PMN-MDSCs: Two Sides of the Same Coin
12. Dendritic Cells in GBM
13. Therapeutic Targeting of DCs
14. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brat, D.J.; Verhaak, R.G.W.; Aldape, K.D.; Yung, W.K.A.; Salama, S.R.; Cooper, L.D.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kim, B.Y.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune–vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; DeChant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.-W.; Liu, L.; Gong, C.-Y.; Shi, H.-S.; Zeng, Y.-H.; Wang, X.-Z.; Zhao, Y.-W.; Wei, Y.-Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Ohri, C.M.; Shikotra, A.; Green, R.H.; Waller, D.A.; Bradding, P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 2009, 33, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Liu, L.; Che, G.-W.; Yu, N.; Dai, F.; You, Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 2010, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohri, C.M.; Shikotra, A.; Green, R.H.; Waller, D.A.; Bradding, P. Tumour necrosis factor-alpha expression in tumour islets confers a survival advantage in non-small cell lung cancer. BMC Cancer 2010, 10, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.-Z.; Xu, S.-L.; Xin, Y.-H.; Yu, S.-C.; Ping, Y.-F.; Chen, L.; Xiao, H.-L.; Wang, B.; Yi, L.; Wang, Q.-L.; et al. Tumor-Associated Microglia/Macrophages Enhance the Invasion of Glioma Stem-like Cells via TGF-β1 Signaling Pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, L.; Zhang, I.Y.; Liang, J.; Wang, H.; Ouyang, M.; Wu, S.; Da Fonseca, A.C.C.; Weng, L.; Yamamoto, Y.; et al. RAGE Expression in Tumor-Associated Macrophages Promotes Angiogenesis in Glioma. Cancer Res. 2014, 74, 7285–7297. [Google Scholar] [CrossRef] [Green Version]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses1. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef]
- Tabatabai, G.; Frank, B.; Mohle, R.; Weller, M.; Wick, W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-beta-dependent HIF-1alpha-mediated induction of CXCL12. Brain 2006, 129, 2426–2435. [Google Scholar] [CrossRef] [Green Version]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.J.; Ebsworth, K.; Berahovich, R.D.; Penfold, M.E.T.; Liu, S.-C.; Al Omran, R.; Kioi, M.; Chernikova, S.B.; Tseng, D.; Mulkearns-Hubert, E.E.; et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer 2014, 110, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; De Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Korzhevskii, D.E.; Kirik, O.V. Brain Microglia and Microglial Markers. Neurosci. Behav. Physiol. 2016, 46, 284–290. [Google Scholar] [CrossRef]
- Sousa, C.; Biber, K.; Michelucci, A. Cellular and Molecular Characterization of Microglia: A Unique Immune Cell Population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.L.; Goodsall, A.L.; Hickey, W.F.; Sedgwick, J.D. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995, 154, 4309–4321. [Google Scholar]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.-I.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathol. 2015, 36, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.-P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Poon, C.C.; Gordon, P.M.K.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.-Y.; Ling, X.; Caruso, H.G.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2018, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, H.; Minasi, S.; Ugolini, A.; Trevisi, G.; Napoletano, C.; Zizzari, I.G.; Gessi, M.; Giangaspero, F.; Mangiola, A.; Nuti, M.; et al. Immunohistochemical Characterization of Immune Infiltrate in Tumor Microenvironment of Glioblastoma. J. Pers. Med. 2020, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-Associated Microglia/Macrophages Display an Expression Profile Different from M1 and M2 Polarization and Highly Express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Maleszewska, M.; Steranka, A.; Smiech, M.; Kaza, B.; Pilanc, P.; Dabrowski, M.; Kaminska, B. Sequential changes in histone modifications shape transcriptional responses underlying microglia polarization by glioma. Glia 2021, 69, 109–123. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. Evidence for Lactate as a Neuronal Energy Source. J. Neurosci. 2011, 31, 7477. [Google Scholar] [CrossRef] [Green Version]
- Díaz-García, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017, 26, 361–374.e4. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the complex metabolic nature of astrocytes. Front. Cell. Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nat. Cell Biol. 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 2010, 13, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Pérez, L.A.; Choi, B.D.; Archer, G.E.; Cui, X.; Flores, C.; Johnson, L.A.; Schmittling, R.J.; Snyder, D.; Herndon, J.E.; Bigner, D.D.; et al. Myeloablative Temozolomide Enhances CD8+ T-Cell Responses to Vaccine and Is Required for Efficacy against Brain Tumors in Mice. PLoS ONE 2013, 8, e59082. [Google Scholar] [CrossRef] [Green Version]
- Hudson, A.L.; Parker, N.R.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Chen, J.; Wheeler, H.R.; Howell, V.M. Glioblastoma Recurrence Correlates With Increased APE1 and Polarization Toward an Immuno-Suppressive Microenvironment. Front. Oncol. 2018, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Côté, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro-Oncology 2011, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, L.; Werba, G.; Tiwari, S.; Ly, N.N.G.; Nguy, S.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Daley, D.; Barilla, R.; et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016, 150, 1659–1672.e5. [Google Scholar] [CrossRef] [Green Version]
- Majety, M.; Runza, V.; Lehmann, C.; Hoves, S.; Ries, C.H. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J. 2017, 285, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; AlShehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 2020, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.M.; Daguindau, E.; Rech, J.C.; Chinnaswamy, K.; Zhang, J.; Hura, G.L.; Griesenauer, B.; Bolten, Z.; Robida, A.; Larsen, M.; et al. From proteomics to discovery of first-in-class ST2 inhibitors active in vivo. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A.; et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D.I. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.-P.; He, H.-Y.; Wang, H.; Li, W.-S.; Luo, L.; Huang, Z.-C.; Guo, Y. Glioma-Derived ADAM10 Induces Regulatory B Cells to Suppress CD8+ T Cells. PLoS ONE 2014, 9, e105350. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nat. Cell Biol. 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Shand, F.H.W.; Ueha, S.; Otsuji, M.; Koid, S.S.; Shichino, S.; Tsukui, T.; Kosugi-Kanaya, M.; Abe, J.; Tomura, M.; Ziogas, J.; et al. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 7771–7776. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Akasaki, Y.; Liu, G.; Chung, N.H.C.; Ehtesham, M.; Black, K.L.; Yu, J.S. Induction of a CD4+ T Regulatory Type 1 Response by Cyclooxygenase-2-Overexpressing Glioma. J. Immunol. 2004, 173, 4352–4359. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, B.; Rayman, P.; Ireland, J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology 2011, 13, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-Oncology 2015, 18, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Petersen-Baltussen, H.M.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Increase in Both CD14-Positive and CD15-Positive Myeloid-Derived Suppressor Cell Subpopulations in the Blood of Patients With Glioma But Predominance of CD15-Positive Myeloid-Derived Suppressor Cells in Glioma Tissue. J. Neuropathol. Exp. Neurol. 2015, 74, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro-Oncology 2016, 18, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.-F.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008, 83, 1136–1144. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neuro-Oncology 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef]
- Mastio, J.; Condamine, T.; Dominguez, G.; Kossenkov, A.; Donthireddy, L.; Veglia, F.; Lin, C.; Wang, F.; Fu, S.; Zhou, J.; et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J. Exp. Med. 2019, 216, 2150–2169. [Google Scholar] [CrossRef] [Green Version]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.-L.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Goldstein, M.; Heylmann, D.; Kaina, B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE 2012, 7, e39956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Horn, L.A.; Ciavattone, N.G. Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy. Front. Oncol. 2019, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; DeSantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Lan, J.; Li, R.; Yin, L.-M.; Deng, L.; Gui, J.; Chen, B.-Q.; Zhou, L.; Meng, M.-B.; Huang, Q.-R.; Mo, X.-M.; et al. Targeting Myeloid-derived Suppressor Cells and Programmed Death Ligand 1 Confers Therapeutic Advantage of Ablative Hypofractionated Radiation Therapy Compared With Conventional Fractionated Radiation Therapy. Int. J. Radiat. Oncol. 2018, 101, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Colwell, N.A.; Heimberger, A.B. Translational Immunotherapy of Brain Tumors; Sampson, J.H., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 63–82. [Google Scholar]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Wu, C.; Muroski, M.E.; Miska, J.; Lee-Chang, C.; Shen, Y.; Rashidi, A.; Zhang, P.; Xiao, T.; Han, Y.; Lopez-Rosas, A.; et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomed.: Nanotechnol. Biol. Med. 2019, 16, 126–137. [Google Scholar] [CrossRef]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Massara, M.; Persico, P.; Bonavita, O.; Poeta, V.M.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [Green Version]
- Zadora, P.; Dabrowski, W.; Czarko, K.; Smolen, A.; Kotlinska-Hasiec, E.; Wiorkowski, K.; Sikora, A.; Jarosz, B.; Kura, K.; Rola, R.; et al. Preoperative neutrophil–Lymphocyte count ratio helps predict the grade of glial tumor–A pilot study. Neurol. Neurochir. Polska 2015, 49, 41–44. [Google Scholar] [CrossRef]
- Bambury, R.M.; Teo, M.Y.; Power, D.G.; Yusuf, A.; Murray, S.; Battley, J.E.; Drake, C.; O’Dea, P.; Bermingham, N.; Keohane, C.; et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J. Neuro-Oncol. 2013, 114, 149–154. [Google Scholar] [CrossRef]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.-A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil–lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neuro-Oncol. 2017, 132, 463–471. [Google Scholar] [CrossRef]
- Schernberg, A.; Nivet, A.; Dhermain, F.; Ammari, S.; Escande, A.; Pallud, J.; Louvel, G.; Deutsch, E. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin. Transl. Radiat. Oncol. 2018, 10, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, A.; Cederarv, M.; Wolmer-Solberg, N.; Tammik, C.; Stragliotto, G.; Peredo, I.; Fornara, O.; Xu, X.; Dzabic, M.; Taher, C.; et al. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. OncoImmunology 2015, 5, e1075693. [Google Scholar] [CrossRef] [Green Version]
- Hor, W.S.; Huang, W.L.; Lin, Y.S.; Yang, B.C. Cross-talk between tumor cells and neutrophils through the Fas (APO-1, CD95)/FasL system: Human glioma cells enhance cell viability and stimulate cytokine production in neutrophils. J. Leukoc. Biol. 2003, 73, 363–368. [Google Scholar] [CrossRef]
- Chio, C.C.; Wang, Y.S.; Chen, Y.L.; Lin, S.J.; Yang, B.C. Down-regulation of Fas-L in glioma cells by ribozyme reduces cell apoptosis, tumour-infiltrating cells, and liver damage but accelerates tumour formation in nude mice. Br. J. Cancer 2001, 85, 1185–1192. [Google Scholar] [CrossRef]
- Iwatsuki, K.-C.; Kumara, E.; Yoshimine, T.-C.; Nakagawa, H.-C.; Sato, M.; Hayakawa, T. Elastase expression by infiltrating neutrophils in gliomas. Neurol. Res. 2000, 22, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; De Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477. [Google Scholar] [CrossRef] [Green Version]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.K.; Waziri, A. Neutrophil Degranulation and Immunosuppression in Patients with GBM: Restoration of Cellular Immune Function by Targeting Arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef] [Green Version]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013, 62, 1745–1756. [Google Scholar] [CrossRef]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Chai, E.; Zhang, L.; Li, C. LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression. Cancer Manag. Res. 2019, 11, 7307–7315. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A Clonogenic Bone Marrow Progenitor Specific for Macrophages and Dendritic Cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Segura, E.; Amigorena, S. Identification of human inflammatory dendritic cells. Oncoimmunology 2013, 2, e23851. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-López, M.; Labiano, S.; Morales-Kastresana, A.; Rodríguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Coisne, C.; Enzmann, G.; Rottapel, R.; Engelhardt, B. Alpha4beta1 integrin mediates the recruitment of immature dendritic cells across the blood-brain barrier during experimental autoimmune encephalomyelitis. J. Immunol. 2010, 184, 7196–7206. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.D.; McMahon, E.J.; Schreiner, B.; Bailey, S.L. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann. N. Y. Acad. Sci. 2007, 1103, 179–191. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhao, Q.; Gabrusiewicz, K.; Kong, L.-Y.; Xia, X.; Wang, J.; Ott, M.; Xu, J.; Davis, R.E.; Huo, L.; et al. Author Correction: FGL2 promotes tumor progression in the CNS by suppressing CD103+ dendritic cell differentiation. Nat. Commun. 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nat. Cell Biol. 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H., 2nd. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Wei, H.; Wang, S.; Zhang, D.; Hou, S.; Qian, W.; Li, B.; Guo, H.; Kou, G.; He, J.; Wang, H.; et al. Targeted Delivery of Tumor Antigens to Activated Dendritic Cells via CD11c Molecules Induces Potent Antitumor Immunity in Mice. Clin. Cancer Res. 2009, 15, 4612–4621. [Google Scholar] [CrossRef] [Green Version]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.-I.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Chen, Y.; Hao, K.; Zheng, M.; Chen, B.; Li, K.; Wang, Y.; Zhang, W.; Zhang, Y.; Mou, X.; et al. CD103+ Cell Growth Factor Flt3L Enhances the Efficacy of Immune Checkpoint Blockades in Murine Glioblastoma Model. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.C.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.T.; Wildes, T.J.; Drake, J.A.; Moore, G.L.; Dean, B.D.; Abraham, R.S.; Mitchell, D.A. Lin−CCR2+ hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2021, 10, 18. https://doi.org/10.3390/cells10010018
De Leo A, Ugolini A, Veglia F. Myeloid Cells in Glioblastoma Microenvironment. Cells. 2021; 10(1):18. https://doi.org/10.3390/cells10010018
Chicago/Turabian StyleDe Leo, Alessandra, Alessio Ugolini, and Filippo Veglia. 2021. "Myeloid Cells in Glioblastoma Microenvironment" Cells 10, no. 1: 18. https://doi.org/10.3390/cells10010018
APA StyleDe Leo, A., Ugolini, A., & Veglia, F. (2021). Myeloid Cells in Glioblastoma Microenvironment. Cells, 10(1), 18. https://doi.org/10.3390/cells10010018