Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression

 ,
,
Abstract
:1. Introduction
2. Cytokines
2.1. Interleukin-10
2.2. Transforming Growth Factor-β
2.3. Type-II Inflammatory Cytokines
3. Metabolites
3.1. Tryptophan and Kynurenine
3.2. Adenosine
3.3. Nitric Oxide
3.4. L-Arginine
3.5. Prostaglandin-E2
4. Impairment of IFN-I Signalling
5. Hypoxia
6. Extracellular Vesicles
7. Exclusion of T Cells from the Tumour Bed and Disruption of T Cell Homeostasis
8. Inhibitory Receptors
8.1. T Cell Immunoglobulin and Mucin Domain-Containing 3 (TIM-3)
8.2. Lymphocyte Activation Gene-3 (LAG-3)
8.3. T cell Immunoreceptor with Ig and ITIM Domain (TIGIT)
8.4. Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4)
8.5. Programmed Cell Death Protein 1 (PD-1)
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Damotte, D.; Warren, S.; Arrondeau, J.; Boudou-Rouquette, P.; Mansuet-Lupo, A.; Biton, J.; Ouakrim, H.; Alifano, M.; Gervais, C.; Bellesoeur, A.; et al. The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J. Transl. Med. 2019, 17, 357. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Huang, R.-R.; Wen, D.-R.; Paul, E.; Wünsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Balakrishnan, L.; Sharma, K.; Khan, A.A.; Advani, J.; Gowda, H.; Tripathy, S.P.; Suar, M.; Pandey, A.; Gandotra, S.; et al. A network map of Interleukin-10 signaling pathway. J. Cell Commun. Signal. 2016, 10, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, D.F.; Zlotnik, A.; Vieira, P.; Mosmann, T.R.; Howard, M.; Moore, K.W.; Garra, A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 1991, 146, 3444. [Google Scholar]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815. [Google Scholar]
- Kim, B.-G.; Joo, H.-G.; Chung, I.-S.; Chung, H.Y.; Woo, H.-J.; Yun, Y.-S. Inhibition of interleukin-10 (IL-10) production from MOPC 315 tumor cells by IL-10 antisense oligodeoxynucleotides enhances cell-mediated immune responses. Cancer Immunol. Immunother. 2000, 49, 433–440. [Google Scholar] [CrossRef]
- Steinbrink, K.; Jonuleit, H.; Müller, G.; Schuler, G.; Knop, J.R.; Enk, A.H. Interleukin-10–Treated Human Dendritic Cells Induce a Melanoma-Antigen–Specific Anergy in CD8+ T Cells Resulting in a Failure to Lyse Tumor Cells. Blood 1999, 93, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8(+) T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312.e295. [Google Scholar] [CrossRef] [PubMed]
- Loercher, A.E.; Nash, M.A.; Kavanagh, J.J.; Platsoucas, C.D.; Freedman, R.S. Identification of an IL-10-Producing HLA-DR-Negative Monocyte Subset in the Malignant Ascites of Patients with Ovarian Carcinoma That Inhibits Cytokine Protein Expression and Proliferation of Autologous T Cells. J. Immunol. 1999, 163, 6251. [Google Scholar] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, D.; Wu, P.; Wang, Z.; Huang, J. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PLoS ONE 2015, 10, e0139598. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, R.; Cao, Y.; Gu, Y.; Lin, C.; Liu, X.; Lv, K.; He, X.; Fang, H.; Jin, K.; et al. Poor Clinical Outcomes and Immunoevasive Contexture in Intratumoral IL-10-Producing Macrophages Enriched Gastric Cancer Patients. Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Zhang, L.; Yang, X.; Li, H.; Ma, B.; Walton, S.; Wu, X.; Yuan, J.; Wang, T.; Liu, X. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum. Vaccines Immunother. 2020, 16, 2328–2332. [Google Scholar] [CrossRef]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 Elicits IFNg-Dependent Tumor Immune Surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef] [Green Version]
- Naing, A.; Wong, D.J.; Infante, J.R.; Korn, W.M.; Aljumaily, R.; Papadopoulos, K.P.; Autio, K.A.; Pant, S.; Bauer, T.M.; Drakaki, A.; et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): A multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019, 20, 1544–1555. [Google Scholar] [CrossRef]
- Teixeira, A.F.; ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-b directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O’Brien, D.; Ahmad, R.; et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 Is Essential for TGF-β to Convert Naive CD4+CD25- Cells to CD25+Foxp3+ Regulatory T Cells and for Expansion of These Cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of Multiple Cytokine Combinations and TGF-β on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-R.; Kwak, Y.; Yang, T.; Han, J.H.; Park, S.-H.; Ye, M.B.; Lee, W.; Sim, K.-Y.; Kang, J.-A.; Kim, Y.-C.; et al. Myeloid-Derived Suppressor Cells Are Controlled by Regulatory T Cells via TGF-b during Murine Colitis. Cell Rep. 2016, 17, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.R.; Wang, Y.X.; Guo, S.; Han, S.Y.; Li, H.H.; Jin, S.F. Prognostic significance of in situ and plasma levels of transforming growth factor β1, -2 and -3 in cutaneous melanoma. Mol. Med. Rep. 2015, 11, 4508–4512. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shen, C.; Wang, X.; Lai, Y.; Zhou, K.; Li, P.; Liu, L.; Che, G. Prognostic value of TGF-β in lung cancer: Systematic review and meta-analysis. BMC Cancer 2019, 19, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jiang, W.; Huang, W.; Ye, M.; Zhu, X. Prognostic Values of Transforming Growth Factor-Beta Subtypes in Ovarian Cancer. BioMed Res. Int. 2020, 2020, 2170606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, J.; Mao, K.; Deng, H.; Yang, Y.; Zhou, E.; Liu, J. Role of transforming growth factor-β1 in triple negative breast cancer patients. Int. J. Surg. 2017, 45, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef]
- Sow, H.S.; Ren, J.; Camps, M.; Ossendorp, F.; ten Dijke, P. Combined Inhibition of TGF-β Signaling and the PD-L1 Immune Checkpoint Is Differentially Effective in Tumor Models. Cells 2019, 8, 320. [Google Scholar] [CrossRef] [Green Version]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.M.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Biswas, T.; Gu, X.; Yang, J.; Ellies, L.G.; Sun, L.-Z. Attenuation of TGF-β signaling supports tumor progression of a mesenchymal-like mammary tumor cell line in a syngeneic murine model. Cancer Lett. 2014, 346, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. OncoImmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8+ Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [Green Version]
- Kabata, H.; Flamar, A.-L.; Mahlakõiv, T.; Moriyama, S.; Rodewald, H.-R.; Ziegler, S.F.; Artis, D. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol. 2020, 13, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.-S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza-Gonzalez, A.; Xu, K.; Wu, T.-C.; Aspord, C.; Tindle, S.; Marches, F.; Gallegos, M.; Burton, E.C.; Savino, D.; Hori, T.; et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J. Exp. Med. 2011, 208, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.B.; Gartner, J.G.; Leonard, W.J.; Ellison, C.A. Lack of Functional TSLP Receptors Mitigates Th2 Polarization and the Establishment and Growth of 4T1 Primary Breast Tumours but has Different Effects on Tumour Quantities in the Lung and Brain. Scand. J. Immunol. 2013, 78, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Sugaya, M.; Suga, H.; Oka, T.; Kawaguchi, M.; Miyagaki, T.; Fujita, H.; Sato, S. Thymic Stromal Chemokine TSLP Acts through Th2 Cytokine Production to Induce Cutaneous T-cell Lymphoma. Cancer Res. 2016, 76, 6241–6252. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- De Monte, L.; Wörmann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Ameri, A.H.; Moradi Tuchayi, S.; Zaalberg, A.; Park, J.H.; Ngo, K.H.; Li, T.; Lopez, E.; Colonna, M.; Lee, R.T.; Mino-Kenudson, M.; et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Pastille, E.; Wasmer, M.-H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, I.; Radosavljevic, G.; Mitrovic, M.; Lisnic Juranic, V.; McKenzie, A.N.J.; Arsenijevic, N.; Jonjic, S.; Lukic, M.L. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur. J. Immunol. 2011, 41, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Piazza, M.; Nowell, C.S.; Koch, U.; Durham, A.-D.; Radtke, F. Loss of Cutaneous TSLP-Dependent Immune Responses Skews the Balance of Inflammation from Tumor Protective to Tumor Promoting. Cancer Cell 2012, 22, 479–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochman, Y.; Leonard, W.J. The Role of Thymic Stromal Lymphopoietin in CD8+ T Cell Homeostasis. J. Immunol. 2008, 181, 7699–7705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shane, H.L.; Klonowski, K.D. A Direct and Nonredundant Role for Thymic Stromal Lymphopoietin on Antiviral CD8 T Cell Responses in the Respiratory Mucosa. J. Immunol. 2014, 192, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Turkoz, A.; Manivasagam, S.; Yockey, L.J.; Turkoz, M.; Kopan, R. Elevated Epidermal Thymic Stromal Lymphopoietin Levels Establish an Antitumor Environment in the Skin. Cancer Cell 2012, 22, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Cunningham, T.J.; Manivasagam, S.; Ngo, K.H.; Moradi Tuchayi, S.; Reddy, R.; Meyers, M.A.; DeNardo, D.G.; Yokoyama, W.M. Thymic stromal lymphopoietin blocks early stages of breast carcinogenesis. J. Clin. Investig. 2016, 126, 1458–1470. [Google Scholar] [CrossRef] [Green Version]
- Kienzl, M.; Hasenoehrl, C.; Valadez-Cosmes, P.; Maitz, K.; Sarsembayeva, A.; Sturm, E.; Heinemann, A.; Kargl, J.; Schicho, R. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. OncoImmunology 2020, 9, 1776059. [Google Scholar] [CrossRef]
- Gao, X.; Wang, X.; Yang, Q.; Zhao, X.; Wen, W.; Li, G.; Lu, J.; Qin, W.; Qi, Y.; Xie, F.; et al. Tumoral Expression of IL-33 Inhibits Tumor Growth and Modifies the Tumor Microenvironment through CD8+ T and NK Cells. J. Immunol. 2015, 194, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Ohno, T.; Nishii, N.; Bhingare, A.; Tachinami, H.; Kashima, Y.; Nagai, S.; Saito, H.; Nakae, S.; Azuma, M. Endogenous IL-33 exerts CD8+ T cell antitumor responses overcoming pro-tumor effects by regulatory T cells in a colon carcinoma model. Biochem. Biophys. Res. Commun. 2019, 518, 331–336. [Google Scholar] [CrossRef]
- Yang, Q.; Li, G.; Zhu, Y.; Liu, L.; Chen, E.; Turnquist, H.; Zhang, X.; Finn, O.J.; Chen, X.; Lu, B. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur. J. Immunol. 2011, 41, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-C.; Hwang, Y.-S.; Chen, Y.-Y.; Liu, C.-L.; Shen, C.-N.; Hong, W.-H.; Lo, S.-M.; Shen, C.-R. Interleukin-4 Supports the Suppressive Immune Responses Elicited by Regulatory T Cells. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; McLoed, A.G.; Saxon, J.A.; Habermann, A.C.; Connelly, L.; Dulek, D.; Peebles, R.S.; Fingleton, B.; et al. Interleukin-5 Facilitates Lung Metastasis by Modulating the Immune Microenvironment. Cancer Res. 2015, 75, 1624–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salomé, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.-S.; et al. ILC2-modulated T cell–to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.; Mulders, P.; Punt, C.J.; et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, M.; Do, M.H.; Chou, C.; Stamatiades, E.G.; Nixon, B.G.; Shi, W.; Zhang, X.; Li, P.; Gao, S.; et al. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 2020, 587, 121–125. [Google Scholar] [CrossRef]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-β suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef]
- Lorvik, K.B.; Hammarström, C.; Fauskanger, M.; Haabeth, O.A.W.; Zangani, M.; Haraldsen, G.; Bogen, B.; Corthay, A. Adoptive Transfer of Tumor-Specific Th2 Cells Eradicates Tumors by Triggering an In Situ Inflammatory Immune Response. Cancer Res. 2016, 76, 6864–6876. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, M.; Ito, T.; Tumes, D.J.; Endo, Y.; Onodera, A.; Hashimoto, K.; Motohashi, S.; Yamashita, M.; Nishimura, T.; Ziegler, S.F.; et al. Memory type 2 helper T cells induce long-lasting antitumor immunity by activating natural killer cells. Cancer Res. 2011, 71, 4790–4798. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Wagage, S.; John, B.; Krock, B.L.; Hall, A.O.H.; Randall, L.M.; Karp, C.L.; Simon, M.C.; Hunter, C.A. The Aryl Hydrocarbon Receptor Promotes IL-10 Production by NK Cells. J. Immunol. 2014, 192, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ye, Z.; Kijlstra, A.; Zhou, Y.; Yang, P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2014, 177, 521–530. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Riesenberg, R.; Weiler, C.; Spring, O.; Eder, M.; Buchner, A.; Popp, T.; Castro, M.; Kammerer, R.; Takikawa, O.; Hatz, R.A.; et al. Expression of Indoleamine 2,3-Dioxygenase in Tumor Endothelial Cells Correlates with Long-term Survival of Patients with Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishio, T.; Goto, S.; Tahara, K.; Tone, S.; Kawano, K.; Kitano, S. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martine, E.D.C.; Arjan, A.v.d.L.; Corine, J.H.; Jeroen, J.W.M.J.; Adri, Z.; Ruud, D.; Peter, J.M.V.; Bob, L.; Gert, J.O. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef]
- Astigiano, S.; Morandi, B.; Costa, R.; Mastracci, L.; D’Agostino, A.; Ratto, G.B.; Melioli, G.; Frumento, G. Eosinophil Granulocytes Account for Indoleamine 2,3-Dioxygenase-Mediated Immune Escape in Human Non Small Cell Lung Cancer. Neoplasia 2005, 7, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Suda, T.; Furuhashi, K.; Suzuki, M.; Fujie, M.; Hahimoto, D.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 2010, 67, 361–365. [Google Scholar] [CrossRef]
- Feder-Mengus, C.; Wyler, S.; Hudolin, T.; Ruszat, R.; Bubendorf, L.; Chiarugi, A.; Pittelli, M.; Weber, W.P.; Bachmann, A.; Gasser, T.C.; et al. High expression of indoleamine 2,3-dioxygenase gene in prostate cancer. Eur. J. Cancer 2008, 44, 2266–2275. [Google Scholar] [CrossRef] [Green Version]
- Ino, K.; Yoshida, N.; Kajiyama, H.; Shibata, K.; Yamamoto, E.; Kidokoro, K.; Takahashi, N.; Terauchi, M.; Nawa, A.; Nomura, S.; et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 2006, 95, 1555–1561. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandapathil, M.; Hilldorfer, B.; Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Ren, J.; Lang, S.; Jackson, E.K.; Gorelik, E.; Whiteside, T.L. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J. Biol. Chem. 2010, 285, 7176–7186. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic Regulation of the Expansion and Immunosuppressive Activity of CD11b+Gr1+ Cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.P.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-Deficient Mice Are Resistant to Carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Linden, J. Adenosine A2A Receptors Intrinsically Regulate CD8+ T Cells in the Tumor Microenvironment. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Flores-Santibáñez, F.; Fernández, D.; Meza, D.; Tejón, G.; Vargas, L.; Varela-Nallar, L.; Arredondo, S.; Guixé, V.; Rosemblatt, M.; Bono, M.R.; et al. CD73-mediated adenosine production promotes stem cell-like properties in mouse Tc17 cells. Immunology 2015, 146, 582–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yan, J.; Putheti, P.; Wu, Y.; Sun, X.; Toxavidis, V.; Tigges, J.; Kassam, N.; Enjyoji, K.; Robson, S.C.; et al. Isolated CD39 Expression on CD4+ T Cells Denotes both Regulatory and Memory Populations. Am. J. Transplant. 2009, 9, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.; Lin, X.; Gao, Y.; Zhu, Y. Prognositic value of CD73-adenosinergic pathway in solid tumor: A meta-analysis and systematic review. Oncotarget 2017, 8, 57327–57336. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K.; et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.K.; Sin, J.I.; Choi, J.; Park, S.H.; Lee, T.S.; Choi, Y.S. Overexpression of CD73 in epithelial ovarian carcinoma is associated with better prognosis, lower stage, better differentiation and lower regulatory T cell infiltration. J. Gynecol. Oncol. 2012, 23, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Supernat, A.; Markiewicz, A.; Welnicka-Jaskiewicz, M.; Seroczynska, B.; Skokowski, J.; Sejda, A.; Szade, J.; Czapiewski, P.; Biernat, W.; Zaczek, A. CD73 expression as a potential marker of good prognosis in breast carcinoma. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 103–107. [Google Scholar] [CrossRef]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e2419. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Ekmekcioglu, S.; Grimm, E.A.; Roszik, J. Targeting iNOS to increase efficacy of immunotherapies. Hum. Vaccines Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-N.; Hsieh, F.-J.; Cheng, Y.-M.; Chang, K.-J.; Lee, P.-H. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in angiogenesis and clinical outcome of human gastric cancer. J. Surg. Oncol. 2006, 94, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Amunni, G.; Villanucci, A.; Boddi, V.; Baroni, G.; Taddei, A.; Taddei, G.L. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in ovarian cancer: Correlation with clinical outcome. Gynecol. Oncol. 2004, 92, 806–812. [Google Scholar] [CrossRef]
- Ambs, S.; Merriam, W.G.; Bennett, W.P.; Felley-Bosco, E.; Ogunfusika, M.O.; Oser, S.M.; Klein, S.; Shields, P.G.; Billiar, T.R.; Harris, C.C. Frequent nitric oxide synthase-2 expression in human colon adenomas: Implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998, 58, 334–341. [Google Scholar] [PubMed]
- Chhatwal, V.J.S.; Ngoi, S.S.; Chan, S.T.F.; Chia, Y.W.; Moochhala, S.M. Aberrant expression of nitric oxide synthase in human polyps, neoplastic colonic mucosa and surrounding peritumoral normal mucosa. Carcinogenesis 1994, 15, 2081–2085. [Google Scholar] [CrossRef] [PubMed]
- Anttila, M.A.; Voutilainen, K.; Merivalo, S.; Saarikoski, S.; Kosma, V.-M. Prognostic significance of iNOS in epithelial ovarian cancer. Gynecol. Oncol. 2007, 105, 97–103. [Google Scholar] [CrossRef]
- Ropponen, K.M.; Kellokoski, J.K.; Lipponen, P.K.; Eskelinen, M.J.; Alanne, L.; Alhava, E.M.; Kosma, V.M. Expression of Inducible Nitric Oxide Synthase in Colorectal Cancer and Its Association with Prognosis. Scand. J. Gastroenterol. 2000, 35, 1204–1211. [Google Scholar] [CrossRef]
- Bingisser, R.M.; Tilbrook, P.A.; Holt, P.G.; Kees, U.R. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734. [Google Scholar]
- Olivier, H.; James, K.L. Inhibition of MHC II Gene Transcription by Nitric Oxide and Antioxidants. Curr. Pharm. Des. 2004, 10, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Rivoltini, L.; Carrabba, M.; Huber, V.; Castelli, C.; Novellino, L.; Dalerba, P.; Mortarini, R.; Arancia, G.; Anichini, A.; Fais, S.; et al. Immunity to cancer: Attack and escape in T lymphocyte–tumor cell interaction. Immunol. Rev. 2002, 188, 97–113. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Ambs, S.; Merriam, W.G.; Ogunfusika, M.O.; Bennett, W.P.; Ishibe, N.; Hussain, S.P.; Tzeng, E.E.; Geller, D.A.; Billiar, T.R.; Harris, C.C. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med. 1998, 4, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zhu, C.; Li, F.; Hegazi, R.; He, K.; Babyatsky, M.; Bauer, A.J.; Plevy, S.E. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J. Biol. Chem. 2004, 279, 10776–10783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, P.; Alfarano, M.G.; Svider, P.F.; Parikh, F.; Lu, G.; Kidwai, S.; Xiong, H.; Sikora, A.G. iNOS expression in CD4+ T cells limits Treg induction by repressing TGFβ1: Combined iNOS inhibition and Treg depletion unmask endogenous antitumor immunity. Clin. Cancer Res. 2014, 20, 6439–6451. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Munder, M.; Eichmann, K.; Morán, J.M.; Centeno, F.; Soler, G.; Modolell, M. Th1/Th2-Regulated Expression of Arginase Isoforms in Murine Macrophages and Dendritic Cells. J. Immunol. 1999, 163, 3771–3777. [Google Scholar]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bron, L.; Jandus, C.; Andrejevic-Blant, S.; Speiser, D.E.; Monnier, P.; Romero, P.; Rivals, J.-P. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, E85–E93. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Ma, Z.; Lian, J.; Yang, M.; Wuyang, J.; Zhao, C.; Chen, W.; Liu, C.; Zhao, Q.; Lou, C.; Han, J.; et al. Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol. Res. Pract. 2019, 215, 152383. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chen, W.; Chen, J.; Zheng, Q.; Dong, J.; Zhu, Y. The Oncogenic Role of ARG1 in Progression and Metastasis of Hepatocellular Carcinoma. BioMed Res. Int. 2018, 2018, 2109865. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.-J.; Zabaleta, J.; Ochoa, A.C. l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Martí i Líndez, A.-A.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Núñez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K.; et al. Mitochondrial arginase-2 is a cell-autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight 2020, 4. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Tsai, F.Y.-C.; Bauer, T.M.; Muigai, L.; Liang, Y.; Bennett, M.K.; Orford, K.W.; Fu, S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. J. Clin. Oncol. 2017, 35, 3005. [Google Scholar] [CrossRef]
- Yau, T.; Cheng, P.N.; Chan, P.; Chan, W.; Chen, L.; Yuen, J.; Pang, R.; Fan, S.T.; Poon, R.T. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Investig. New Drugs 2013, 31, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Kalinski, P. Regulation of Immune Responses by Prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Parikh, A.; Shapiro, G.I.; Varga, A.; Naing, A.; Meric-Bernstam, F.; Ataman, Ö.; Reyderman, L.; Binder, T.A.; Ren, M.; et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 2020, 8, e000222. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kim, H.T.; Nellore, A.; Patsoukis, N.; Petkova, V.; McDonough, S.; Politikos, I.; Nikiforow, S.; Soiffer, R.; Antin, J.H.; et al. Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/β-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J. 2014, 4, e178. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guan, K.; Zhou, Y.; Wu, J.; Wang, Y.; Wang, W. Prostaglandin E2 signal inhibits T regulatory cell differentiation during allergic rhinitis inflammation through EP4 receptor. World Allergy Organ. J. 2019, 12, 100090. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.M.; Kong, W.; Ganea, D. Prostaglandin E2 inhibits Tr1 cell differentiation through suppression of c-Maf. PLoS ONE 2017, 12, e0179184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youlin, K.; Weiyang, H.; Simin, L.; Xin, G. Prostaglandin E(2) Inhibits Prostate Cancer Progression by Countervailing Tumor Microenvironment-Induced Impairment of Dendritic Cell Migration through LXRα/CCR7 Pathway. J. Immunol. Res. 2018, 2018, 5808962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierlich, P.; Lex, V.; Technau, A.; Keupp, A.; Morper, L.; Glunz, A.; Sennholz, H.; Rachor, J.; Sauer, S.; Marcu, A.; et al. Prostaglandin E2 in a TLR3- and 7/8-agonist-based DC maturation cocktail generates mature, cytokine-producing, migratory DCs but impairs antigen cross-presentation to CD8+ T cells. Cancer Immunol. Immunother. 2020, 69, 1029–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzai, A.C.; Wagner, T.; Audsley, K.M.; Newnes, H.V.; Barrett, L.W.; Barnes, S.; Wylie, B.C.; Stone, S.; McDonnell, A.; Fear, V.S.; et al. Diverse Anti-Tumor Immune Potential Driven by Individual IFNα Subtypes. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.F.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-α Production by Plasmacytoid Dendritic Cells Favors Regulatory T-cell Expansion That May Contribute to Breast Cancer Progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, S.; Zhou, M.; Pan, Z.; Sun, R.; Chen, M.; Jiang, H.; Shi, B.; Luo, H.; Li, Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Su, T.; Zhang, Y.; Valerie, K.; Wang, X.Y.; Lin, S.; Zhu, G. STING activation in cancer immunotherapy. Theranostics 2019, 9, 7759–7771. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef] [Green Version]
- Provance, O.K.; Lewis-Wambi, J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019, 21, 59. [Google Scholar] [CrossRef] [Green Version]
- Effern, M.; Glodde, N.; Braun, M.; Liebing, J.; Boll, H.N.; Yong, M.; Bawden, E.; Hinze, D.; van den Boorn-Konijnenberg, D.; Daoud, M.; et al. Adoptive T Cell Therapy Targeting Different Gene Products Reveals Diverse and Context-Dependent Immune Evasion in Melanoma. Immunity 2020, 53, 564–580.e569. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Brooks, J.M.; Menezes, A.N.; Ibrahim, M.; Archer, L.; Lal, N.; Bagnall, C.J.; von Zeidler, S.V.; Valentine, H.R.; Spruce, R.J.; Batis, N.; et al. Development and Validation of a Combined Hypoxia and Immune Prognostic Classifier for Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 5315–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Wu, S.; Chen, X.; Ye, Y.; Weng, Y.; Pan, Y.; Chen, Z.; Chen, L.; Qiu, X.; Qiu, S. Characterization of Hypoxia Signature to Evaluate the Tumor Immune Microenvironment and Predict Prognosis in Glioma Groups. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B.; Wang, X.; Guan, G.; Xin, Y.; Sun, Y.D.; Wang, J.H.; Guo, Y.; Zang, Y.J. Macrophage transcriptome modification induced by hypoxia and lactate. Exp. Ther. Med. 2019, 18, 4811–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Hammami, A.; Charpentier, T.; Smans, M.; Stäger, S. IRF-5-Mediated Inflammation Limits CD8+ T Cell Expansion by Inducing HIF-1α and Impairing Dendritic Cell Functions during Leishmania Infection. PLOS Pathog. 2015, 11, e1004938. [Google Scholar] [CrossRef]
- Yang, M.; Ma, C.; Liu, S.; Sun, J.; Shao, Q.; Gao, W.; Zhang, Y.; Li, Z.; Xie, Q.; Dong, Z.; et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology 2009, 128, e237–e249. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Chen, K.; Cheng, Y.; Liu, S.; Xia, M.; Chen, Y.; Zhu, H.; Li, Z.; Cao, X. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity 2019, 50, 600–615.e615. [Google Scholar] [CrossRef] [Green Version]
- Blengio, F.; Raggi, F.; Pierobon, D.; Cappello, P.; Eva, A.; Giovarelli, M.; Varesio, L.; Bosco, M.C. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology 2013, 218, 76–89. [Google Scholar] [CrossRef]
- Weigert, A.; Weichand, B.; Sekar, D.; Sha, W.; Hahn, C.; Mora, J.; Ley, S.; Essler, S.; Dehne, N.; Brüne, B. HIF-1α is a negative regulator of plasmacytoid DC development in vitro and in vivo. Blood 2012, 120, 3001–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 2016, 7, 75763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhong, M.; Wang, C.; Xu, Y.; Gao, W.-Q.; Zhang, Y. CCL5-deficiency enhances intratumoral infiltration of CD8+ T cells in colorectal cancer. Cell Death Dis. 2018, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Vuillefroy de Silly, R.; Dietrich, P.-Y.; Walker, P.R. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? OncoImmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [Green Version]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-N.; Yang, J.-F.; Huang, D.-J.; Ni, H.-H.; Zhang, C.-X.; Zhang, L.; He, J.; Gu, J.-M.; Chen, H.-X.; Mai, H.-Q.; et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8+ T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Yan, Y.; Keng, S. Exosomes in the ascites of ovarian cancer patients: Origin and effects on anti-tumor immunity. Oncol. Rep. 2011, 25, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, S.; Zheng, X.; Zheng, P.; Fu, Y.; Wu, C.; Lu, B.; Ju, J.; Jiang, J. Immune suppressed tumor microenvironment by exosomes derived from gastric cancer cells via modulating immune functions. Sci. Rep. 2020, 10, 14749. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human Tumor-Derived Exosomes Selectively Impair Lymphocyte Responses to Interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Piranlioglu, R.; Achyut, B.R.; Korkaya, H.; Liu, Y.; Arbab, A.S. The critical immunosuppressive effect of MDSC-derived exosomes in the tumor microenvironment. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.-C.; Guo, S.-C. Role of extracellular vesicles in tumour microenvironment. Cell Commun. Signal. 2020, 18, 163. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- De Sousa Andrade, L.N.; Otake, A.H.; Cardim, S.G.B.; da Silva, F.I.; Ikoma Sakamoto, M.M.; Furuya, T.K.; Uno, M.; Pasini, F.S.; Chammas, R. Extracellular Vesicles Shedding Promotes Melanoma Growth in Response to Chemotherapy. Sci. Rep. 2019, 9, 14482. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A.M. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front. Immunol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivarcsi, A.; Müller, A.; Hippe, A.; Rieker, J.; van Lierop, A.; Steinhoff, M.; Seeliger, S.; Kubitza, R.; Pippirs, U.; Meller, S.; et al. Tumor immune escape by the loss of homeostatic chemokine expression. Proc. Natl. Acad. Sci. USA 2007, 104, 19055–19060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, J.M.; Gomez, A.C.; Aguilar, A.; Salgado, R.; Balko, J.M.; Bravo, L.; Doimi, F.; Bretel, D.; Morante, Z.; Flores, C.; et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018, 8, 4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, F.; Ping, Y.; Wang, L.; Chen, X.; Wang, D.; Cao, L.; Zhao, S.; Li, B.; Kalinski, P.; et al. Local production of the chemokines CCL5 and CXCL10 attracts CD8 + T lymphocytes into esophageal squamous cell carcinoma. Oncotarget 2015, 6, 24978. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Alonso, M.; Hirsch, T.; Wildmann, C.; van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 2017, 8, 793. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Froemming, A.; Vater, A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Bouzin, C.; Brouet, A.; De Vriese, J.; DeWever, J.; Feron, O. Effects of Vascular Endothelial Growth Factor on the Lymphocyte-Endothelium Interactions: Identification of Caveolin-1 and Nitric Oxide as Control Points of Endothelial Cell Anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.A.; Darrah, P.A.; Foulds, K.E.; Hoffer, E.; Caffrey-Carr, A.; Norenstedt, S.; Perbeck, L.; Seder, R.A.; Kedl, R.M.; Loré, K. Monocytes Acquire the Ability to Prime Tissue-Resident T Cells via IL-10-Mediated TGF-b Release. Cell Rep. 2019, 28, 1127–1135.e1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Bevan, M.J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, A.P.; Braun, A.; Ritchie, S.C.; Carbone, F.R.; Mackay, L.K.; Gebhardt, T.; Inouye, M. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 2019, 14, e0210495. [Google Scholar] [CrossRef]
- Harly, C.; Kenney, D.; Wang, Y.; Ding, Y.; Zhao, Y.; Awasthi, P.; Bhandoola, A. A Shared Regulatory Element Controls the Initiation of Tcf7 Expression During Early T Cell and Innate Lymphoid Cell Developments. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef] [Green Version]
- Danilo, M.; Chennupati, V.; Silva, J.G.; Siegert, S.; Held, W. Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 2018, 22, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Wiesel, M.; Crouse, J.; Bedenikovic, G.; Sutherland, A.; Joller, N.; Oxenius, A. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol. 2012, 42, 320–329. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Lord, G.M.; Dardalhon, V.; Lee, D.H.; Sabatos-Peyton, C.A.; Glimcher, L.H.; Kuchroo, V.K. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur. J. Immunol. 2010, 40, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015, 6, 6072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8⁺ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Ngiow, S.F.; Sullivan, J.M.; Teng, M.W.L.; Kuchroo, V.K.; Smyth, M.J.; Anderson, A.C. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology 2013, 2, e23849. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, Y.; Zhang, J.-P.; Liang, J.; Li, L.; Zheng, L. Tim-3 Expression Defines Regulatory T Cells in Human Tumors. PLoS ONE 2013, 8, e58006. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Qin, S.; Dong, B.; Yi, M.; Chu, Q.; Wu, K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Patnaik, A.; Moreno, V.; Stein, M.; Jankowska, A.M.; Mendizabal, N.V.d.; Liu, Z.T.; Koneru, M.; Calvo, E. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J. Clin. Oncol. 2019, 37, 12. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, W.M.D.; Forde, P.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, S.; et al. Abstract CT183: Phase (Ph) I/II study of MBG453± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019, 79, CT183. [Google Scholar] [CrossRef]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134, 570. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-m.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef] [PubMed]
- Maçon-Lemaître, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Bruniquel, D.; Borie, N.; Hannier, S.; Triebel, F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 1998, 48, 116–124. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [Green Version]
- Zelba, H.; Bedke, J.; Hennenlotter, J.; Mostböck, S.; Zettl, M.; Zichner, T.; Chandran, A.; Stenzl, A.; Rammensee, H.-G.; Gouttefangeas, C. PD-1 and LAG-3 Dominate Checkpoint Receptor–Mediated T-cell Inhibition in Renal Cell Carcinoma. Cancer Immunol. Res. 2019, 7, 1891–1899. [Google Scholar] [CrossRef]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.B.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4+CD25highFoxp3+ Regulatory T Cells That Are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.-W.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Ma, S.-R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. OncoImmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.R.; Peinado, P.; Fuentes-Antrás, J.; Pérez-Segura, P.; Pandiella, A.; Amir, E.; Ocaña, A. Prognostic Value of Lymphocyte-Activation Gene 3 (LAG3) in Cancer: A Meta-Analysis. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.; Ulahannan, S.V.; Chu, Q.; Patel, M.; George, B.; Auguste, A.; Leo-Kress, T.; Stadermann, K.B.; Kraemer, N.; Elgadi, M.; et al. Abstract 779: A phase I study of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb: Biomarker analyses from the microsatellite stable metastatic colorectal cancer (MSS mCRC) cohort. Cancer Res. 2020, 80, 779. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Lakhani, N.J.; Johnson, M.L.; Park, H.; Wang, D.; Yap, T.A.; Dowlati, A.; Maki, R.G.; Lynce, F.; Ulahannan, S.V.; et al. First-in-human study of REGN3767 (R3767), a human LAG-3 monoclonal antibody (mAb), ± cemiplimab in patients (pts) with advanced malignancies. J. Clin. Oncol. 2019, 37, 2508. [Google Scholar] [CrossRef]
- Casati, C.; Camisaschi, C.; Rini, F.; Arienti, F.; Rivoltini, L.; Triebel, F.; Parmiani, G.; Castelli, C. Soluble Human LAG-3 Molecule Amplifies the In vitro Generation of Type 1 Tumor-Specific Immunity. Cancer Res. 2006, 66, 4450–4460. [Google Scholar] [CrossRef] [Green Version]
- Prigent, P.; El mir, S.; Dréano, M.; Triebel, F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur. J. Immunol. 1999, 29, 3867–3876. [Google Scholar] [CrossRef]
- El mir, S.; Triebel, F. A Soluble Lymphocyte Activation Gene-3 Molecule Used as a Vaccine Adjuvant Elicits Greater Humoral and Cellular Immune Responses to Both Particulate and Soluble Antigens. J. Immunol. 2000, 164, 5583–5589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Jilisihan, B.; Wang, W.; Tang, Y.; Keyoumu, S. Soluble LAG3 acts as a potential prognostic marker of gastric cancer and its positive correlation with CD8+T cell frequency and secretion of IL-12 and INF-γ in peripheral blood. Cancer Biomark. 2018, 23, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Legat, A.; Maby-El Hajjami, H.; Baumgaertner, P.; Cagnon, L.; Abed Maillard, S.; Geldhof, C.; Iancu, E.M.; Lebon, L.; Guillaume, P.; Dojcinovic, D.; et al. Vaccination with LAG-3Ig (IMP321) and Peptides Induces Specific CD4 and CD8 T-Cell Responses in Metastatic Melanoma Patients—Report of a Phase I/IIa Clinical Trial. Clin. Cancer Res. 2016, 22, 1330–1340. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Q.; Wang, Z.; Zhang, H.; Zeng, H.; Huang, Q.; Chen, Y.; Jiang, W.; Lin, Z.; Qu, Y.; et al. Intratumoral TIGIT+ CD8+ T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J. Immunother. Cancer 2020, 8, e000978. [Google Scholar] [CrossRef]
- Xu, D.; Zhao, E.; Zhu, C.; Zhao, W.; Wang, C.; Zhang, Z.; Zhao, G. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology 2020, 225, 151915. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, J.; Chen, Y.; Cui, J.; Lei, Y.; Cui, Y.; Jiang, N.; Jiang, W.; Chen, L.; Chen, Y.; et al. Combined evaluation of the expression status of CD155 and TIGIT plays an important role in the prognosis of LUAD (lung adenocarcinoma). Int. Immunopharmacol. 2020, 80, 106198. [Google Scholar] [CrossRef]
- Duan, X.; Liu, J.; Cui, J.; Ma, B.; Zhou, Q.; Yang, X.; Lu, Z.; Du, Y.; Su, C. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol. Med. Rep. 2019, 20, 3773–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, D.; Duong, S.; Wingerath, J.; Woller, N.; Manns, M.P.; Timrott, K.; Kleine, M.; Ramackers, W.; Roessler, S.; Nahnsen, S.; et al. Transcriptome profiling identifies TIGIT as a marker of T cell exhaustion in liver cancer. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.-F.; Joller, N.; Tan, D.J.; Teng, M.W.L.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepletier, A.; Madore, J.; O’Donnell, J.S.; Johnston, R.L.; Li, X.-Y.; McDonald, E.; Ahern, E.; Kuchel, A.; Eastgate, M.; Pearson, S.-A.; et al. Tumor CD155 expression is associated with resistance to anti-PD1 immunotherapy in metastatic melanoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity 2020, 53, 805–823.e815. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-h.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Stamper, C.C.; Zhang, Y.; Tobin, J.F.; Erbe, D.V.; Ikemizu, S.; Davis, S.J.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4–Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Liu, Q.; Deng, G.; Zhang, J.; Liang, N.; Xie, J.; Zhang, J. The prognostic value of cytotoxic T-lymphocyte antigen 4 in cancers: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 42913. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Salmons, J.; Nowak, A.K.; Rozali, E.N.; Khong, A.; Dick, I.M.; Harken, J.A.; Robinson, B.W.; Lake, R.A. Synergistic Effect of CTLA-4 Blockade and Cancer Chemotherapy in the Induction of Anti-Tumor Immunity. PLoS ONE 2013, 8, e61895. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kim, D.W.; Bassett, R.L.; Cain, S.; Washington, E.; Hwu, W.J.; Kim, K.B.; Papadopoulos, N.E.; Homsi, J.; Hwu, P.; et al. A phase II study of ipilimumab plus temozolomide in patients with metastatic melanoma. Cancer Immunol. Immunother. 2017, 66, 1359–1366. [Google Scholar] [CrossRef]
- Qian, J.M.; Martin, A.M.; Martin, K.; Hammoudeh, L.; Catalano, P.J.; Hodi, F.S.; Cagney, D.N.; Haas-Kogan, D.A.; Schoenfeld, J.D.; Aizer, A.A. Response rate and local recurrence after concurrent immune checkpoint therapy and radiotherapy for non-small cell lung cancer and melanoma brain metastases. Cancer 2020, 126, 5274–5282. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Chaput-Gras, N.; Lanoy, E.; Larive, A.; Mateus, C.; Routier, E.; Sun, R.; Tao, Y.G.; Massard, C.; Bahleda, R.; et al. Dose escalation phase 1 study of radiotherapy in combination with anti-cytotoxic-T-lymphocyte-associated antigen 4 monoclonal antibody ipilimumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, A.A.; Yu, T.F.-Y.; Leach, D.R.; Allison, J.P. CTLA-4 blockade synergizes with tumor-derived granulocyte– macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 10067–10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kojima, Y.; Uno, T.; Hayakawa, Y.; Teng, M.W.L.; Yoshizawa, H.; Yagita, H.; Gejyo, F.; Okumura, K.; Smyth, M.J. Combination Therapy of Established Tumors by Antibodies Targeting Immune Activating and Suppressing Molecules. J. Immunol. 2010, 184, 5493–5501. [Google Scholar] [CrossRef] [Green Version]
- Redmond, W.L.; Linch, S.N.; Kasiewicz, M.J. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol. Res. 2014, 2, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, S.; Massi, D.; Mandalà, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Pembrolizumab: First Global Approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Shergold, A.L.; Millar, R.; Nibbs, R.J.B. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol. Res. 2019, 145, 104258. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- López-Soto, A.; Gonzalez, S.; Folgueras, A.R. IFN Signaling and ICB Resistance: Time is on Tumor’s Side. Trends Cancer 2017, 3, 161–163. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2019, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef] [Green Version]





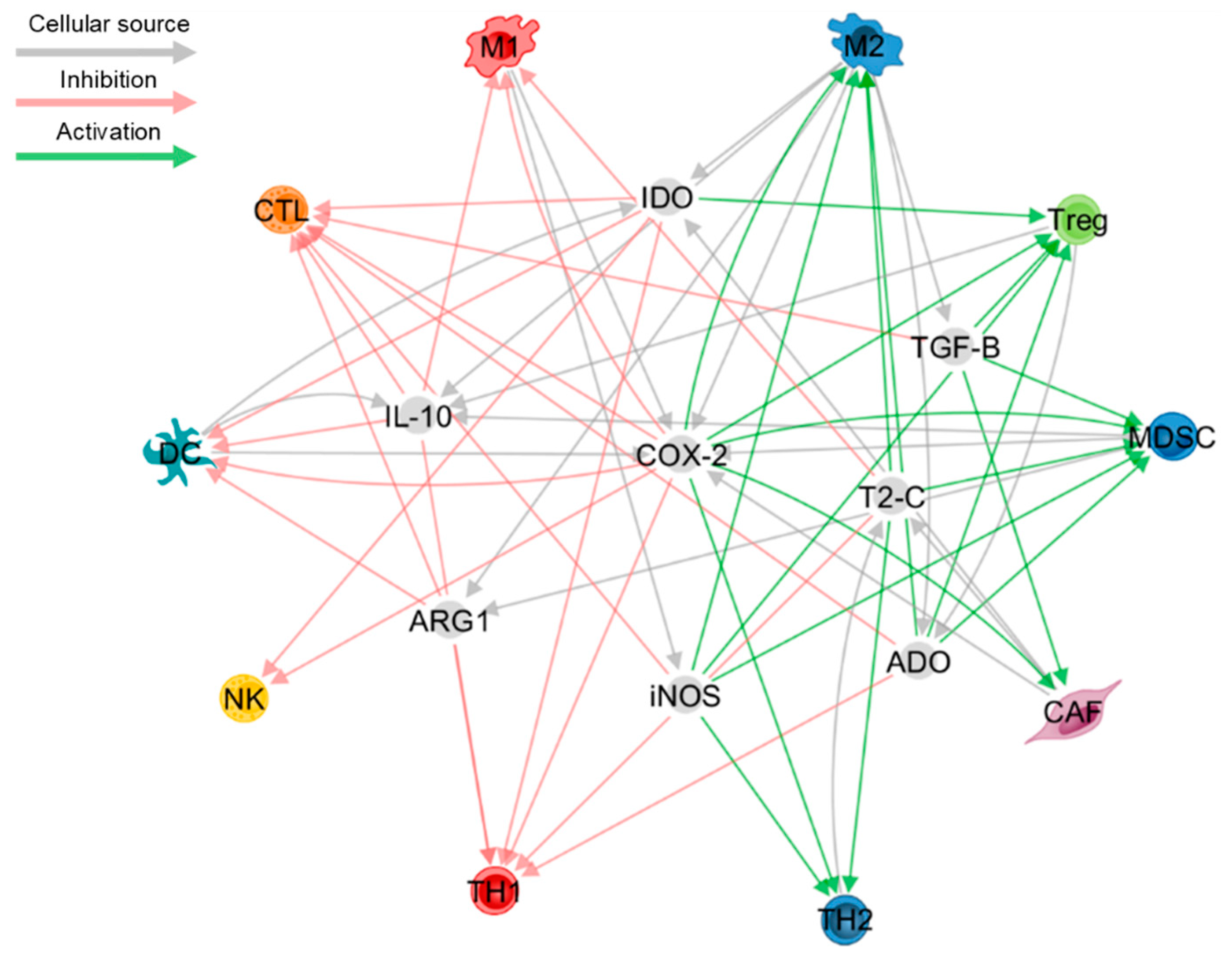{kind=link}
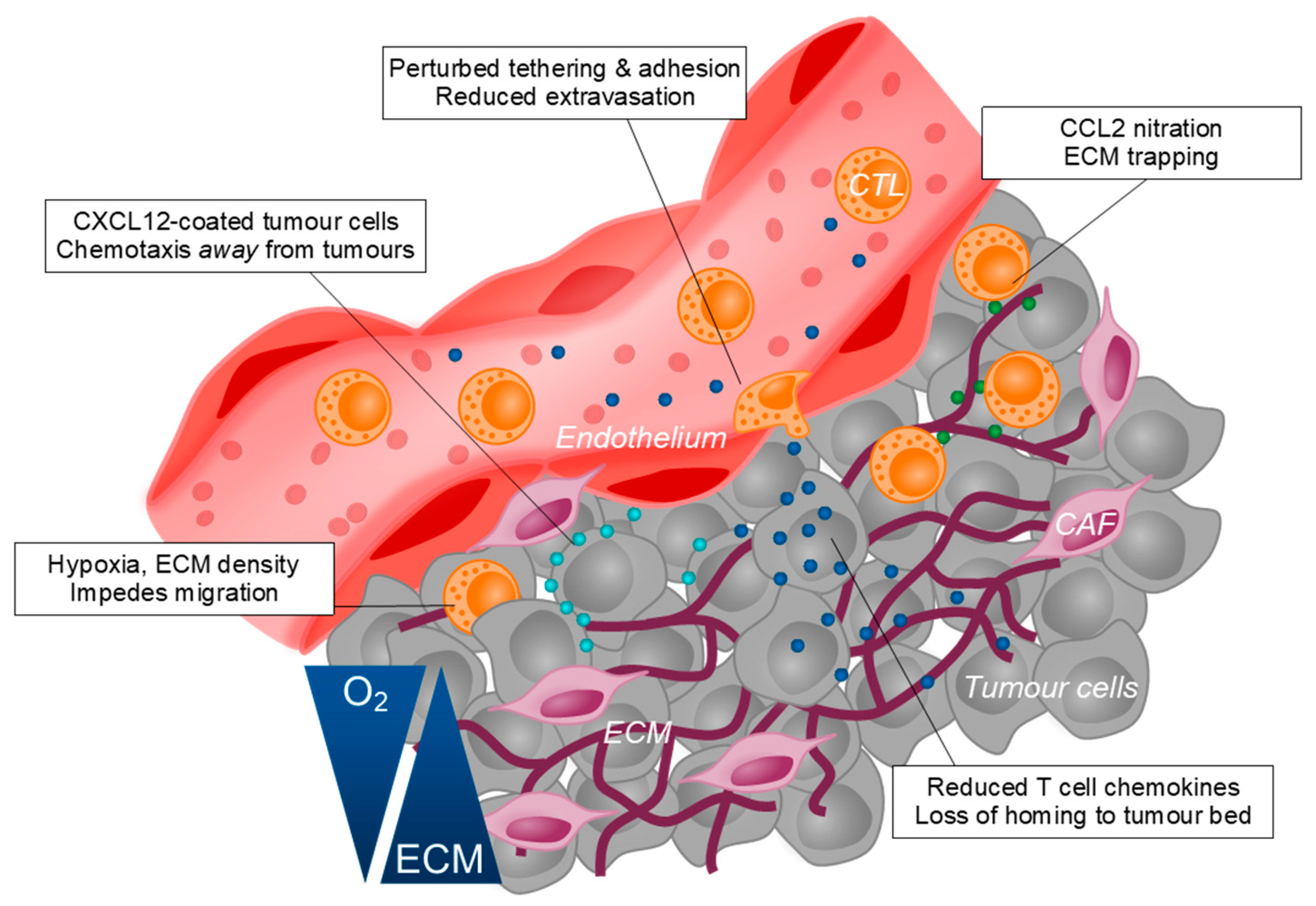{kind=link}
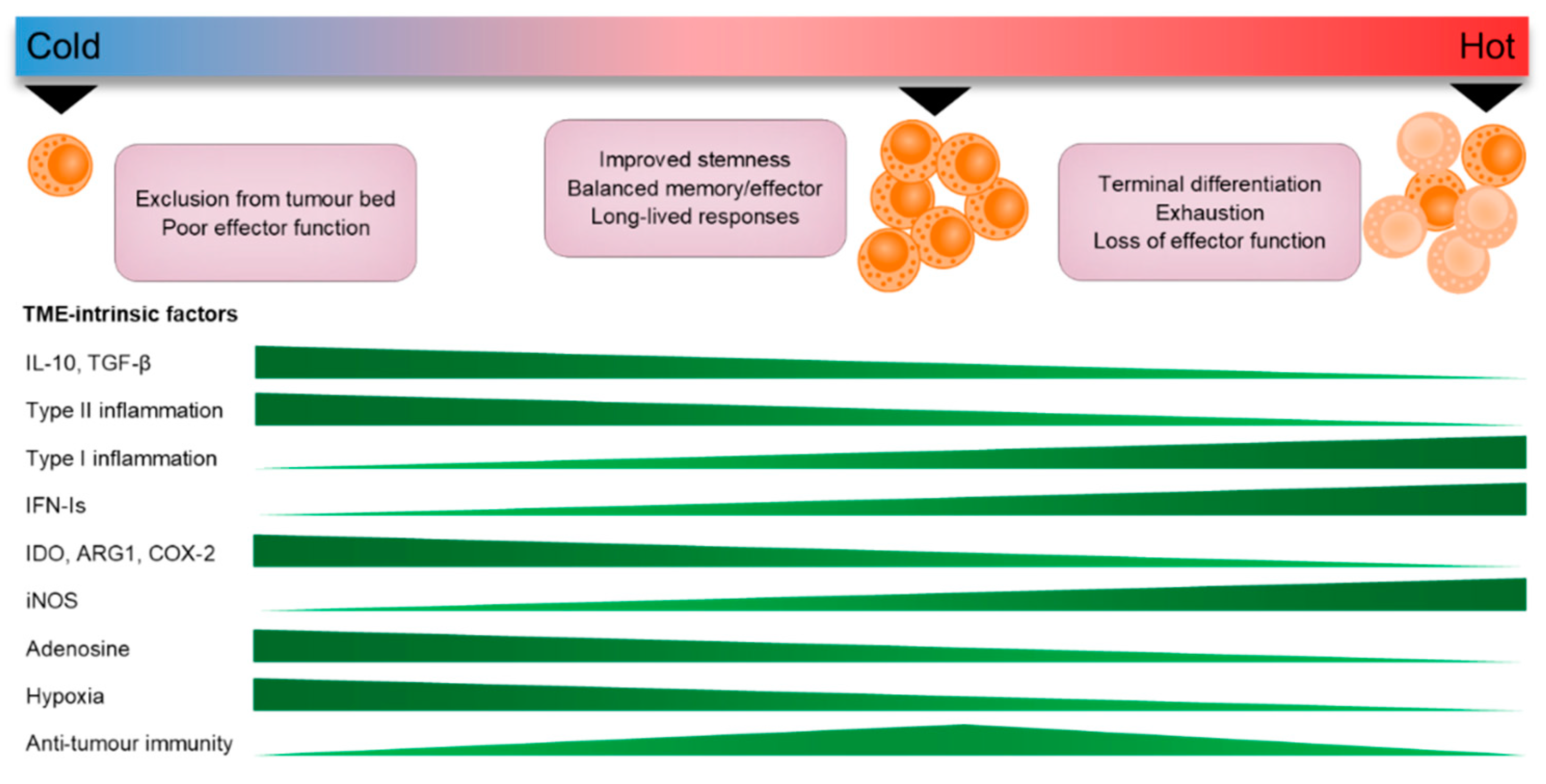{kind=link}
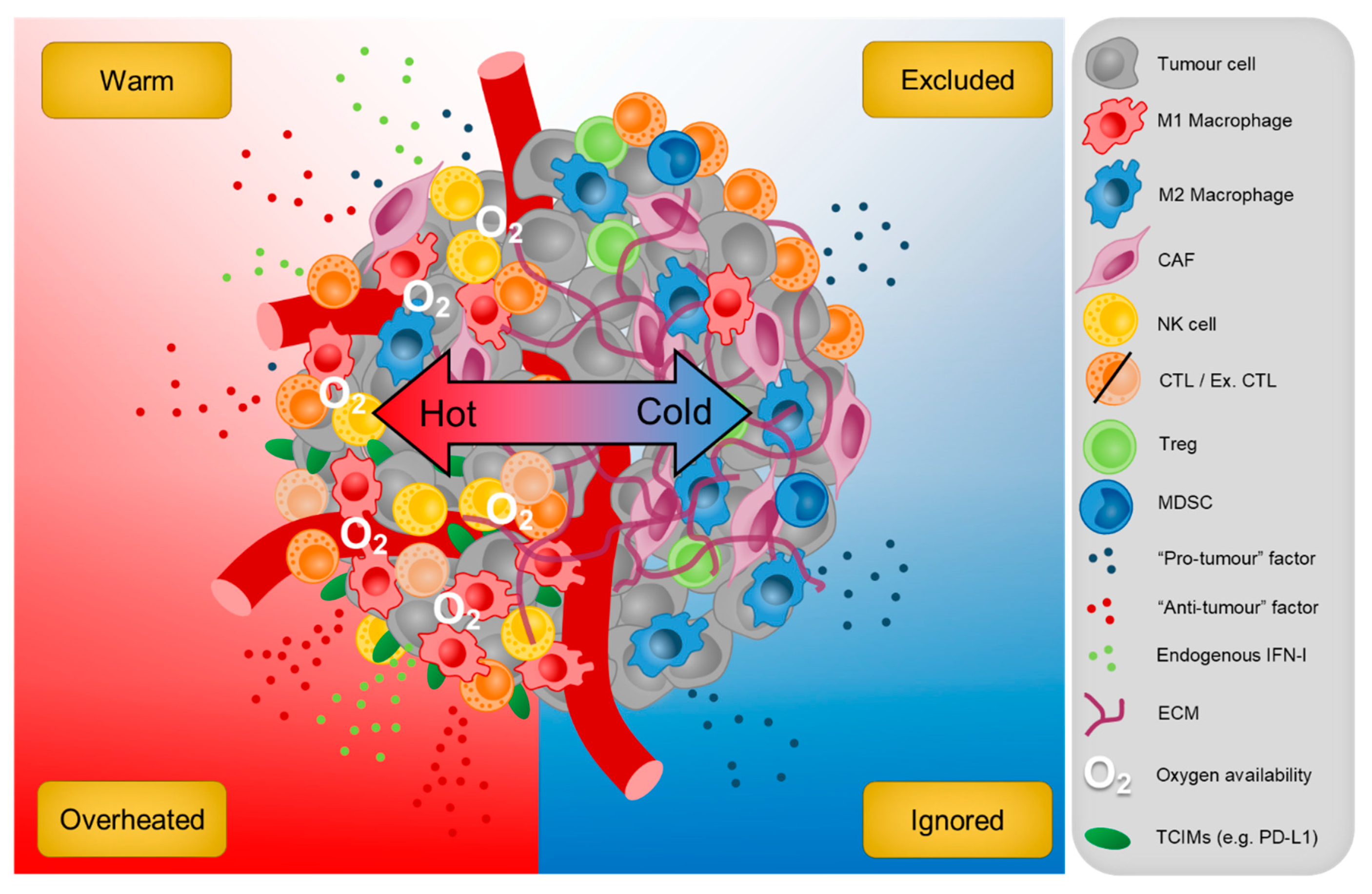{kind=link}
| Product | Description | Clinical Stage | Cancer(s) | Trial No. |
|---|---|---|---|---|
| A. TIM-3 | ||||
| Sym023 (Symphogen) | IgG1 mAb | 1 | Advanced solid cancer or lymphoma | NCT03489343 NCT03311412 |
| TSR-022 (Tesaro) | Humanised IgG4 mAb | 2 | HCC, melanoma, advanced solid cancers | NCT03680508 NCT02817633 NCT03307785 |
| LY3321367 (Eli Lilly and Co) | IgG1k, Fc silent | 1 | Melanoma, MSI-H and advanced solid cancer | NCT03099109 NCT02791334 |
| MBG453 (Novartis) | Humanised IgG4 mAb | 2 | AML, CMML-2, MDS, GBM | NCT04150029 NCT04266301 NCT03946670 NCT03066648 NCT03940352 NCT02608268 NCT03961971 |
| BGB-A425 (BeiGene) | IgG1, variant, Fc silent | 1/2 | Advanced/ Metastatic solid cancer | NCT03744468 |
| ICAGN02390 (Incyte) | IgG1k, N297A, Fc silent | 1 | Advanced solid cancer, melanoma | NCT03652077 NCT04370704 |
| BMS-986258 (Bristol-Myers Squibb) | IgG1, Fc silent | 1/2 | Advanced solid cancer | NCT03446040 |
| RO7121661 (Hoffman-La Roche) | PD-1/TIM-3 bispecific Ab | 1 | Melanoma, NSCLC, SCLC, ESCC, urothelial cancer | NCT03708328 NCT03869190 |
| B. LAG-3 | ||||
| IMP321 * (Immunopet) | LAG-3-Ig fusion protein | 1/2 | Breast cancer, advanced solid cancers, NSCLC, HNSCC | NCT02614833 NCT03252938 NCT03625323 |
| Relatlimab # (Bristol-Myers Squibb) | IgG4 mAb | 2 | Uveal melanoma, CRC, sarcoma, melanoma | NCT04552223 NCT03642067 NCT04095208 NCT03470922 |
| LAG525 (Novartis) | IgG4 mAb | 2 | Breast cancer, advanced solid cancer, melanoma | NCT03499899 NCT02460224 NCT03742349 NCT03484923 |
| MK-4280 (Merck) | Humanised IgG4 | 1/2 | HL, NHL, BCL, NSCLC, RCC, advanced solid cancer | NCT03598608 NCT02720068 NCT03516981 NCT04626479 NCT04626518 |
| REGN3767 (Regeneron) | IgG4 mAb | 1 | BC, advanced cancers | NCT03005782 NCT01042379 |
| TSR-033 (Tesaro) | Humanised IgG4 mAb | 1 | Advanced solid cancers | NCT03250832 NCT02817633 |
| Sym022 (Symphogen) | Fc inert mAb | 1 | Advanced solid cancer, lymphoma | NCT04641871 NCT03311412 |
| INCAGN02385 (InCyte) | Fc-engineered IgG1k | 1/2 | Melanoma | NCT04370704 |
| MGD013 (MacroGenics) | Humanised LAG3-PD-1 bispecific Ab | 1/2 | GC, HCC, advanced solid cancers | NCT04212221 NCT03219268 NCT04178460 |
| FS118 (F-star) | Humanised LAG3-PD-L1 bispecific Ab | 1 | Advanced solid cancer | NCT03440437 |
| C. TIGIT | ||||
| BMS-986207 (Bristol-Myers Squibb) | IgG1 mAb, FcγR null | 1/2 | EC, OC, Myeloma, advanced solid cancer, | NCT04570839 NCT02913313 NCT04150965 |
| IBI939 (Innovent) | IgG1 mAb | 1 | Advanced cancer | NCT04353830 |
| BGB-A1217 (BeiGene) | Humanized IgG1 mAb | 1 | Metastatic solid tumours | NCT04047862 |
| Tiragolumab † (Genentech) | IgG1 mAb | 1–3 | NSCLC, SCLC, ESCC, BC | NCT03563716 NCT04300647 NCT04543617 NCT04584112 |
| Etigilmab (Mereo BioPharma) | Humanised IgG1 mAb | 1 | Advanced solid cancer | NCT03119428 |
| Vibostolimab (Merck) | Humanised IgG1 mAb | 1/2 | GC, NSCLC, PC, melanoma, | NCT02964013 NCT02861573 NCT04305054 NCT04303169 NCT04165070 NCT04305041 |
| Domvanalimab (Arcus Biosciences) | Humanised IgG1 mAb | 1 | Advanced solid cancer, NSCLC | NCT03628677 NCT04262856 |
| ASP8374 (Potenza) | IgG4 mAb | 1 | Advanced solid cancer | NCT03260322 NCT03945253 |
| Product | Combination(s) | Cancer Type(s) | Seminal Study |
|---|---|---|---|
| A. CTLA-4 | |||
| Ipilimumab | Nil | Melanoma | NCT00094653 |
| (Bristol-Myers Squibb) | Surgery | Melanoma | NCT00636168 |
| Nivolumab | Melanoma | NCT01844505 | |
| Nivolumab | RCC | NCT02231749 | |
| Nivolumab | MSI-H/dMMR CRC | NCT02060188 | |
| Nivolumab | HCC | NCT01658878 | |
| Nivolumab and limited Chemotherapy | NSCLC | NCT03215706 | |
| Nivolumab | Malignant pleural mesothelioma | NCT02899299 | |
| B. PD-1 | |||
| Cemiplimab (Regeneron) | Nil | Cutaneous SCC | NCT02760498 |
| Pembrolizumab | Nil | Melanoma | NCT01866319 |
| (Merck) | Surgery | Melanoma | NCT02362594 |
| Nil | NSCLC | NCT01295827 | |
| NCT02220894 | |||
| Doublet platinum-based Chemotherapy | NSCLC | NCT02039674 | |
| Carboplatin and paclitaxel | NSCLC | NCT02775435 | |
| Axitinib | RCC | NCT02853331 | |
| Nil | MSI-H/dMMR CRC | NCT02563002 | |
| Nil | SCLC | NCT02054806 | |
| NCT02628067 | |||
| Platinum and FU | HNSCC | NCT01848834 | |
| Nil | HNSCC | NCT02358031 | |
| Nil | Gastric cancer | NCT02335411 | |
| Nil | ESCC | NCT02564263 | |
| Nil | HCC | NCT02702414 | |
| Nil | MCC | NCT02267603 | |
| Lenvatinib | Endometrial cancer | NCT02501096 | |
| Nil | cSCC | NCT03284424 | |
| Paclitaxel or gemcitabine and carboplatin | TNBC | NCT02819518 | |
| Nil | Urothelial cancer | NCT02335424 | |
| NCT02625961 | |||
| Nil | PMBCL | NCT02576990 | |
| Nil | Classical HL | NCT02453594 | |
| NCT02684292 | |||
| Nivolumab | Nil | NSCLC | NCT01642004 |
| (Bristol-Myers Squibb) | Nil | RCC | NCT01668784 |
| Nil | Classical HL | NCT02181738 | |
| NCT01592370 | |||
| Nil | HNSCC | NCT02105636 | |
| Nil | Urothelial Carcinoma | NCT02387996 | |
| Nil | MSI-H/dMMR CRC | NCT02060188 | |
| Nil | HCC | NCT01658878 | |
| Surgery | Melanoma | NCT02388906 | |
| Ipilimumab | SCLC | NCT01928394 | |
| Nil | ESCC | NCT02569242 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armitage, J.D.; Newnes, H.V.; McDonnell, A.; Bosco, A.; Waithman, J. Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells 2021, 10, 56. https://doi.org/10.3390/cells10010056
Armitage JD, Newnes HV, McDonnell A, Bosco A, Waithman J. Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells. 2021; 10(1):56. https://doi.org/10.3390/cells10010056
Chicago/Turabian StyleArmitage, Jesse D., Hannah V. Newnes, Alison McDonnell, Anthony Bosco, and Jason Waithman. 2021. "Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression" Cells 10, no. 1: 56. https://doi.org/10.3390/cells10010056
APA StyleArmitage, J. D., Newnes, H. V., McDonnell, A., Bosco, A., & Waithman, J. (2021). Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells, 10(1), 56. https://doi.org/10.3390/cells10010056